DOI:
10.1039/B515930J
(Paper)
CrystEngComm, 2006,
8, 75-83
Synthesis and solid state structure for a series of poly(1-pyrrolylmethyl)benzene derivatives. Control of the interplaying π–π and C–H⋯π interactions?†
Received
9th November 2005
, Accepted 9th January 2006
First published on 12th January 2006
Abstract
The syntheses of new hexa- and 1,2,4,5-tetra-1-pyrrolylmethyl-benzene (1 and 2, respectively) compounds and their crystal structures, along with those for the related disubstituted o-, m- and p-di-1-pyrrolylmethyl-benzene (3–5) derivatives are reported. The arrangements of molecules in the 1-D structure for 2–5 are controlled by the interplay of two different weak interactions: π–π and C–H⋯π
(Ph) interactions. The absence of such interactions in the packing of 1 seems to be related to the arrangement of the pyrrolylmethyl arms with respect to the benzene core, which prevents the π cloud of the aromatic ring to be part of any intermolecular interaction. In addition, C–H⋯π
(pyrrolyl) interactions are found in the 2-D structure for most of the compounds in the solid state.
Introduction
Non-bonding intermolecular interactions are of fundamental importance for understanding molecular recognition phenomena, biological processes and physical and chemical properties of new materials.1 The understanding and control of these intra- and intermolecular forces and of the supramolecular self-assembly process is a prerequisite for the progress of crystal engineering.2 Among these, the stacked (facial) arrangements of aromatic–aromatic molecules are of particular interest as π–π interactions (Fig. 1). The latter interactions are intermolecular forces similar to hydrogen bonding, whose nature is still a matter of discussion.3 In the arrangement of π–π stacking aromatic rings, one can generally distinguish two different situations (Fig. 1, top), that is, face-to-face (I) and offset or slipped (II). The distance between the arene planes is taken as the criterion to suggest π stacking and this lies between 3.3 to 3.8 Å.3 Several theoretical3f studies and geometrical analysis of crystal structures3d show that the stacked arrangement (I) is unfavorable and the offset (II) is favorable. A simple picture of the electronic distribution of benzene possessing a positively charged core (σ framework) surrounded by a negatively charged periphery (π electrons), rationalizes these results on an electrostatic basis (Fig. 1, bottom).3a,4 Thus, it is assumed that the offset displacement of parallel aromatic rings is the result of dominant π–σ attraction over π–π repulsion. For example, larger systems such as pyrene or coronene show an offset π stacking with the hydrogens roughly over ring centers. However, it has been found that in the case of crystal structures containing benzyl groups, the observed offset π stacking of the arene rings seems to be due to the combination of π stacking plus C–H⋯π
(Ph) interactions between methylene hydrogens and the aromatic ring, which has been named as hybrid interactions
(Fig. 2).5 This raises the question of whether stacking of the aromatic rings prevails over or is influenced by other weak interactions such as, e.g., C–H⋯π hydrogen bonds.
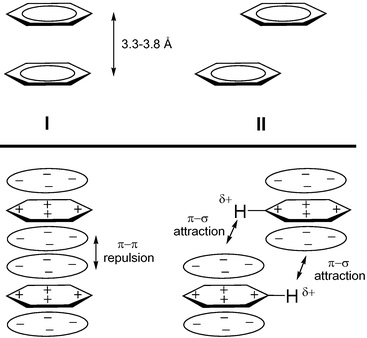 |
| | Fig. 1 Typical arrangements of π–π interacting aromatic rings in the solid state (above bold line) and schematic illustration of electrostatic interactions according to the Hunter and Sanders’ model (below bold line). Adapted from ref. 3d with permission from The Royal Society of Chemistry. | |
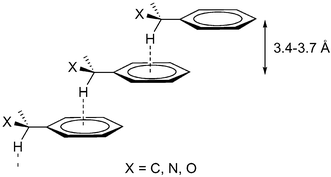 |
| | Fig. 2 Hybrid interactions found in a CSD search for X-benzyl groups (see text for details). | |
Over the recent years there has been increasing interest in polysubstituted benzene derivatives, the so-called “multi-armed” molecules, where all substituents are usually the same.6 An important consideration in the design of such compounds is how the arms will arrange themselves around the benzene unit. Various studies have shown that the most favoured arrangement, on steric grounds, is one where all adjacent arms are on opposite sides of the plane of the core unit. Pioneering work by MacNicol already addressed this point and even predicted that tri-substituted aromatic compounds would form π–π stacked dimers.6a,b However, it is suprising that even though many of such multi-armed molecules have been prepared as molecular platforms for supramolecular chemistry and their crystal structures have been reported since MacNicol's work, their crystal packings and intermolecular interactions have hardly been described in detail.6h,7
We wish to report here the syntheses and crystal structures of two new multi-armed molecules, which have been prepared as part of our studies on modification of polymer structure by using multi-pyrrolyl monomers.8 The new molecules, hexa(1-pyrrolylmethyl)benzene (1) and 1,2,4,5-tetra(1-pyrrolylmethyl)benzene (2), contain a benzene core to which six or four pyrrolylmethyl arms, respectively, are appended (Fig. 3). We report the X-ray crystal structures of 1–2 and those for the previously synthesized family of di-pyrrolyl monomers, namely di(1-pyrrolylmethyl)-o-benzene (3), di(1-pyrrolylmethy)l-m-benzene (4) and di(1-pyrrolylmethyl)-p-benzene (5)
(Fig. 3).9 A comparison of the X-ray structures and crystal packings shows that whereas packing for 1 seems to be dominated by van der Waals or electrostatic interactions other than π–π interactions, those for 2–5 are controlled by a combination of π–π and C–H⋯π
(Ph) interactions.
