Photochemistry of cyclohexane on Cu(111)
Dai
Yamaguchi
a,
Taketoshi
Matsumoto
ab,
Kazuya
Watanabe
ab,
Noriaki
Takagi†
a and
Yoshiyasu
Matsumoto
*ab
aThe Graduate University for Advanced Studies (SOKENDAI), Department of Photoscience, Hayama, Kanagawa, 240-0193, Japan. E-mail: matsumoto@ims.ac.jp
bNational Institutes of Natural Sciences, Institute for Molecular Science, Okazaki, Aichi, 444-8585, Japan
First published on 17th November 2005
Abstract
The photochemistry of cyclohexane on Cu(111) and its excitation mechanism have been studied by temperature-programmed desorption, ultraviolet and X-ray photoelectron spectroscopy. Cyclohexane weakly adsorbed on Cu(111) has been known to show a broadened and redshifted CH stretching band, i.e., CH vibrational mode softening. Although no dehydrogenation takes place thermally on this surface and by the irradiation of photons at 5.0 eV, adsorbed cyclohexane is dissociated to cyclohexyl and hydrogen by the irradiation of photons at 6.4 eV. This is a marked contrast to cyclohexane in the gas phase where the onset of absorption is located at 7 eV. When the surface irradiated by 6.4-eV photons is further annealed, cyclohexyl is dehydrogenated to form cylcohexene that desorbs at 230 K. The systematic measurements of photochemical cross sections at 6.4 eV with linearly polarized light as a function of incident angle indicate that the electronic transition from the highest occupied band of cyclohexane to a partially occupied hybridized band near the Fermi level is responsible for the photochemistry. The hybridized band is formed by the interactions between the electronic states of cyclohexane and the metal substrate. The role of the hybridized band in the photochemistry and the CH vibrational mode softening is discussed.
1. Introduction
In contrast to unsaturated hydrocarbons such as ethylene, acetylene and benzene, the understanding of the adsorption structures and adsorbate-substrate interactions of small saturated hydrocarbons on metal surfaces is much less satisfying. Generally small saturated hydrocarbons adsorb weakly on the surfaces of late transition metals and noble metals. A puzzling feature in the adsorption of saturated hydrocarbons is a broad CH stretching band significantly redshifted from the other symmetric and anti-symmetric CH stretching bands, which was first observed for cyclohexane on Ni(111) and Pt(111).1 This peculiar feature is called CH vibrational mode softening.The origin of the CH vibrational mode softening has been extensively discussed in the past. Raval et al.2–4 suggested that a hydrogen-bonding-like interaction exists between a metal surface and CH groups which lie in close proximity to it. This interaction is understood in terms of two-way electron transfer from a bonding CH σ orbital to unoccupied metal orbitals and from occupied metal orbitals to an anti-bonding CH σ* orbital. On the other hand, Wöll and coworkers found a new peak referred to M* resonance below the Rydberg resonance peak in the spectra of C 1s near edge X-ray absorption fine structure (NEXAFS) of saturated hydrocarbons on metal surfaces.5–7 This indicates the existence of hybrid orbitals between the hydrocarbon and the metal substrate. With the aid of ab initio calculations on cyclopropane8,9 and cyclohexane on Cu(111)9,10 they showed that the hybridization is due to the back-donation of electron into the adsorbed alkane instead of the two-way electron transfer.
Another peculiar observation relevant to the interactions between saturated hydrocarbon and metal is the photochemistry of methane on metal surfaces.11–16 The first excited state of methane where the anti-bonding CH σ* state is completely mixed in the Rydberg series is located at ∼10 eV above the ground state.17 In spite of the high-lying excited state of methane, methane is photolyzed to methyl and hydrogen by the irradiation of 6.4-eV photons when it adsorbs on Pt(111),11–14 Pd(111)15 and Cu(111)16 surfaces. The careful measurements of photochemical cross sections with various incident angles and polarizations of the UV light strongly indicate that the excitation responsible for the photochemistry is from the highest occupied molecular orbital (HOMO) state to the unoccupied state near the Fermi level. The excited state involved in the photochemistry was proposed to be the hybridized state between the lowest unoccupied molecular orbital with the CH anti-bonding character and metal unoccupied orbitals.11 This hybridization scheme was supported later by ab initio calculations.18
Thus, the interactions of saturated hydrocarbon with metal surfaces now start to become clear. The peculiar phenomena, i.e., the CH vibrational mode softening and the UV-photochemistry of methane, appear to have the same cause. That is, the existence of the partially occupied band around the Fermi level, which is caused by the hybridization of electronic states between hydrocarbon and metal. If so, cyclohexane on metal surfaces is also expected to show a transition from the HOMO state to the hybridized state, leading to photochemistry as in the case of methane. This motivates us to study the photochemistry of cyclohexane on metal surfaces by photons whose energies are below the absorption threshold of cyclohexane in the gas phase.
