Asymmetric organocatalytic conjugate addition of malonates to enones using a proline tetrazole catalyst
Kristian Rahbek Knudsen, Claire E. T. Mitchell and Steven V. Ley*
Department of Chemistry, University of Cambridge, Lensfield Rd, Cambridge, UK CB2 1EW. E-mail: svl1000@cam.ac.uk; Fax: + 44 (0)1223 336442
First published on 28th November 2005
Abstract
5-Pyrrolidin-2-yltetrazole performs as a useful organocatalyst for the asymmetric addition of malonates to a range of enones, with good to excellent enantioselectivities.
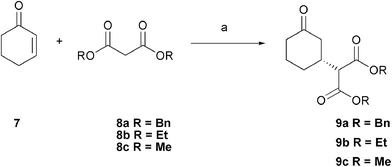
The asymmetric conjugate addition of carbon nucleophiles to electron-poor alkenes is an important transformation in modern synthetic chemistry.1 Malonates are an easily accessible source of donors as the two electron-withdrawing esters enable enolate formation under mild conditions. Dimethylmalonate and, to a lesser extent, diethylmalonate, are particularly valuable units due to their facile direct decarboxylation under Krapcho conditions.2
A variety of transition metal catalysts have been developed for the asymmetric conjugate addition of 1,3-dicarbonyl compounds to enones.3 The most general systems reported to date include the BINOL–heterobimetallic complexes developed by Shibasaki et al. which gave excellent enantioselectivities for the addition of malonate and other closely related carbon nucleophiles to cyclic enones;4 these have been used on a kilogram scale.5 Jacobsen and co-workers have investigated the use of an aluminium–salen catalyst for the addition of methylcyanoacetate to a range of enones also in high yields and in good to excellent enantioselectivity.6 Proline rubidium salts have been used similarly to catalyse the addition of di-iso-propyl and di-tert-butyl malonates to both acyclic and cyclic α,β-unsaturated enones; here the reaction proceeded with low to good enantioselectivities (35–88% ee).7 Proline 1 itself (Fig. 1) has been employed for the addition of 1,3-diketones to methylvinylketone and subsequent cyclization to 3-hydroxycoumarins.8Cinchona alkaloid-derived phase transfer catalysts have also been employed in the addition of malonates to chalcone and closely related substrates, with good enantioselectivity.9 Recently, use of a bifunctional hydrogen-bonding organocatalyst for the addition of malonitrile to α,β-unsaturated imides with good yields and enantioselectivities has been reported.10 However, the addition of less reactive species such as malonates and β-ketoesters could not be achieved.
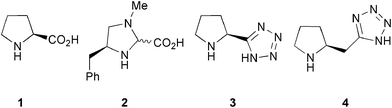 | ||
| Fig. 1 Proline and related organocatalysts. | ||
Imidazoline catalyst 2 has been observed to provide good to excellent enantioselectivities for the conjugate addition of malonates to acyclic α,β-unsaturated enones (59–99%).11 Nevertheless, although the addition of dibenzyl and diethyl malonates proceeded with excellent enantioselectivities, the addition of methyl malonate proceeded with lower selectivity (71% ee) and reaction times were typically between 5 and 12 days. In addition, the malonates were employed as the reaction solvent, and thus were used in approximately 8–16 fold excess. Since these malonates are not especially volatile, their eventual removal is problematic.
We and others have previously reported on the tetrazole analogue of proline 3 as a more soluble and effective catalyst in a variety of transformations.12 We have shown that tetrazole 3 is an improved catalyst for the conjugate addition of nitroalkanes to both cyclic and acyclic enones,13 using the achiral meso base 2,5-dimethylpiperazine 5 (Fig. 2) as an additive under conditions adapted from those previously developed by Hanessian and Pham for the addition of nitroalkanes to cyclic enones catalysed by proline.14 Moderate to excellent enantioselectivities were obtained (58–98% ee). In connection with an ongoing synthesis project in our group, we required a scalable method for the asymmetric addition of malonates to a wide range of enones both cyclic and acyclic.