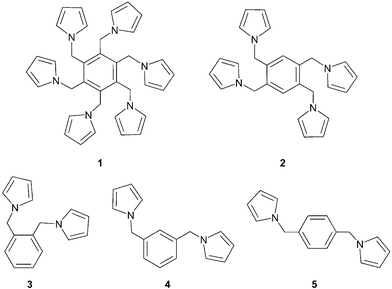 |
| | Fig. 3 Schematic drawing of compounds 1–5. | |
Results
Whereas disubstituted benzene derivatives with bulky substituents are easily accessible, the corresponding polysubstituted derivatives can be synthetically challenging, due to steric repulsions.
However, the new hexa(1-pyrrolylmethyl)benzene (1) and 1,2,4,5-tetra(1-pyrrolylmethyl)benzene (2) compounds were easily prepared in high yields (92–96%) by direct reaction of the pyrrole potassium salt in anhydrous THF with the corresponding hexabromide derivative, according to the general Scheme 1. Compounds 1–2 have been fully characterized by standard methods and their structures has been confirmed by X-ray diffraction.
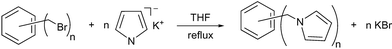 |
| | Scheme 1 | |
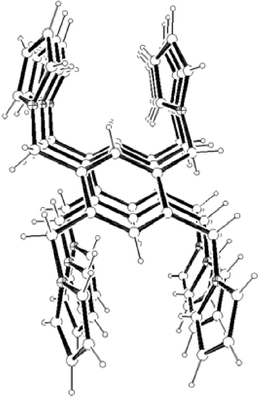
Crystals of 1 suitable for X-ray analysis were obtained from an acetonitrile solution. Fig. 4 shows a perspective view of one of the two crystallographically independent centrosymmetric molecules. The pyrrolylmethyl substituents alternate “a(bove)” and “b(elow)” the plane of the benzene ring in an ababab pattern, which minimises steric constraints, and similar to other hexasubstituted benzenes.6 Thus, as can be seen in Fig. 4, three arms of the molecule are above the benzene plane and three below it. Taking the distance between the benzene plane (C1a–C3ai) and the N atoms of the pyrrolylmethyl arms as a reference, the N atoms are 1.12–1.18 Å above (N8a, N20a, N14ai) or below (N8ai, N20ai, N14a) the benzene plane in 1.
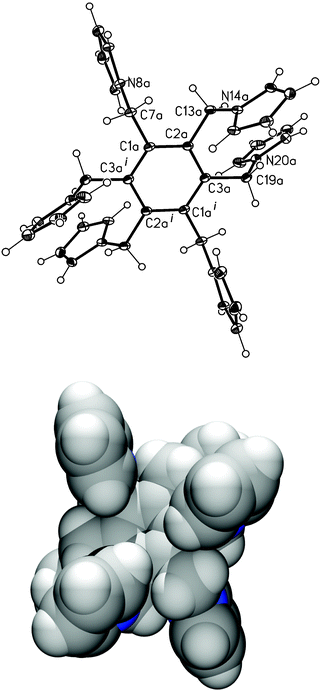 |
| | Fig. 4 View of the molecular structure of one of the two independent centrosymmetric molecules (molecule 1a) of compound 1 with numbering scheme (top, thermal displacement ellipsoids set at 20% probability level; superscript i refers equivalent position −x
+ 2, −y
+ 1, −z
+ 1) and a space filling diagram (bottom). | |
X-ray analysis of crystals for 2, which were obtained from a THF solution, shows a surprising arrangement of the pyrrolylmethyl substituents in the solid state (Fig. 5). The adjacent pyrrolylmethyl arms are not on opposite sides of the benzene plane as would be expected on steric grounds but nearly in plane, N8 being 0.27 Å above the benzene ring (C1–C3j) and that for N14j 0.24 Å below it. In addition, the pyrrolyl rings containing N8 and N14j or N8j and N14 both form dihedral angles of 19.24°. The latter is the result of a pair of bifurcated intramolecular C–H⋯N hydrogen bonds (C3j–H⋯N8 or C3–H⋯N8j 2.485 Å, 120.29° and C3j–H⋯N14j or C3–H⋯N14 2.495 Å, 102.38°). This is an uncommon conformation which is also observed in the solid state structure of the trisubstituted benzyl 3,5-bis(1H-benzotriazol-1-ylmethyl)-phenyl ether and which presents an analogous intramolecular C–H⋯N hydrogen bond.10 However, in our case, the nitrogen atom in pyrrolylmethyl rings does not have any “free” lone pair so that these intramolecular C–H⋯N hydrogen bonds seem to be of π-bond nature, that is, C–H⋯π
(pyrrolyl) hydrogen bonds as shown in Fig. 5
(C3j–H⋯MPy(N8) or C3–H⋯MPy(N8j) 2.777 Å, 126.30° and C3j–H⋯MPy(N14j) or C3–H⋯MPy(N14) 2.869 Å, 125.59°; MPy
= centroid of pyrrolyl ring).
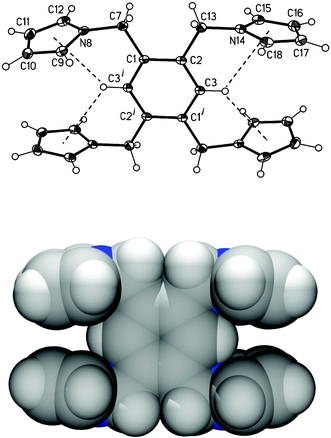 |
| | Fig. 5 View of the molecular structure of centrosymmetric compound 2 showing numbering scheme (top, thermal displacement ellipsoids set at 20% probability level; superscript j refers equivalent position −x
+ 1, −y
+ 1, −z
+ 1) and a space filling diagram (bottom). | |
The solid structure of 1 consists of closely packed parallel offset aromatic rings along the shortest c-axis giving infinite columns as shown in Fig. 6. The interplanar distance of 7.928 Å is much longer than the separations observed for common π-stacked aromatic rings and, consequently, there are no π–π interactions among molecules of 1 in the solid state.3 Thus, no intermolecular contacts are observed in the one-dimensional structure (1-D). Nevertheless, the ring normal and the vector between the ring centroids form an angle of 30.5° and centroid–centroid distance between arene rings is 9.204 Å
(Table 1). The two-dimensional structure of 1 is then constructed by weak C(sp2)–H⋯π
(pyrrolyl) hydrogen bonds between the pyrrole hydrogen and rings of contiguous 1-D columns (Fig. 7). Tables 1–2 list the intermolecular distances and angles of interest.