Cyclohexane is one of the most extensively studied saturated hydrocarbons on metal surfaces. As stated earlier, cyclohexane on various metal surfaces shows the CH vibrational mode softening. In particular, the adsorption structure on Cu(111) was studied in detail by infrared reflection absorption spectroscopy (IRAS),2,4,9,10 high-resolution electron energy loss spectroscopy (HREELS)2–4 and low energy electron diffraction (LEED).4 According to those studies, cyclohexane in small coverages retains the chair configuration and adsorbs on Cu(111) in C3v symmetry with the carbon skeleton parallel to the surface. The stretching mode of the CH bonds pointing to the surface shows the redshift of Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ∼ 130 cm−1, which is much smaller than those on other single crystal surfaces such as Pt(111)
(Δ
∼ 310 cm−1) and Ru(001)
(Δ
∼ 340 cm−1).1,19–21 Thus, the adsorbate–substrate interactions on Cu(111) are not strong such that no dehydrogenation occurs thermally.
∼ 130 cm−1, which is much smaller than those on other single crystal surfaces such as Pt(111)
(Δ
∼ 310 cm−1) and Ru(001)
(Δ
∼ 340 cm−1).1,19–21 Thus, the adsorbate–substrate interactions on Cu(111) are not strong such that no dehydrogenation occurs thermally.
The studies on nonthermal excitation of cyclohexane on metals are scarce. Koel and coworkers studied electron-induced chemistry of cyclohexane on Pt(111)22 and Au(111)23 surfaces. They confirmed that cyclohexyl is formed by bombarding the surface by electrons at ∼30 eV. The cross section for electron-induced dissociation of cyclohexane in multilayers is significantly higher than that in the first layer. This indicates that the excited state lifetime is substantially reduced by the interaction with the metal surfaces.
In this paper, we report the photochemistry of cyclohexane on Cu(111) with photons at 5.0 and 6.4 eV. Cyclohexane in the gas phase is transparent in the range of photon energy.24 However, we show that cyclohexane is dissociated to produce cyclohexyl and hydrogen by the irradiation of 6.4 eV photons. The systematic measurements of photochemical cross sections with linearly polarized light indicates that the excitation occurs by the transition from the HOMO state of cyclohexane to the hybridized state between cyclohexane and surface Cu atoms. We also discuss the implication of the electronic state mixing for the CH vibrational mode softening.
2. Experimental
Experiments were mainly performed in an ultrahigh vacuum (UHV) apparatus (base pressure ≤2 × 10−10 Torr) that has been described in detail elsewhere.11 Briefly, it is equipped with a four-grids retardation electron analyzer for LEED, an X-ray gun and a hemisphere electron analyzer for X-ray photoelectron spectroscopy (XPS), a differentially pumped quadrupole mass spectrometer (QMS), an ion gun, a high precision sample manipulator, and laser inlet/outlet ports with CaF2 windows. The measurements of ultraviolet photoelectron spectroscopy (UPS) were performed in a different UHV apparatus made of μ-metal equipped with a He-discharge lamp and a hemisphere electron analyzer.A Cu(111) crystal with dimensions of 10 mm (diameter) × 1.0 mm (thickness) was cleaned by the repeating of many cycles of Ar-ion sputtering at room temperature, flashing to 1000 K, and annealing at 850 K. The sample could be cooled to 100 K with liquid nitrogen and resistively heated above 1000 K. The surface cleanness was examined by XPS.
Cyclohexane (Wako, HPLC grade) was purified by the freeze–pump–thaw method prior to introducing to the UHV chamber. The surface was exposed to cyclohexane at the surface temperature of 150 K to make a saturated first layer or at 100 K to make multilayers. The exposure of 6.0 L (1 L = 10−6 Torr s) at 150 K was used to make a perfectly saturated first layer of cyclohexane. The saturated adsorption was confirmed by temperature-programmed desorption (TPD) and XPS measurements.
Unless noted specifically, nonpolarized ultraviolet light from an excimer laser (ArF: 193 nm, 6.4 eV and KrF: 248 nm, 5.0 eV) was directed onto the surface at 100 K. Laser pulses (9 mJ cm−2, 15 ns) at the repetition rate of 10 Hz did not raise the surface temperature over 130 K during photon irradiation. The maximum transient temperature jump due to the pulse-laser heating was estimated to be 17 K at the incident angle of 0°, which does not exceed the desorption temperature of cyclohexane, 180 K. The fraction of cyclohexane desorbed by this temperature jump was estimated to be less than 10−5.25 Thus, laser-induced thermal desorption was negligible. For measurements of the polarization-dependent cross sections of photochemical processes, the incident light was polarized with a polarizer composed of five pairs of UV-grade fused quartz plates placed at the Brewster angle at 193 nm. Post-irradiation XPS was used to quantify the coverages of hydrocarbon adsorbates as a function of the number of accumulated photons incident on the unit surface area, Nph.
3. Results and discussion
3.1. Adsorption state
The adsorption state and structure of cyclohexane on Cu(111) have been studied by various techniques in the past.2,4,9,10 Here, we confirmed the adsorption state of cyclohexane on the Cu(111) surface by TPD and UPS. Fig. 1 shows TPD results as a function of cyclohexane coverage, θA, recorded by monitoring the mass signals at m/z = 56. The clean surface at 150 K was exposed to cyclohexane. The desorption peak from the surface exposed to 2.0 L cyclohexane appears at ∼175 K and its peak intensity is saturated with the exposure of 6.0 L. We refer this saturation coverage to 1 monolayer (1 ML). The peak temperature increases with coverage while the leading edges of the desorption peaks coincide with each other. This is a typical feature of the zeroth order desorption kinetics. Thus, cyclohexane may form islands and the adsorbates at the periphery are less bound than those inside the islands. The H2-TPD results (not shown) do not show any desorption peaks except for the peak at ∼175 K due to the cracking of cyclohexane in the ionization chamber of the QMS. Furthermore, the intensity of the C 1s peak is negligible after the sample is annealed over the desorption temperature of cyclohexane. Thus, we confirmed the previous results that cyclohexane only desorbs and does not proceed any chemical reactions on Cu(111) when the surface is annealed. | ||
| Fig. 1 TPD results of cyclohexane from Cu(111) as a function of exposure. The surface at 150 K was exposed to (a) 2, (b) 4, and (c) 6 L of cyclohexane. Signals were detected at m/z = 56. The heating rate was 2 K s−1. | ||
Fig. 2 shows UP spectra of cyclohexane on Cu(111) as a function of θA. The spectrum taken from the clean surface shows a peak of the s,p-derived surface state at the binding energy Eb = 0.32 eV and the peaks of d bands in 2.5 < Eb < 4.8 eV. The peak at Eb = 1.02 eV is due to the d bands excited by the He Iβ resonance line. The intensities of these bands decrease with the increase of θA and they are completely quenched at θA = 1 ML. On the other hand, the new peaks at 5.2, 6.2, 7.5, and 9.0 eV grow with the increase of θA. These are the bands of cyclohexane originating in CH and CC bonding orbitals. The spectrum at θA = 1.5 ML is identical to those obtained from the condensed layers of cyclohexane on Ni(111)26 and Ru(001).19 As stated earlier, the extra peak originating in the adsorption-induced state due to the hybridization between the orbitals of cyclohexane and the copper substrate may exist near the Fermi level. However, we could not detect the feature in the UP spectra. This may be due to the small density-of-states of the band below the Fermi level.
 | ||
| Fig. 2 He I UP spectra as a function of the coverage of cyclohexane adsorbed on Cu(111) at 100 K. (a) 0, (b) 0.25, (c) 0.5, (d) 1.0, (e) 1.5 ML. The surface state at Eb = 0.36 eV and the Cu d band in Eb = 2.5–4.8 eV completely disappear when the first layer of cyclohexane is completed. | ||
Together with the results of IRAS,2,4,9,10 HREELS,2–4 and LEED4 done in the past, we summarize the adsorption state and structure of cyclohexane on Cu(111). Below θA ≤ 0.14 ML cyclohexane in the chair configuration adsorbs in C3ν symmetry with its carbon skeleton parallel to the surface. In 0.14 ≤ θA ≤ 1 ML, cyclohexane tilts its carbon skeleton towards the surface normal. Cyclohexane adsorbed on Cu(111) simply desorbs and does not decompose thermally.
3.2. Photochemistry
Fig. 3 shows the post-irradiation TPD results as a function of the number of photons irradiated, Nph. After the Cu(111) surface covered with 1 ML cyclohexane was irradiated by the desired number of 6.4 eV photons, cyclohexane and H2-TPD results were recorded by monitoring the mass signals at m/z = 56 and 2, respectively. The desorption peak of cyclohexane at 180 K decreases with Nph. On the other hand, a new peak appears at 350 K in the H2-TPD result. This peak is attributed to the associative desorption of hydrogen.27 Thus, it is clear that cyclohexane is dehydrogenated by the irradiation of 6.4 eV photons. | ||
| Fig. 3 TPD results of cyclohexane (m/z = 56) from Cu(111) with 1 ML cyclohexane (a) before and (b) after irradiation of 6.4-eV photons. (c) Post-irradiation TPD of H2 (m/z = 2). The surface at 150 K was initially exposed to 6 L cyclohexane. The number of photons irradiated was 1.5 × 1020 cm−2. The heating rate was 2 K s−1. | ||
The post-irradiation TPD measurements were performed at m/z = 78 (benzene), 67 (cyclohexene), and 80 (cyclohexadiene). No detectable signals were obtained for benzene and cyclohexadiene, but the desorption peak of cylcohexene was observed at 230 K as shown in Fig. 4(b). At this mass number the desorption peak of cyclohexane at 175 K was also observed because of its cracking in the mass chamber. To examine whether or not cyclohexene is a primary photo-product, we measured TPD at m/z = 67 from the Cu(111) surface covered with cyclohexene only. As shown in Fig. 4(c), cyclohexene desorbs from the Cu(111) surface at 185 K. This desorption temperature is lower than that observed in the post-irradiation TPD by 45 K. Therefore, cyclohexene is not the primary photo-product. Consequently, we conclude that the primary photo-product is cyclohexyl that is further dehydrogenated to produce cyclohexene by heating. This finding is consistent with the electron-induced chemistry of cyclohexane on Au(111),23 where cyclohexyl was identified by IRAS. The C 1s XPS measurements after cyclohexene is desorbed indicate no hydrocarbons are left over. Thus, cyclohexene converted from cyclohexyl does not proceed further dehydrogenation but desorbs molecularly at 230 K.
 | ||
| Fig. 4 TPD results of cyclohexene from Cu(111) recorded at m/z = 67 (a) before and (b) after irradiation of 6.4-eV photons. The number of photons irradiated was 1.5 × 1020 cm−2. The surface at 150 K was initially exposed to 6 L cyclohexane. (c) TPD result of cyclohexene taken from Cu(111) covered with cyclohexene. The surface at 100 K was exposed to 4 L cyclohexene. The desorption peak of cyclohexene at 230 K in (b) is due to the dehydrogenation of cyclohexyl produced by photodissociation of cyclohexane. | ||
The progress of the photochemistry was monitored by C 1s XPS as a function of Nph. In Fig. 5, the open circle represents the result measured right after the sample was irradiated by the desired number of 6.4 eV photons, corresponding to the sum of coverages of cyclohexyl and unreacted cyclohexane. The closed circle represents the coverage of hydrocarbon after the photo-irradiated surface was annealed to 180 K for 1 min. This procedure is to remove unreacted cyclohexane and keep only the product, cyclohexyl, on the surface. Thus, the difference between the two results plotted with the open triangle is the coverage of unreacted cyclohexane. The coverage of cyclohexane decreases with Nph, but is not completely reduced to zero, i.e., it levels off at θA(∞) ∼ 0.3 ML. Assuming the first order process for dissociation and desorption of cyclohexane with the effective cross sections of σdis and σdes, respectively, we fitted the coverages of unreacted cyclohexane, θA(Nph), and cyclohexyl, θB(Nph), to the following equations, respectively,
| θA(Nph) = (θA(0) − θA(∞))exp(−σTNph) + θA(∞) | (3.1) |
 | (3.2) |
 | ||
| Fig. 5 Changes in the adsorbate coverages as a function of the number of photons irradiated at 6.4 eV. Coverages were estimated from the integrated area of the C 1s peak in XPS. Coverages of (a) hydrocarbons (cyclohexane and cyclohexyl) right after UV irradiation (open circles), (b) unreacted cyclohexane (open triangles), and (c) cyclohexyl (closed circles). The solid curves are the fitting results. | ||
The studies on the electron-induced chemistry at 30 eV indicate that cyclohexane in multilayers show substantially larger cross sections than one in the first layer.22,23 We also examined how the photochemical cross section at 6.4 eV is changed by cyclohexane in multilayers. Following the same procedure to estimate the coverage of cyclohexyl cited in the previous paragraph, we plot in Fig. 6 the coverage of cyclohexyl produced on the surface with θA(0) = 2 ML (closed circles) in addition to those on the surface with θA(0) = 1 ML (open circles) as a function of Nph. The growth of the cyclohexyl coverage is not affected by the existence of the multilayer. This suggests that only cyclohexane in the first layer is photo-active and molecules in the multilayer do not contribute to the product formation. This is a marked difference from the electron-induced chemistry. The direct interaction of cyclohexane with the surface is essential for the photochemistry at 6.4 eV to occur.
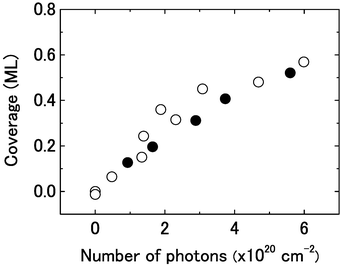 | ||
| Fig. 6 Comparison of the cyclohexyl yields for the initial cyclohexane coverage of 1 ML (open circles) with those of 2 ML (closed circles) as a function of the number of photons irradiated at 6.4 eV. | ||
Irradiation with 5.0-eV photons was also performed, but no appreciable photochemistry was observed. The depletion of the C 1s intensity of cyclohexane was under the experimental error, i.e <5%. Thus, we estimated σT to be <6 × 10−23 cm2 at 5.0 eV.
3.3. Excitation mechanism
Cyclohexane in the gas phase shows an absorption band to the series of Rydberg states in the energy range above 7 eV. The absorption cross section is 2 × 10−22 cm2 at 7.0 eV and increases with photon energy.24 Thus, isolated cyclohexane is basically transparent in the photon energy range utilized in the current study. If the photochemistry observed in this study is due to the electronic excitation of cyclohexane, the excited state involved in the transition has to be lowered by the hybridization between the electronic states of cyclohexane and the metal substrate. Otherwise, the photochemistry is induced by the electronic excitation of the substrate, i.e., the substrate-mediated excitation.In order to clarify this point, we measured how the cross section depends on the polarization (p or s) and the incident angle γ of the excitation light.11 If the excitation is due to the optical transition in the adsorbate, the cross section should be proportional to |![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) ·
·![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) |2, where is a transition dipole moment fixed to the molecular frame and is the electric vector of the excitation light at the surface. On the other hand, if the excitation is due to hot electron transfer from the surface to cyclohexane following the creation of electron-hole pairs owing to absorption of photons by the substrate, the cross section is proportional to the absorbance of the substrate. Fig. 7 shows the variations of the electric field strength, 〈Ei2〉/cos γ
(i
=
x,y,z), and the substrate absorbance, Ai(i
= p,s), as a function of the incident angle estimated by Fresnel’s equations28 with the refractive index of n
= 0.96 and the extinction coefficient k
= 1.37.29 Here, the electric field strength is divided by cos γ, since this correction factor is necessary when calculated results are directly compared with experimental ones normalized to the intercepted irradiance. Note that this method is useful only if adsorbate has a transition dipole component along the surface normal, since the incident-angle dependence of the electric field strength along the surface plane is very similar to that of absorbance of the substrate.
|2, where is a transition dipole moment fixed to the molecular frame and is the electric vector of the excitation light at the surface. On the other hand, if the excitation is due to hot electron transfer from the surface to cyclohexane following the creation of electron-hole pairs owing to absorption of photons by the substrate, the cross section is proportional to the absorbance of the substrate. Fig. 7 shows the variations of the electric field strength, 〈Ei2〉/cos γ
(i
=
x,y,z), and the substrate absorbance, Ai(i
= p,s), as a function of the incident angle estimated by Fresnel’s equations28 with the refractive index of n
= 0.96 and the extinction coefficient k
= 1.37.29 Here, the electric field strength is divided by cos γ, since this correction factor is necessary when calculated results are directly compared with experimental ones normalized to the intercepted irradiance. Note that this method is useful only if adsorbate has a transition dipole component along the surface normal, since the incident-angle dependence of the electric field strength along the surface plane is very similar to that of absorbance of the substrate.
 | ||
| Fig. 7 Dependence of the electric field strength 〈Ei2〉/cos γ (i = x,y,z) and the substrate absorbance Ai (i = p,s) on the incident angle (γ) for p- and s-polarization light (hν = 6.4 eV) impinging on a copper surface. The results are calculated from Fresnel’s equations with n = 0.96 and k = 1.37. | ||
Fig. 8 shows a plot of σT measured with p- and s-polarized light as a function of the incident angle. The cross sections are normalized at γ = 0°. It is remarkable that the cross sections with p-polarized light deviates from the absorbance of the substrate. This indicates clearly that the electronic transition in the adsorbate plays a central role in the photochemistry. According to the UP spectra (Fig. 2), the edge of the HOMO level is located around 5 eV. Thus, since the photochemistry takes place at 6.4 eV, but not at 5.0 eV, the appreciable density of states of the unoccupied electronic states responsible for the electronic transition has to be located at right above the Fermi level.
 | ||
| Fig. 8 Incident angle dependence of photochemical cross-sections obtained with p- (open circles) and s-polarized light (closed circles). The cross sections were determined by the post-irradiation XPS as a function of the number of photons irradiated. The dotted and dashed–dotted curves are calculated absorbance of a copper substrate for p- and s-polarized light, respectively. The solid and dashed curves represent the best fitting result of the observed cross sections by p- and s-polarized light in terms of the transition dipole and the electric field strengths at the surface. The data are scaled by the values at γ = 0°. | ||
Assuming that the oriented transition dipoles are equally distributed with three equivalent azimuthal directions as a result of the symmetry of the substrate, i.e., C3, we can describe the cross sections as,28
| σp ∝ [0.5 sin2θ〈Ex2〉 + cos2θ〈Ez2〉]/cos γ, | (3.3) |
| σs ∝ [0.5 sin2θ〈Ey2〉]/cos γ, | (3.4) |
The photochemistry of cyclohexane on Cu(111) is very similar to that of methane on metal surfaces with respect to a number of points in the following.11–16 (i) The hydrocarbons are dehydrogenated by irradiation of UV photons that are not absorbed in the gas phase. (ii) The photochemistry is induced by the excitation of electronic states of the adsorbates from HOMO to the unoccupied states just above the Fermi level. (iii) The hydrocarbons at the initial coverage of 1 ML is not completely dissociated, i.e., the photochemistry is quenched at a certain coverage of the photo-product.
The similarities originate in the electronic structure and the interactions with the metal substrates common in saturated hydrocarbons. The excited state of saturated hydrocarbons with the CH anti-bonding character is located at the energy range close to the ionization potential. Thus, the excited state is buried in the Rydberg series. Ab initio calculations indicate that this excited state of the hydrocarbons is hybridized with the substrate electronic states.9,10,18 In fact, Wöll and coworkers have shown that the Rydberg resonance located at 287.7 eV is strongly quenched and a distinct new resonance at 285.1 eV, the M* resonance, in the NEXAFS spectra of cyclohexane adsorbed on Cu(111).5–7 The M* resonance was attributed to the transition from the C 1s state to the band caused by the hybridization between the electronic states of cyclohexane and the metal substrate (referred to the M* band hereafter). Furthermore, the occurrence of the M* resonance correlates with the CH vibrational mode softening. Since the broad M* band is located close to the Fermi level, a part of M* band is occupied, i.e., back-donation from filled metal states to the band with the CH anti-bonding character of cyclohexane. Thus, the electronic excitation from the HOMO band to the unoccupied part of the M* band is possible by 6.4 eV photons. Consequently, the CH vibrational mode softening and the UV photochemistry of the saturated hydrocarbons have the origin in common, i.e., the M* band.
Although no CH vibrational mode softening occurs in the adsorption of methane on Pt(111), the symmetric CH stretching band was observed in IRAS spectra which should not appear if methane retains Td symmetry.30 The appearance of this band is also a manifestation of the interactions of the saturated hydrocarbon with the metal substrate.
The excited-state potential surface along the CH coordinate at the equilibrium distance of the softened CH bond of cyclohexane in the ground state is likely repulsive because of electron occupation in the M* band with the CH anti-bonding character and the hole occupation in the band with the CH bonding character. Thus, upon the excitation from the HOMO band to the M* band, the CH bond starts elongating. As the CH bond is elongated, the hydrogen atom mainly gains kinetic energy on the excited-state potential surface. However, CH dissociation will not be completed on the potential energy surface, since the excited state is likely quenched rapidly back to the ground state potential surface via electron transfer from the M* band to the bulk and electron back transfer from the bulk to the CH bonding band. Thus, only a fraction of excited cyclohexane adsorbate that gains enough kinetic energy in the excited state to overcome the CH dissociation barrier in the ground state potential surface can be dissociated.
The photochemistry does not further proceed to dehydrogenate cyclohexyl. This result can be understood in terms of the adsorption structure of cyclohexane and cyclohexyl. Since cyclohexane is adsorbed in the tilted configuration at 1 ML,4 the only one CH bond is in close proximity to the metal surface. Thus, this CH bond is dissociated by UV irradiation. Produced cyclohexyl also likely adsorbs in a tilted configuration as in the case of Pt(111),31 in which the carbon atom lost a hydrogen atom directly bonds to the surface. In the tilted configuration, no CH bonds of cyclohexyl can be in the close proximity to the surface, resulting in the interactions between CH moieties and the metal substrate weak so that the M* band no longer exists. This prohibits the photochemistry at 6.4 eV to occur.
The photochemistry is quenched when ∼0.7 ML of cyclohexane is dehydrogenated. This may be caused by the changes in the adsorption structure of cyclohexane while the photochemistry progresses. Cyclohexane produces a cyclohexyl and a hydrogen via the photochemistry. The photo-products remained on the surface may cause the repulsive interactions with unreacted cyclohexane. This makes the distance between cyclohexane and the metal surface longer, resulting in the weaker interactions between cyclohexane and the metal surface. Since the interactions responsible for the photochemistry and the CH vibrational mode softening are realized in the subtle balance between dispersive attraction and Pauli repulsion, the small repulsive force from the adsorbates surrounding unreacted cyclohexane makes a profound effect on the photochemistry and the CH vibrational mode softening.
Another possible origin for the quenching of the photochemistry is increase in the dissociation barrier. When the photochemistry progresses, the number of empty sites decreases with increase of the coverages of cyclohexyl and hydrogen. Therefore, it is more difficult for unreacted cyclohexane to find out the sites available for its photo-products to be adsorbed. Since the interactions with the substrate atoms are essential for lowering the dissociation barrier for cyclohexane compared with that in the gas phase, the poisoning by the preoccupied photo-products makes the dissociation barrier higher.
4. Conclusion
Cyclohexane weakly adsorbed on Cu(111) is dissociated by 6.4-eV photons to produce cyclohexyl and hydrogen. The incident-angle dependence of photochemical cross sections measured with linearly polarized light implies that the excitation responsible for the photochemistry is not photon absorption by the substrate, but the electronic transition from the HOMO band of cyclohexane to a partially occupied band of the adsorption system located near the Fermi level. This is the same excitation mechanism found in the photochemistry of methane on a number of metal surfaces. The important implication of the photochemistry is that the partially occupied band near the Fermi level are also responsible for the CH vibrational mode softening and the M* resonance observed in the NEXAFS measurements. The partially occupied state is formed by the interactions between cyclohexane and metal substrate. Because the existence of the interactions is evident from the simplest saturated hydrocarbon, i.e., methane, to cyclic and long-chained alkanes, such as n-octane,32 it seems that they are ubiquitous in the adsorption systems of saturated hydrocarbons on metal surfaces.Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research on Priority Area (417), Creative Scientific Research Collaboratory on Electron Correlation-Toward a New Research Network between Physics and Chemistry (13NP0201) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and KAKENHI (17105001) from Japan Society for the Promotion of Science (JSPS).References
- J. E. Demuth, H. Ibach and S. Lehwald, Phys. Rev. Lett., 1978, 40, 1044–1047 CrossRef CAS.
- M. A. Chesters, S. F. Parker and R. Raval, J. Electron Spectrosc. Relat. Phenom., 1986, 39, 155–162 CrossRef CAS.
- R. Raval and M. A. Chesters, Surf. Sci. Lett., 1989, 219, L505–L514 CrossRef CAS.
- R. Raval, S. F. Parker and M. A. Chesters, Surf. Sci., 1993, 289, 227–236 CrossRef CAS.