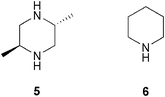 | ||
| Fig. 2 Bases employed. | ||
Initial results using the conditions optimised for the addition of nitroalkanes to enones were promising, giving the product 9a in 65% conversion and 77% ee (Table 1, entry 1). Malonates (pKa diethyl malonate = 13) are less acidic than nitroalkanes (pKa MeNO2 = 10), so the use of the stronger base piperidine (6) was investigated next. The reaction of cyclohexenone 7 with dibenzylmalonate 8a in CH2Cl2 in combination with 6 as the base afforded product 9a in 63% conversion and an improved ee after 2 days (entry 2). Changing the solvent to CHCl3 gave a higher conversion of 87% whilst maintaining the enantioselectivity (entry 3). On the other hand, use of the non-chlorinated solvent MeCN led to a decrease in both enantioselectivity and yield (entry 4). Diethyl malonate 8b as the nucleophile was also found to give improved results using piperidine 6 rather than 5 (entries 5, 6), giving 89% conversion and 92% ee in CH2Cl2 and similar ee's but lower overall conversion in CHCl3 (entry 7).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Malonate | Catalyst | Solvent | Base | Conv.b (%) | eec (%) |
| a 7 (0.2 mmol), catalyst (15 mol%), 8a–c (0.2 mmol), base (0.2 mmol), solvent 1 mL, rt, 2 d.b Estimated by 1H NMR.c Determined by chiral HPLC or GC. Absolute configuration determined from ref. 17. | ||||||
| 1 | 8a | 3 | CH2Cl2 | 5 | 65 | 77 |
| 2 | 8a | 3 | CH2Cl2 | 6 | 63 | 82 |
| 3 | 8a | 3 | CHCl3 | 6 | 87 | 81 |
| 4 | 8a | 3 | MeCN | 6 | 80 | 70 |
| 5 | 8b | 3 | CH2Cl2 | 5 | 65 | 79 |
| 6 | 8b | 3 | CH2Cl2 | 6 | 89 | 92 |
| 7 | 8b | 3 | CHCl3 | 6 | 69 | 89 |
| 8 | 8b | 1 | CHCl3 | 6 | 62 | 38 |
| 9 | 8b | 4 | CHCl3 | 6 | 40 | 0 |
| 10 | 8b | 4 | CH2Cl2 | 6 | 50 | −17 |
| 11 | 8c | 3 | CH2Cl2 | 6 | 85 | 83 |
| 12 | 8c | 3 | CHCl3 | 6 | 87 | 85 |
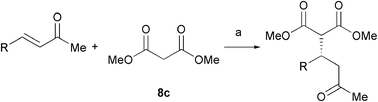
Proline 1, on the other hand, gave only 38% ee in 62% conversion (entry 8). While the homologated tetrazole 4 was effective as a catalyst in the asymmetric Michael addition of ketones to nitroolefins,15 in this work the Michael addition of diethyl malonate to cyclohexenone in CHCl3 provided only the racemic product 9b (entry 9). Interestingly, in CH2Cl24 showed reversed selectivity, favouring formation of the opposite enantiomer of the product, albeit in low ee (entry 10). This switch in enantioselectivity was not observed in the addition of 2-nitropropane to 7 when catalysed by 4. However, use of dimethyl malonate gave the product 9c in good conversion and enantioselectivity in both CH2Cl2 and CHCl3 (entries 11, 12), with a slightly higher ee of 85% being observed in chloroform.
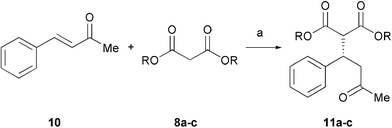
We next investigated the addition of malonate to 4-phenyl-3-buten-2-one 10 as a less reactive acceptor in these Michael addition reactions. Reaction of 10 with dibenzyl malonate 8a (1.5 eq.) gave the product 11a in 59% conversion and 83% ee over 3 days (Table 2, entry 1). However, the use of base 6 in this reaction gave the product in higher yield while maintaining the enantioselectivity (entry 2). For diethyl malonate 8b, the added base 5 gave the product in good ee but with low conversion (entry 3). By changing the base to piperidine 6 we obtained good results both with CH2Cl2 and CHCl3 (entries 4, 5), the reaction in CHCl3 providing a slightly improved ee of 89%. Use of proline 1 gave 57% ee. The reversal of enantioselectivity observed in the addition of diethyl malonate to cyclohexenone with the homologated tetrazole 4 (Table 1, entry 10) was not observed for the acyclic enone 10 (entry 8). In the case of