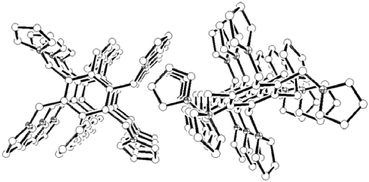 |
| | Fig. 6 A molecular packing diagram of 1 along the c-axis. Hydrogen atoms are omitted for clarity. | |
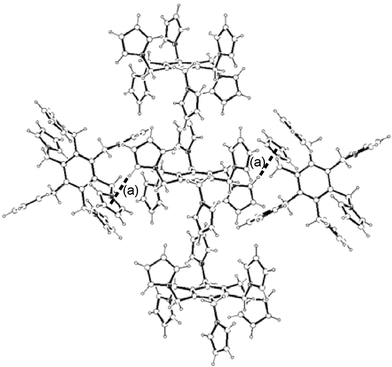 |
| | Fig. 7 2-D structure of 1 and its association as columns supported by intermolecular C–H⋯π
(pyrrolyl) interactions (a). Interactions are shown as dotted lines. Only one molecule per column is shown to the right and to the left of the central one. See Table 2 for metric parameters. | |
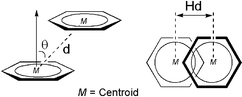
Table 1 Arene–arene packing geometries (Å, °), involved in the 1-D supramolecular construction in 1–5a
|
|
| Compound |
Interplanar |
d
b
|
Hdc |
θ
d
|
| distance |
angle |
|
See embedded Chart at the top for nomenclature.
Ring centroid to ring centroid distance.
Horizontal displacement between the two ring centroids.
Ring normal to vector between the ring centroids angle.
|
|
1
|
7.928 |
0.00 |
9.204 |
4.676 |
30.5 |
|
2
|
4.282 |
0.00 |
5.747 |
3.833 |
41.8 |
|
3
|
3.906 |
0.00 |
5.815 |
4.308 |
47.8 |
|
4
|
3.769 |
0.00 |
5.658 |
4.220 |
48.2 |
|
5
|
3.599 |
0.00 |
5.675 |
4.388 |
50.6 |
Table 2 Geometrical parameters of C–H⋯π hydrogen bonds (Å, °), involved in the supramolecular construction in 1–5
| Compound |
D–H⋯Aa |
d(D–H) |
d(H⋯A)b |
d(D⋯A) |
∠(DHA) |
|
D = donor, A = acceptor.
Not normalised.
M
= centroid of pyrrolyl (Py) or phenyl (Ph) rings.
Centroid for N14A–C15A to C18A.
Centroid for N8–C9 to 12.
Centroid for N14–C15 to C18.
Centroid for N8A–C9A to C12A. Symmetry codes: (a)
=
x, 3/2 −
y, 1/2 +
z; (b) and (c)
x
− 1, y, z; (d) and (i)
−x, y
− 1/2, −z
+ 3/2; (e) and (f)
x, y
− 1, z; (g)
−x, −y
+ 2, −z; (h)
−x, −y, −z
+ 1.
|
|
1
|
(a) C(21B)–H(21B)⋯MPycd |
0.95 |
2.69 |
3.615 |
164 |
|
2
|
(b) C(18)–H(18)⋯MPhc |
0.95 |
2.68 |
3.627 |
174 |
|
3
|
(c) C(12)–H(12)⋯MPhc |
0.93 |
3.03 |
3.954 |
172 |
| (d) C(17)–H(17)⋯MPyce |
0.93 |
2.95 |
3.876 |
171 |
|
4
|
(e) C(15)–H(15)⋯MPhc |
0.95 |
3.12 |
3.982 |
151 |
| (f) C(12)–H(12)⋯MPhc |
0.95 |
2.74 |
3.679 |
169 |
| (g) C(13)–H(13A)⋯MPycf |
0.99 |
2.67 |
3.408 |
132 |
|
5
|
(h) C(9A)–H(9A)⋯MPhc |
0.95 |
2.79 |
3.711 |
163 |
| (i) C(7A)–H(4B)⋯MPycg |
0.99 |
2.87 |
3.802 |
158 |
The solid state structure of 2 consists also of closely packed parallel offset aromatic rings along the shortest a-axis giving infinite columns (Fig. 8), but unlike compound 1, no 2-D structure is formed. More importantly, the interplanar distance (4.282 Å) is much shorter than that for 1 and is slightly over the limit to be considered π–π interactions (Table 1). The ring normal and the vector between the ring centroids and centroid–centroid distance between arene rings (Table 1) are clearly distinct from that for the hexasubstituted compound 1. The main reason for this seems to be a net of intermolecular hydrogen bonds (Fig. 9 and Table 2) between pyrrolyl C(sp2)–H and the π cloud of arene rings in the same direction, giving the observed 1-D infinite columns, that is, a C–H⋯π network.1c,11 In order to shed some light on the importance of the observed hydrogen bond network in 2, we have now determined the structure of the related di(1-pyrrolylmethyl)benzene monomers 3–5
(Fig. 3) by X-ray crystallography. The comparison of all five structures 1–5 is also of interest in order to study the solid state conformations of the pyrrolylmethyl moieties and their possible influence in crystal packings.
 |
| | Fig. 8 A molecular packing diagram of 2 along the a-axis. | |
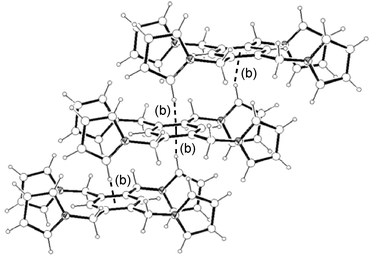 |
| | Fig. 9 1-D structure of 2 and its association as columns supported by intermolecular C–H⋯π
(phenyl) interactions. Interactions are shown as dotted lines. See Table 2 for metric parameters. | |
The o-, m- and p-substituted pyrrolylmethyl compounds 3–5 gave suitable crystals for X-ray analysis. The structures are presented in Fig. 10–12. All conformations are normal and comparable to that of related compounds.6 Thus, the pyrrolylmethyl substituents arrange in an ab pattern with respect to the plane of the benzene ring, as expected. Although intramolecular C–H⋯π
(pyrrolyl) hydrogen bonds seem to be also present in 3–5
(see Fig. 10–12 for metric parameters), these are weaker than that for 2
(vide supra).
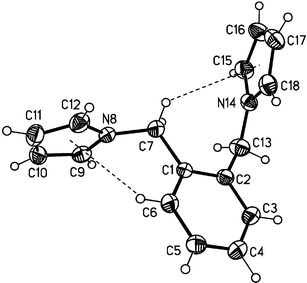 |
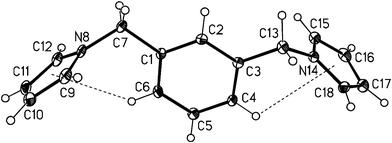
| | Fig. 10 View of compound 3 molecular structure with numbering scheme. Thermal displacement ellipsoids are drawn at the 20% probability level. Intramolecular C–H⋯π
(pyrrolyl) hydrogen bonds are shown as dashed lines (C6–H⋯MPy(N8), 3.028 Å/119.25°; C7–H⋯MPy(N14), 3.289 Å/109.04°; MPy
= centroid of pyrrole ring). | |
 |
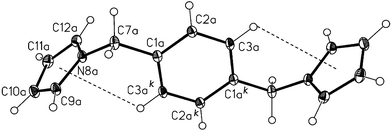
| | Fig. 11 View of compound 4 molecular structure with numbering scheme. Thermal displacement ellipsoids are drawn at the 20% probability level. Intramolecular C–H⋯π
(pyrrolyl) hydrogen bonds are shown as dashed lines (C6–H⋯MPy(N8), 2.911 Å/119.83°; C4–H⋯MPy(N14), 3.145 Å/114.55°; MPy
= centroid of pyrrole ring). | |
 |
| | Fig. 12 View of one of the two independent centrosymmetric molecules (molecule 5a) of compound 5 molecular structure with numbering scheme. Superscript k refers equivalent position −x, −y
+ 1, −z
+ 2. Thermal displacement ellipsoids are drawn at the 20% probability level. Intramolecular C–H⋯π
(pyrrolyl) hydrogen bonds are shown as dashed lines (C3ak–H⋯MPy(N8a) and C3a–H⋯MPy(N8ak), 3.040 Å/117.04°; MPy
= centroid of pyrrole ring). | |
Consideration of the molecular packing for compounds 3–5 shows that the molecules are held together by a variety of intermolecular interactions. As for 1 and 2, the disubstituted derivatives also form infinite columns of parallel displaced (offset) aromatic rings along the shortest crystallographic axes (Fig. 13, 14 and 15). However the interplanar distances in 3–5
(Table 1) are not only much shorter than in the hexasubstituted compound 1 but significantly shorter than that for 2, which qualifies them to be considered weak π–π interactions.3 As in 2, the columns in 3–5 are also held by a net of intermolecular C–H⋯π hydrogen bonds between pyrrolyl C(sp2)–H and the π cloud of benzene rings in the same direction (interactions (c), (e–f) and (h) in Fig. 16, 17 and 18, respectively; Table 2). The 2-D structure in these disubstituted compounds is then formed by C–H⋯π(pyrrolyl) interactions (interactions (d), (g) and (i) in Fig. 16, 17 and 18, respectively). Whereas that for 3 corresponds to pyrrolyl C(sp2)–H hydrogen bonds, those for 4–5 are C(sp3)–H and should be considerably weaker.1c
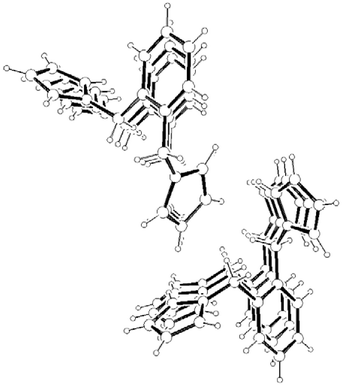 |
| | Fig. 13 A packing diagram of 3 along the b-axis. | |
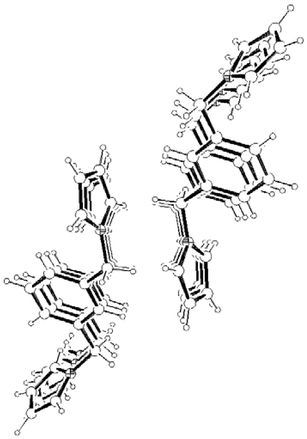 |
| | Fig. 14 A packing diagram of 4 along the b-axis. | |
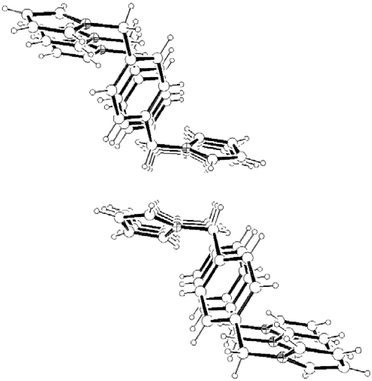 |
| | Fig. 15 A packing diagram of 5 along the b-axis. | |
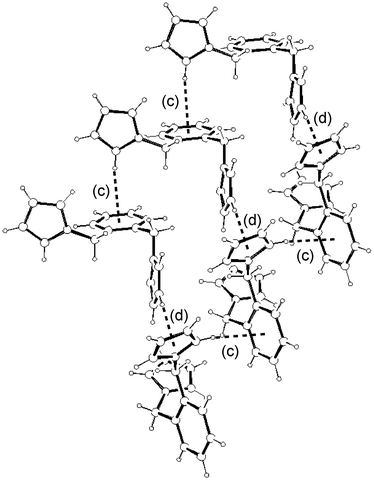 |
| | Fig. 16 2-D Structure of 3 and its association supported by intermolecular C–H⋯π
(phenyl (c) and pyrrolyl (d)) interactions. Interactions are shown as dotted lines. See Table 2 for metric parameters. | |
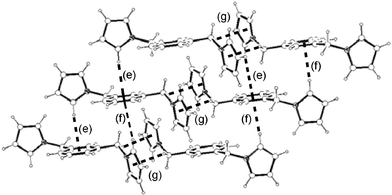 |
| | Fig. 17 2-D structure of 4 and its association supported by intermolecular C–H⋯π
(phenyl (e–f) and pyrrolyl (g)) interactions. Interactions are shown as dotted lines. See Table 2 for metric parameters. | |
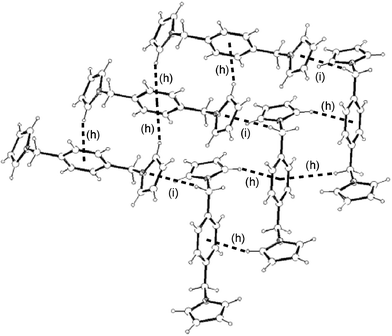 |
| | Fig. 18 2-D structure of 5 and its association supported by intermolecular C–H⋯π
(phenyl (h) and pyrrolyl (i)) interactions. Interactions are shown as dotted lines. See Table 2 for metric parameters. | |
Discussion
The unusual conformation of 2 in the solid state (Fig. 5) merits some discussion. As mentioned in the Results section, the observed conformation seems to be the result of a pair of bifurcated intramolecular C–H⋯π
(pyrrolyl) hydrogen bonds. It could be argued that the adjacent arms in 2 appear to twist as far away as possible from each other due to steric repulsion and therefore imposing the observed conformation. However, the existence of a similar conformation in the trisubstituted benzene derivative benzyl 3,5-bis(1H-benzotriazol-1-ylmethyl)-phenyl ether10 suggests that the intramolecular hydrogen bonding is not negligible and must contribute to the conformation of 2 in the solid state. An important consequence of the observed intramolecular C–H⋯π
(pyrrolyl) hydrogen bonding in 2 is that the benzene ring is available e.g., to act as hydrogen acceptor, as can be seen in the space filling model in Fig. 5
(bottom). In contrast, the aromatic ring in the hexasubstituted 1 is protected and not available due to the ababab pattern adopted by the arms in this molecule (Fig. 4). These molecular conformations have a strong influence on the packing of molecules in the solid state, as discussed below.
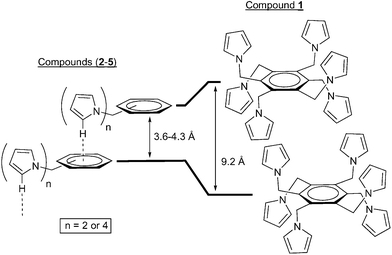
As can be seen in Table 1, the usual arrangement of these compounds is an offset stacking with the arene rings parallelly displaced. The interplanar distances between the planes defined by the aromatic rings and their horizontal displacements (Hd) for the di- and tetrasubstituted 1-pyrrolylmethyl derivatives 2–5 are within the range of 3.6–4.3 and 3.8–4.4 Å, respectively. The interplanar distances, which are taken as the criterion for the existence of π–π interactions, are at the upper limit for the disubstituted compounds 3–5 but above the limit for the tetrasubstituted compound 2. According to Hunter and Sanders' model (Fig. 1, bottom), such offset stacking structures have a contribution from π–σ attraction, the more so with increasing the offset (given by the distance Hd angle θ in Table 1). The fact that the interplanar distance in the tetrasubstituted compound 2 is above the limit to be considered a π–π interaction, suggests that the observed net of intermolecular C–H⋯π hydrogen bonds (Fig. 9 and Table 2) between pyrrolyl C(sp2)–H and the π cloud of arene rings have a stronger influence than the stacking of aromatic rings in this particular case. Interestingly, the ring planes in 2–5 are offset such that a pyrrolyl carbon atom lies almost over the center of the arene ring of a close molecule and its hydrogen atom is roughly over ring centers. In this way, each molecule is connected with its two neighbours (a lower and an upper one)
via one or two C–H⋯π
(Ph) hydrogen bonds. Thus, the molecules in the crystals of 2–5 are arranged in such a way as to optimize the net of hydrogen bonds and this certainly has a stabilization effect and possibly structure determining function.1c Although the distances of the π–π aromatic rings in 2–5 are taken as the criterion for defining the existence of π–π interactions (vide supra), it is important to note that no atom–atom contacts are observed between those aromatic rings in any of the compounds reported in this work. The much larger interplanar distance for the bulkier compound 1
(7.9 Å) is consistent with the absence of C–H⋯π
(Ph) interactions (Fig. 7). Whereas the entire face of the aromatic ring can serve as a hydrogen bond acceptor in 2–5
(Figs. 9, 16, 17, 18 and Table 2), that for the hexasubstituted 1 cannot because the pyrrolylmethyl substituents are blocking both sides of the ring and this prevents the formation of hydrogen bonding (steric control).12
The above considerations demonstrate that the C–H⋯π
(Ph) network does have an influence in the stacking interactions of poly(1-pyrrolylmethyl)benzene derivatives (Fig. 19). When the hydrogen bond network is avoided e.g., as in the case of 1, by blocking the arene ring, the molecules adopt arrangements mainly dominated by van der Waals or electrostatic interactions of the pyrrolyl rings. In this situation the arene rings, although still parallel, separate well apart to optimize other intermolecular forces (Fig. 19 right). However, when the π clouds of the aromatic rings are available to act as proton acceptors, such as in 2–5, hydrogen bonds are then observed in addition to the π–π interactions (Fig. 19 left). The existence of such a C–H⋯π
(Ph) network explains the much shorter arene interplanar distances observed in case of the di- and tetrasubstituted benzene compounds and this certainly stabilizes the offset π–π interactions.
 |
| | Fig. 19 | |
As in carbocyclic aromatics, the π faces of heterocyclic compounds can accept hydrogen bonds.1c In particular, examples for nitrogen containing heterocycles giving CH(aliphatic)⋯π(heterocyclic) interactions have been observed in protein structures,13 smaller molecules14 and have been modelled using ab initio calculations.11a Calculations show that hydrogen bonds are mainly directed at the heteroatom lone pair rather than to the π face. It is important to note that in all studied examples the nitrogen atom have a free lone pair of electrons available for the formation of conventional hydrogen bonds. However, as mentioned above, the nitrogen atom in pyrrolylmethyl rings in our compounds does not have any “free” lone pair so that the C–H⋯π(pyrrolyl) interactions are directed to the center of the ring rather than to the N atom and are, in general, directional (Table 2). Although these very weak hydrogen bonds seem to have a minor significance for the observed stacking interactions, it is interesting to see whether those are proper hydrogen bonds or rather van der Waals contacts. A search of the Cambridge Structural Database (Version 1.7, February 2005) for C–H⋯π(heterocycles) reveals 104 hits, 89 of which corresponding to C–H⋯π(N containing heterocycles). Scattergrams for the geometry of these intermolecular C–H⋯π(heterocycle) interactions show characteristic features of weak hydrogen bonds, that is, as the length decreases, the interactions approach linearity (see supporting information).†
Conclusions
Two new multi-armed molecules hexa(1-pyrrolylmethyl)benzene (1) and 1,2,4,5-tetra(1-pyrrolylmethyl)benzene (2) have been synthesized and their crystal structures have been determined and compared with those for the related disubstituted o-, m- and p-di(1-pyrrolylmethyl)benzene (3–5) compounds. Each of the di- or tetrasubstituted benzene molecules 2–5 packs in a unique array, the formation of which is dependent upon an interplay of weak intermolecular interactions which are instrumental in the formation of weakly bound columns: offset π stacking plus C–H⋯π interactions (interplanar distances 3.6–4.3 Å). The persubstitution of the benzene unit has a marked effect on the crystal packing of 2, which also packs in columns, but in the absence of π stacking or C–H⋯π interactions the molecules separate well apart (7.9 Å). The absence of this hydrogen bond network and π–π interactions in the hexasubstituted 1 can be explained by steric control and suggests that although the π–σ attractions might be controlling the growth of crystals in 2–5, C–H⋯π interactions do play a stabilizing role and possibly structure determining function. When the π clouds of the arene rings are available to act as proton acceptors, such as in 2–5, the rings are offset such that a pyrrolyl hydrogen atom is roughly over ring centers of a neighbouring molecule and displaced to a distance so that C–H⋯π interactions can take place. The uncommon arrangement of the pyrrolylmethyl arms in the molecular structure of 2, due to two bifurcated intramolecular C–H⋯π
(pyrrolyl) hydrogen bonds, seems to facilitate the formation of intermolecular C–H⋯π interactions in this polysubstituted derivative and shows the importance of the molecular arrangement in the crystal packing. C(pyrrolyl)–H⋯π(pyrrolyl) interactions are also found in the 2-D structure for most of the compounds. The results reported show that small changes in the structure of a family of related compounds can have a significant impact on their crystal structures. These observations also serve in the understanding of the basic principles of supramolecular chemistry of π systems and, consequently, for the progress of crystal engineering.
Experimental
General
All manipulations were carried out under a N2 atmosphere. Chemicals were used as follows: THF distilled from Na/benzophenone; pyrrole (Aldrich) distilled; other solvents were of reagent grade quality and were used without further purification; hexakis(bromomethyl)benzene, tetrakis(bromomethyl)benzene and MgSO4 were purchased and used as received; compounds 3–5 were prepared by literature procedures.9 Elemental analyses were performed using a Carlo Erba EA1108 microanalyser. IR spectra were recorded with KBr pellets on a Nicolet 710 FT spectrophotometer. 1H NMR (300.13 MHz) and 13C{1H} NMR (75.47 MHz) spectra were recorded with a Bruker ARX 300 instrument equipped with the appropriate decoupling accessories. Chemical shift values for 1H and 13C{1H} NMR spectra were referenced to SiMe4. Chemical shifts are reported in units of parts per million downfield from SiMe4, and all coupling constants are reported in Hz.
Synthesis of hexa-1-pyrrolylmethyl-benzene (1)
Hexakis(bromomethyl)benzene (640 mg, 1.0 mmol) was added to a suspension of potassium pyrrole (810 mg, 7.7 mmol) in THF (50 ml) and refluxed for 2 h. The reaction mixture was then cooled down and an equal volume of 0.5 M NaCl aqueous solution added. The remaining unreacted pyrrole and potassium bromide were extracted with water. The organic phase was separated, dried over magnesium sulfate and the solvent evaporated after filtration. The resulting light yellow solid was purified by chromatography on silica using CH2Cl2/hexane (80/20) as eluent, affording pure 1 as a white solid [RF(prep)
= 0.82]. Recrystallization from acetonitrile gave microcrystals. Yield 510 mg. (92%). Anal. Calcd. for C36H36N6: C, 78.23; H, 6.57; N, 15.21. Found: C, 78.17; H, 6.72; N, 15.11. IR (cm−1): ν
= 3097 (CPy–H), 2912.3 (C–H), 1494.7 (CPy–H), 1396.4 (C–H), 1382.9 (C–H), 717.5 (CPy–H). 1H NMR (CDCl3): δ 6.44 (s, 12H, N(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2), 6.23 (s, 12H, N(CHCH)2), 5.09 (s, 24H, CH2N). 13C{1H}NMR: 138.1 (s, CAr), 119.9 (s, N(CHCH)2), 109.5 (s, N(CHCH)2), 46.2 (s, CH2N).
CH)2), 6.23 (s, 12H, N(CHCH)2), 5.09 (s, 24H, CH2N). 13C{1H}NMR: 138.1 (s, CAr), 119.9 (s, N(CHCH)2), 109.5 (s, N(CHCH)2), 46.2 (s, CH2N).
Synthesis of 1,2,4,5-tetra-1-pyrrolylmethyl-benzene (2)
The general procedure described above was followed, using 1,2,3,5-tetrakis(bromomethyl)benzene (670 mg, 1.5 mmol). 2 was obtained as a pure white solid after chromatography on silica with CH2Cl2/hexane (70/30)
[RF(prep)
= 0.77]. Recrystallization from THF gave microcrystals. Yield 560 mg. (96%). Anal. Calcd. for C26H26N4: C, 79.16; H, 6.64; N, 14.20. Found: C, 79.29; H, 6.52; N, 14.28. IR (cm−1): ν
= 3097 (CPy–H), 3075 (CAr–H), 2893.3 (C–H), 1498.6 (CPy–H), 1409.9 (C–H), 1373.2 (C–H), 871.8 (CAr–H), 823.5 (CAr–H), 717.2 (CPy–H). 1H NMR (CDCl3): δ 6.65 (s, 2H, CAr–H), 6.65 (s, 8H, N(CHCH)2), 6.20 (s, 8H, N(CHCH)2), 4.90 (s, 8H, CH2N). 13C{1H} NMR: δ
= 135.9 (s, CAr), 129.9(s, CAr), 120.9 (s, N(CHCH)2), 108.9 (s, N(CHCH)2), 50.2 (s, CH2N).
Single crystal X-ray diffraction
Single-crystal data collection for 1, 3, 4 and 5 were performed at −80° with an CAD4 diffractometer using graphite monochromated Cu Kα radiation while the collection for 2 was performed at ambient temperature with a Rigaku AFC5S diffractometer using graphite monochromated Mo Kα radiation. A total of 5048, 1782, 2365, 2299 and 2172 unique reflections were collected for 1, 2, 3, 4 and 5, respectively. The structures were solved by direct methods and refined on F2 by the SHELXL97 program.15 For each compound, non-hydrogen atoms were refined with anisotropic displacement parameters and the hydrogen atoms were treated as riding atoms using the SHELX97 default parameters. Crystallographic parameters for 1–5 are gathered in Table 3.
Table 3 Crystallographic parameters for 1–5
| |
1
|
2
|
3
|
4
|
5
|
|
S
=
[Σ(w(Fo2
−
Fc2)2]/(n
−
p)1/2.
R
=
Σ||Fo| − |Fc||/Σ|Fo|.
Rw
=
[Σw(|Fo2| − |Fc2|)2/Σw|Fo2|2]1/2.
|
| Empirical formula |
C36H36N6 |
C26H26N4 |
C16H16N2 |
C16H16N2 |
C16H16N2 |
| Formula weight |
552.71 |
3 |
394.51 |
236.31 |
236.31 |
236.31 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
P21/n |
P21/c |
|
a/Å |
18.463(2) |
5.7470(6) |
5.815(3) |
9.2058(9) |
14.7475(13) |
|
b/Å |
17.810(2) |
15.511(2) |
8.493(3) |
5.6582(12) |
5.6746(4) |
|
c/Å |
9.2040(10) |
11.798(2) |
27.344(4) |
25.114(2) |
16.6394(14) |
|
β/° |
101.491(8) |
92.911(8) |
90.48(3) |
96.875(8) |
113.980(7) |
|
V/V3 |
2965.9(5) |
1050.3(2) |
1350.3(8) |
1298.7(3) |
1272.30(18) |
|
Z
|
4 |
2 |
4 |
4 |
4 |
|
T/°C |
−80 |
−80 |
23 |
−80 |
−80 |
|
λ
|
1.54184 |
1.54184 |
0.71069 |
1.54184 |
1.54184 |
|
ρ
(g cm−3) |
1.238 |
1.247 |
1.162 |
1.209 |
1.234 |
|
μ/cm−1 |
5.80 |
5.80 |
0.69 |
5.53 |
5.64 |
| Goodness-of-fita on F2 |
1.022 |
1.081 |
0.985 |
1.050 |
1.026 |
|
R
b
[I > 2σ(I)] |
0.0535 |
0.0452 |
0.0562 |
0.0562 |
0.0431 |
|
Rw
c
[I > 2σ(I)] |
0.1376 |
0.1096 |
0.1102 |
0.1351 |
0.1040 |
CCDC reference numbers 286641–286645. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b515930j
Acknowledgements
We thank CICYT (Project MAT2004-01108), Generalitat de Catalunya (2001/SGR/00337) and CSIC (I3P contract to J.G.P.).