- K. Weiss, J. Weckesser and C. Wöll, J. Mol. Struct. (THEOCHEM), 1999, 458, 143–150 CAS.
- C. Wöll, J. Synchrotron Radiat., 2001, 8, 129–135 CrossRef CAS.
- G. Witte, K. Weiss, P. Jakob, J. Braun, K. L. Kostov and C. Wöll, Phys. Rev. Lett., 1998, 80, 121–124 CrossRef CAS.
- C. Wöll, K. Weiss and P. Bagus, Chem. Phys. Lett., 2000, 332, 553–561 CrossRef CAS.
- K. A. Fosser, R. G. Nuzzo, P. S. Bagus and C. Wöll, Angew. Chem., Int. Ed., 2002, 41, 1735–1737 CrossRef CAS.
- K. A. Fosser, R. G. Nuzzo, P. S. Bagus and C. Wöll, J. Chem. Phys., 2003, 118, 5115–5131 CrossRef CAS.
- Y. Matsumoto, Y. A. Gruzdkov, K. Watanabe and K. Sawabe, J. Chem. Phys., 1996, 105, 4775–4788 CrossRef CAS.
- Y. A. Gruzdkov, K. Watanabe, K. Sawabe and Y. Matsumoto, Chem. Phys. Lett., 1994, 227, 243–247 CrossRef CAS.
- K. Watanabe, K. Sawabe and Y. Matsumoto, Phys. Rev. Lett., 1996, 76, 1751–1754 CrossRef CAS.
- Y. A. Gruzdkov, K. Watanabe and Y. Matsumoto, Surf. Sci., 1996, 363, 195–203 CrossRef CAS.
- K. Watanabe and Y. Matsumoto, Surf. Sci., 1997, 390, 250–255 CrossRef CAS.
- K. Watanabe and Y. Matsumoto, Surf. Sci., 2000, 454–456, 262–266 CrossRef CAS.
- M. B. Robin, Higher Excited States of Polyatomic Molecules, Academic Press, Inc., Orlando, FL, 1985, vol. 3 Search PubMed.
- Y. Akinaga, T. Taketsugu and K. Hirao, J. Chem. Phys., 1997, 107, 415–424 CrossRef CAS.
- F. M. Hoffmann, P. A. Thiel and W. H. Weinberg, Surf. Sci., 1983, 130, 173 CrossRef CAS.
- G. D. Waddill and L. L. Kesmodel, Chem. Phys. Lett., 1986, 128, 208–213 CrossRef CAS.
- M. Weldon, P. Uvdal, C. Friend and B. Wiegand, Surf. Sci., 1996, 355, 71–84 CrossRef CAS.
- C. Xu and B. E. Koel, Surf. Sci. Lett., 1993, 292, L803–L809 Search PubMed.
- D. Syomin and B. E. Koel, Surf. Sci., 2002, 489, 61–73 CrossRef.
- J. W. Raymonda, J. Chem. Phys., 1972, 56, 3912–3920 CrossRef CAS.
- D. Burgess Jr., P. C. Stair and E. Weitz, J. Vac. Sci. Technol. A, 1986, 4, 1362–1366 CrossRef.
- W. Huber, P. Zebisch, T. Bornemann and H. P. Steinruck, Surf. Sci., 1990, 239, 353–362 CrossRef CAS.
- G. Anger, A. Winkler and K. D. Rendulic, Surf. Sci., 1989, 220, 1–17 CrossRef CAS.
- X.-L. Zhou, X.-Y. Zhu and J. White, Surf. Sci. Rep., 1991, 13, 73–220 CrossRef CAS.
- Handbook of Optical Constants of Solids, ed. E. D. Palik, Academic, Orlando, FL, 1985 Search PubMed.
- J. Yoshinobu, H. Ogasawara and M. Kawai, Phys. Rev. Lett., 1995, 75, 2176–2179 CrossRef CAS.
- M. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2003, 125, 11131–11135 CrossRef CAS.
- H. Öström, L. Triguero, K. Weiss, H. Ogasawara, M. G. Garnier, D. Nordlund, M. Nyberg, L. G. M. Pettersson and A. Nilsson, J. Chem. Phys., 2003, 118, 3782–3789 CrossRef CAS.
Footnote |
| † Present address: Department of Advanced Materials Science, University of Tokyo, Kashiwa, Chiba, 277-8561, Japan. |
| This journal is © the Owner Societies 2006 |