dimethyl malonate 8c, reactions in CHCl3 led to a significantly improved enantioselectivity of 85% compared to both CH2Cl2 and MeCN (entries 9–11). The reactions were monitored by NMR (conversion) and HPLC (ee) over 5 days at various catalyst loadings. Use of 15 mol% of tetrazole 3 led to a higher conversion of 97%, with the reaction essentially complete within 2 days, but with a slightly decreased 83% ee (entry 12). NMR analysis of the reaction with 10 mol% 3 demonstrated that the reaction reached 89% conversion after 2 days, but did not progress any further. Pleasingly, use of only 5 mol% 3 gave the product in 92% conversion and 85% ee with a slightly prolonged reaction time of 3 days (entry 13). Again, NMR analysis indicated the reaction did not progress to completion even after extended treatment.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Malonate | Catalyst | Solvent | Base | Conv.b (%) | eec (%) |
| a 10 (0.2 mmol), 3 (10 mol%), 8a–c (0.3 mmol), base (0.2 mmol), solvent 1 mL, rt, 3 d.b Measured by 1H NMR.c Determined by chiral HPLC.d 15 mol% of catalyst 3 was used, 2 d.e 5 mol% of catalyst 3 was used. | ||||||
| 1 | 8a | 3 | CH2Cl2 | 5 | 59 | 83 |
| 2 | 8a | 3 | CH2Cl2 | 6 | 90 | 83 |
| 3 | 8b | 3 | CH2Cl2 | 5 | 29 | 87 |
| 4 | 8b | 3 | CH2Cl2 | 6 | 78 | 87 |
| 5 | 8b | 3 | CHCl3 | 6 | 82 | 89 |
| 6 | 8b | 3 | MeCN | 6 | 93 | 85 |
| 7 | 8b | 1 | CHCl3 | 6 | 82 | 57 |
| 8 | 8b | 4 | CH2Cl2 | 6 | 24 | 42 |
| 9 | 8c | 3 | CH2Cl2 | 6 | 97 | 81 |
| 10 | 8c | 3 | CHCl3 | 6 | 89 | 85 |
| 11 | 8c | 3 | MeCN | 6 | 89 | 77 |
| 12 | 8cd | 3 | CHCl3 | 6 | 97 | 83 |
| 13 | 8ce | 3 | CHCl3 | 6 | 92 | 85 |
Next, the addition of a range of malonates to enone 10 under these optimised conditions was investigated (Table 3). Good yields were obtained for all malonates. In contrast to previous work with the imidazolidinone catalyst 2, the reaction was found to be relatively insensitive to the nature of the malonate, with 84% ee obtained with the less sterically bulky dimethyl malonate (entry 3). The more hindered di-iso-propyl malonate reacted more slowly, giving a 68% yield after 3 days, but with a 93% ee (entry 4). Interestingly, preliminary results with mixed malonate 8e gave 67% yield in a 1 ∶ 1.6 dr with 43% ee of both diasteromers.
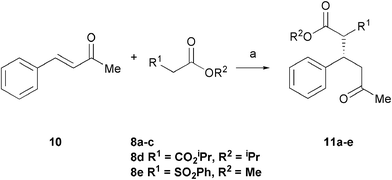
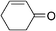
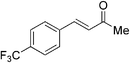
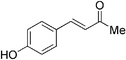
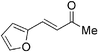
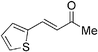
Although the diethyl malonate 8b gave higher enantioselectivity than the dimethyl malonate 8c, the decarboxylation of the latter under Krapcho conditions is more facile,2 and hence the products are more synthetically useful. Therefore, the scope of the addition of dimethyl malonate to a series of enones was tested. For cyclohexenone, the isolated yield was 87% (Table 4, entry 1). Substituted 4-phenyl-3-buten-2-ones were also examined. When the phenyl ring was substituted with an electron-withdrawing p-CF3 group, the product was obtained in 84% yield and 78% ee (entry 2). The p-OH substituted enone afforded a moderate yield of 64% in 62% ee (entry 3). As the pKa of the phenol proton is comparable to that of the malonate, the reaction was also run with two equivalents of base 6 present, which afforded the product in a slightly higher yield of 70% without any significant improvement in enantioselectivity (entry 4). Heterocyclic enones were also found to perform well in the reaction. The furan substituted enone gave 81% ee in a 69% yield (entry 5). The thiophene derived enone gave 82% yield and 84% ee (entry 6).