References
-
(a)
G. R. Desiraju, Crystal Engineering. The Design of Organic Solids, Elsevier Science Publishers B. V., Amsterdam, 1989 Search PubMed;
(b) K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143 CrossRef CAS;
(c)
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 2001 Search PubMed;
(d) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS;
(e) D. Swierczynski, R. Luboradzki, G. Dolgonos, J. Lipkowski and H.-J. Schneider, Eur. J. Org. Chem., 2005, 1172 CrossRef CAS and references therein;
(f) R. Sudha, M. Kohtani and M. F. Jarrold, J. Phys. Chem. B, 2005, 109, 6442 CrossRef CAS;
(g) J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193 RSC.
- D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1 RSC.
-
(a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS;
(b) V. E. Williams, R. P. Lemieux and G. R. J. Thatcher, J. Org. Chem., 1996, 61, 1927 CrossRef CAS;
(c) Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207 CAS;
(d) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC;
(e) O. Takahashi, Y. Kohno, S. Iwasaki, K. Saito, M. Iwaoka, S. Tomoda, Y. Umezawa, S. Tsuboyama and M. Nishio, Bull. Chem. Soc. Jpn., 2001, 74, 2421 CrossRef CAS;
(f) F. Cozzi, R. Annunziata, M. Benaglia, M. Cinquini, L. Raimondi, K. K. Baldridge and J. S. Siegel, Org. Biomol. Chem., 2003, 1, 157 RSC;
(g) M. Piacenza and S. Grimme, ChemPhysChem, 2005, 6, 1 CrossRef;
(h) Y. Kobayashi and S. Kazuhiko, J. Am. Chem. Soc., 2005, 127, 15054 CrossRef CAS.
- Hunter and Sanders' simple model of charge distribution has been criticised by theoretical investigators although it does allow qualitative understanding of the π–π interactions. Even though we use this simple model through this article, the term “π–π interactions” is used for non-covalent interactions between π systems without implying anything concerning the origin of this interaction.
- Z. Ciunik and J. Slawomir, J. Mol. Struct., 1998, 442, 115 CrossRef CAS.
-
(a)
D. D. MacNicol, in Inclusion Compounds, ed. J. L. Atwood, J. ED. Davies and D. D. MacNicol, Academic Press, London, 1984, vol. 2, ch. 5, pp. 124–168 Search PubMed;
(b) A. S. Jessiman, D. D. MacNicol, P. R. Mallison and I. Vallance, J. Chem. Soc., Chem. Commun., 1990, 1619 RSC;
(c) C. M. Hartshorn and P. J. Steel, Aust. J. Chem., 1995, 48, 1587 CAS;
(d) C. M. Hartshorn and P. J. Steel, Angew. Chem., Int. Ed. Engl., 1996, 35, 2655 CrossRef;
(e) F. K. Gormley, J. Gronbach, S. M. Draper and A. P. Davis, J. Chem. Soc., Dalton Trans., 2000, 173 RSC;
(f) G. Hennrich and E. V. Anslyn, Chem.-Eur. J., 2002, 8, 2218 CrossRef CAS;
(g) J. Bloxham, C. J. Moody and A. M. Z. Slawin, Tetrahedron, 2002, 58, 3709 CrossRef CAS;
(h) D. A. McMorran and P. J. Steel, Tetrahedron, 2003, 59, 3701 CrossRef CAS;
(i) P. J. Steel, Acc. Chem. Res., 2005, 38, 243 CrossRef CAS.
-
(a) E. M. D. Keegstra, B.-H. Huisman, E. M. Paardekooper, F. J. Hoogesteger, J. W. Zwikker, L. W. Jenneskens, Huub. Kooijman, A. Schouten, N. Veldman and A. L. Spek, J. Chem. Soc., Perkin Trans. 2, 1995, 229 Search PubMed;
(b) D. L. Reger, R. F. Semeniuc, I. Silaghi-Dumitrescu and M. D. Smith, Inorg. Chem., 2003, 42, 3751 CrossRef CAS.
-
C. Masalles, PhD Thesis, Universitat Ramón Lull, 2002.
- K. I. Chane-Ching, J. C. Lacroix, R. Baudry, M. Jouini, S. Aeiyach, C. Lion and P. C. Lacaze, J. Electroanal. Chem., 1998, 453, 139 CrossRef CAS.
- S. Selvanayagam, D. Velmurugan, K. Ravikumar, M. Dhanasekaran and P. Rajakumar, Acta Crystallogr., 2004, E60, o2165 CAS.
-
(a) U. Samanta, P. Chakrabarti and J. Chandrasekhar, J. Phys. Chem. A, 1998, 102, 8964 CrossRef CAS;
(b) H. Takahashi, S. Tsuboyama, Y. Umezawa, K. Honda and M. Nishio, Tetrahedron, 2000, 56, 6185 CrossRef CAS;
(c) F. Ugozzoli, A. Arduini, C. Massera, A. Pochini and A. Secchi, New J. Chem., 2002, 26, 1717 Search PubMed;
(d) M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- R. Custelcean, M. G. Gorbunova and P. V. Bonnesen, Chem.-Eur. J., 2005, 11, 1459 CrossRef CAS.
- P. Chakrabarti and U. Samanta, J. Mol. Biol., 1995, 251, 9 CrossRef CAS.
-
(a) R. Hunter, R. H. Haueisen and A. Irving, Angew. Chem., Int. Ed. Engl., 1994, 33, 566 CrossRef;
(b) I. Nobeli, S. L. Price, J. P. M. Lommerse and R. Taylor, J. Comput. Chem., 1997, 18, 2060 CrossRef CAS;
(c) H. Suezawa, T. Yoshida, Y. Umezawa, S. Tsuboyama and M. Nishio, Eur. J. Inorg. Chem., 2002, 3148 CrossRef CAS.
-
G. M. Sheldrick, SHELX97. University of Göttingen, Germany, 1997.
Footnote |
| † Electronic supplementary information (ESI) available: CSD search for intermolecular C–H⋯π(heterocycles) interactions (Chart S1, Fig. S1 and S2). See DOI: 10.1039/b515930j |
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.