The mechanism for these reactions has not yet been established, although it is believed that the catalyst and enone form an intermediate iminium ion species. The function of the base is not readily apparent, since changing the base affects not only yield but also the enantioselectivity. Studies on ammonium carbanions have shown that in non-polar solvents the tetrabutylammonium salt and the enolate remain associated through hydrogen bonds.16 It is plausible that in the non-polar solvents required for these reactions, the amine additive and the malonate remain associated, and it is these hydrogen-bonded complexes which are involved in the addition step. Kinetic studies are presently underway to elucidate these pathways. Also, further investigations into the asymmetric addition of mixed malonate type nucleophiles into enone acceptors are ongoing.
In conclusion, we have developed an improved organocatalytic conjugate addition of malonates to enones. The reaction gives good results for a range of substrates furnishing the products in good yield with good to high enantioselectivities. As only 1.5 equivalents of enone are used as coupling partner, the reaction is readily scaled and practical to operate.
We thank the Carlsberg Foundation (to KRK), Avecia (to CETM), the EPSRC (to CETM), and Novartis (through a Research Fellowship to SVL) for financial support.
Notes and references
- M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033 CrossRef CAS; N. Krause and A. Hoffmann-Roder, Synthesis, 2001, 171 CrossRef CAS.
- (a) A. P. Krapcho, Synthesis, 1982, 805 CrossRef CAS; (b) A. P. Krapcho, Synthesis, 1982, 893 CrossRef.
- (a) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033 CrossRef CAS; (b) N. Krause and A. Hoffmann-Roder, Synthesis, 2001, 171 CrossRef CAS; (c) J. Christoffers, Eur. J. Org. Chem., 1998, 1259 CrossRef CAS.
- (a) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS; (b) K. Majima, R. Takita, A. Okada, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 15837 CrossRef CAS; (c) K. Majima, S. Y. Tosaki, T. Ohshima and M. Shibasaki, Tetrahedron Lett., 2005, 46, 5377 CrossRef CAS.
- Y. Xu, K. Ohori, T. Ohshima and M. Shibasaki, Tetrahedron, 2002, 58, 2585 CrossRef CAS.
- (a) M.S. Taylor and E. N. Jacobsen, J. Am. Chem. Soc., 2003, 125, 11204 CrossRef CAS; (b) M. S. Taylor, D. N. Zalatan, A. N. Lerchner and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1313.
- (a) M. Yamaguchi, T. Shiraishi and M. Hirama, Angew. Chem., Int. Ed. Engl., 1993, 32, 1176 CrossRef; (b) M. Yamaguchi, T. Shiraishi and M. Hirama, J. Org. Chem., 1996, 61, 3520–3530 CrossRef CAS.
- D. Gryko, Tetrahedron: Asymmetry, 2005, 16, 1377 CrossRef CAS.
- T. Ooi, D. Ohara, K. Fukumoto and K. Maruoka, Org. Lett., 2005, 7, 3195 CrossRef CAS.
- Y. Hoashi, T. Okino and Y. Takemoto, Angew. Chem., Int. Ed., 2005, 44, 4032 CrossRef CAS.
- N. Halland, P. S Aburel and K. A. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 661 CrossRef CAS.
- (a) A. J. A. Cobb, D. A. Longbottom, D. M. Shaw and S. V. Ley, Chem. Commun., 2004, 1808 RSC; (b) A. J. A. Cobb, D. M. Shaw and S. V. Ley, Synlett, 2004, 558 CAS; (c) A. Hartikka and P. I. Arvidsson, Tetrahedron: Asymmetry, 2004, 15, 1831 CrossRef CAS; (d) N. Momiyama, H. Torii, S. Saito and H. Yamamoto, Proc. Natl. Acad. Sci. USA, 2004, 101, 5374 CrossRef CAS; (e) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983 CrossRef CAS; (f) Y. Yamamoto, N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5962 CrossRef CAS; (g) A. J. A. Cobb, D. M. Shaw, D. A. Longbottom, J. B. Gold and S. V. Ley, Org. Biomol. Chem., 2005, 3, 84 RSC; (h) D. B. Ramachary and C. F. Barbas, Org. Lett., 2005, 7, 1577 CrossRef CAS.
- C. E. T. Mitchell, S. E. Brenner and S. V. Ley, Chem. Commun., 2005, 5346 RSC.
- S. Hanessian and V. Pham, Org. Lett., 2000, 2, 2975 CrossRef CAS.
- C. E. T. Mitchell, A. J. A. Cobb and S. V. Ley, Synlett, 2005, 611 CAS.
- M. T. Reetz, S. Huette and R. Goddard, J. Am. Chem. Soc., 1993, 115, 9339 CrossRef CAS.
- H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, J. Am. Chem. Soc., 1995, 117, 6194 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |