Non-covalent expansion of an organic bilayer involving exo-cavity interplay of tetraphenylphosphonium with para-sulfonato-calix[4]arene
Mohamed
Makha
,
Alexandre N.
Sobolev
and
Colin L.
Raston
*
Chemistry M313, School of Biomedical, Biomolecular and Chemical Sciences, University of Western Australia, Crawley, Perth, WA 6009, Australia. E-mail: clraston@chem.uwa.edu.au; Fax: (+61) 8 6488 1005
First published on 16th November 2005
Abstract
Reaction of equimolar amounts of sodium para-sulfonato-calix[4]arene, tetraphenylphosphonium chloride and ytterbium chloride in water results in the formation of a mineral clay-like structure, where the hydrophobic tetraphenylphosphonium cations interpose a bilayer arrangement based on a 2D coordination polymer of (calix[4]arene)5−/Na+/Yb3+.
Layered materials that mimic the structure and properties of naturally occurring clays1 are of interest in supramolecular chemistry2 and crystal engineering.3 Major research efforts have been directed towards developing solids that intercalate or adsorb ions and small molecules, especially hydrogen, for storage,4 separations5 and catalysis.6 Amphiphilic para-sulfonato-calix[4]arene can serve as a platform to construct inorganic/organic clay-like materials that mimic naturally occurring clays. Atwood et al. pioneered the use of para-sulfonato-calix[4]arene in building-up such structures, initially as a bilayer arrangement with the calixarenes arranged in an up/down fashion.7 Subsequent investigations on the solid state supramolecular complexes of the same calixarene8 revealed the prevalence of this structural motif and perturbations thereof.7 The bilayer arrangement is comprised of hydrophobic layers of adjacent calixarenes engaged in π-stacking interactions, and a hydrophilic domain between these layers containing included water molecules, metal ions and various organic molecules such as crown ethers, amino acids, peptides, and nucleic acids and bases.9,10
Pseudo-polymorphic tetraphenylphosphonium complexes of para-sulfonato-calix[4]arene11 have a phenyl group of the cation that snugly fits into the cavity of the calixarene, which is consistent with electrostatic considerations for the components in the structures, and associated with a description of the bilayer arrangement, Fig. 1.9 Other departures from the bilayer arrangement for complexes of the same calixarene include the formation of spectacular spheroidal and tubular arrays that are formed under special conditions.12 Formation of the above tetraphenylphosphonium complexes is templated by lanthanide ions, but these ions are not incorporated into the structures.11 This is surprising given the higher positive charge density of the metal ions compared to the phosphonium cations, which is expected to favour the cations being drawn into close proximity to the sulfonate groups at the expense of the phosphonium cations.
![Schematic diagram showing the structural types of para-sulfonato-calix[4]arene interaction with Ph4P+ and ytterbium cations, depending on the relative amounts of the components.](/image/article/2006/CC/b513543e/b513543e-f1.gif) | ||
| Fig. 1 Schematic diagram showing the structural types of para-sulfonato-calix[4]arene interaction with Ph4P+ and ytterbium cations, depending on the relative amounts of the components. | ||
Herein we report a profound structural variation based on the same supramolecular tectons, notably tetraphenylphosphonium cations and para-sulfonato-calix[4]arene anions. In this case, an organic clay-like structure, 1, is formed, but now with the same lanthanide cations incorporated into the structure, Fig. 2 and Fig. 3, and interestingly with the calixarene taking on a −5 charge and with water included in the cavity, Fig. 1 and Fig. 4.
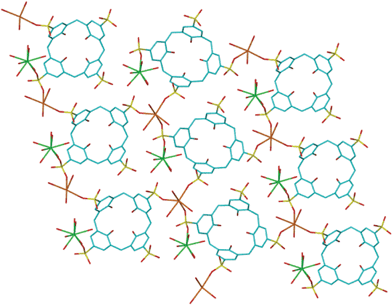 | ||
| Fig. 2 Projection of the 2D coordination polymer and associated metal interactions with the calixarene sulfonato groups in 1. | ||
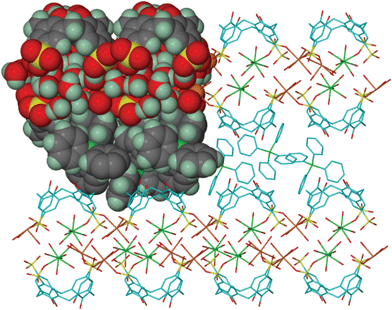 | ||
| Fig. 3 The extended structure of 1 showing the restriction of the coordination polymer within each layer of calixarenes and the inclusion of the phosphonium cations between these layers. | ||
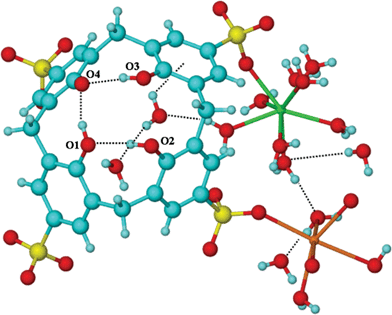 | ||
| Fig. 4 Projection of calixarene and associated water molecules plus metal ions. Important distances (Å): O1⋯H–O2 1.89 (O1⋯O2 2.703), O4⋯O1–H 1.68 (O1⋯O4 2.505), O4⋯H–O3 1.67 (O4⋯O3 2.503), O2⋯O3 2.946; dotted lines represent H-bonds. | ||
Slow evaporation of equimolar mixtures of tetraphenylphosphonium chloride, sodium para-sulfonato-calix[4]arene and ytterbium(III) chloride afforded a crystalline complex, 1,† which was characterised by a single crystal X-ray diffraction study. The reproducibility of the formation of the complex was checked by comparing the unit cell dimensions of different samples that appeared to be of uniform morphology (yield 40%). Complex 1 crystallises in the space group P21/c and has overall composition [{para-sulfonato-calix[4]arene}5−{Na(H2O)3}+{Yb(H2O)7}3+] [(C6H5)4P)+]·5H2O, i.e. the calixarene takes on an overall charge of −5, with one of the phenolic groups being deprotonated. This is not uncommon for bilayer structures based on this calixarene in the cone conformation.13 The structure was of sufficient accuracy to locate all the hydrogen atoms, and clearly showed one of the phenolic oxygen centres to be deprotonated and three hydrogen bonds to the three phenolic OH groups, Fig. 4 (O⋯O = 2.503–2.946 Å). In contrast, the complex formed in the presence of excess phosphonium cations showed all four phenolic OH groups.
Within the bilayers, individual layers of calixarenes form a 2D coordination polymer where sodium cations are bound to three sulfonate groups of three nearest-neighbouring calixarenes (Na–O = 2.313(2), 2.465(2) and 2.284(2) Å). Three water molecules complete the six-coordinate environment around each sodium cation (Na–O = 2.427(2), 2.377(2) and 2.643(2) Å). The ytterbium cation is hepta-aqua with one linkage to a sulfonate group (Yb–O = 2.299(2) Å); an oxygen of the same sulfonate group is also bound to a sodium ion, Fig. 2.
The structure is not heavily hydrated relative to related complexes devoid of the phosphonium cations.11 Five included water molecules, and coordinated water molecules form an intricate hydrogen bonded network, Fig. 4. Two of the included hydrogen bonded water molecules are situated close to the cavity of the calixarene (O⋯O = 2.887(3) Å), one residing close to an aromatic ring of the calixarene at a distance consistent with the formation of a weak π-arene non-classical hydrogen bond interaction (closest OH⋯arene centroid distance = 2.52 Å). The remaining included water molecules are at hydrogen bonding distances from two water ligands coordinated to the ytterbium cation (O⋯O separation = 2.721(3) and 2.613 (3) Å), and one is at a hydrogen bonding distance from a water molecule coordinated to the sodium cation (O⋯O = 2.769 (3) Å).
A striking feature of the structure is the intercalation of tetraphenylphosphonium cations between the layers of calixarenes at the interface of their hydroxyl and phenolate groups, Fig. 3. Thus, the electrostatic attraction of the negatively-charged lower rim of the calixarenes to the organic cations expands the bilayer arrangement. This establishes a precedent for forming supersized hydrophobic bilayers without carrying-out a covalent synthesis, i.e. incorporating pendant moieties on the amphiphile.
The overall structure is reminiscent of the arrangement of clay minerals and ion exchange resins.14 This is unlike the structure of the complex formed in the presence of excess phosphonium cations, where one arm of the cation is in the cavity of the calixarene, Fig. 1.10,11
The tetraphenylphosphonium cations form columnar arrays through continuous phenyl embraces15 involving CH⋯π interactions (CH⋯π = 2.652 Å), Fig. 5. The phosphonium cations are also involved in CH⋯π interactions with the aromatic rings of the calixarenes (CH⋯π = 2.817 Å).
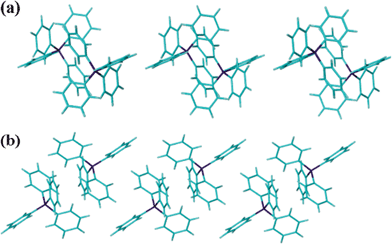 | ||
| Fig. 5 Projections (a) along and (b) orthogonal to the columns of phenyl-embraced phosphonium cations in 1. | ||
The formation of compound 1 has implications for crystal engineering. A simple reduction in the relative amount of phosphonium cation results in exclusion of the phosphonium cations from the cavity of the calixarene. Moreover, the phosphonium cations are now arranged within the bilayers in phenyl-embraced columnar arrays, whereas the complexes formed in the presence of excess phosphonium cations have the calixarenes in columnar arrays. Thus, the ratio of the large cations and anions is critical to the outcome of the interaction of the components. This finding is restricted to the presence of one mole equivalent of aquated ytterbium cations, and clearly a combinatorial approach is needed to map out the structural diversity for this system at various ratios, and also to quantify the effect of changing the nature of the lanthanide ions, as well as the ionic strength of the solution. It may be possible to form either the icosahedron or cuboctahedron arrangements of twelve calixarenes12 with tetraphenylphosphonium ions in the core. Such arrangements would have the phosphonium cations at the base of the calixarenes, as in the present structure. There is also the possibility of larger micelle-like arrangements and further expansion of the bilayers without resorting to covalent chemistry.
We thank the ARC for their support of this work.
Notes and references
- K. T. Holman, A. M. Pivovar, J. A. Swift and M. D. Ward, Acc. Chem. Res., 2001, 34, 107 CrossRef CAS; M. J. Zaworotko, Chem. Commun., 2001, 1 RSC.
- J.-M. Lehn, Supramolecular Chemistry - Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- G. R. Desiraju, Crystal engineering: From molecules to materials, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS; A. M. Beatty, K. E. Granger and A. E. Simpson, Chem.–Eur. J., 2002, 8, 3256.
- C. Liu, Y. Y. Fan, M. Liu, H. T. Cong, H. M. Cheng and M. S. Dresselhaus, Science, 1999, 286, 1127 CrossRef CAS; A. Matsumoto, T. Odani, K. Sada, M. Miyata and K. Tashiro, Nature, 2000, 405, 328 CrossRef CAS; M. Fichtner, Adv. Eng. Mater., 2005, 7, 443–455 CrossRef CAS.
- R. Bishop, Chem. Soc. Rev., 1996, 25, 311 RSC.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., 1999, 111, 58 ( Angew. Chem., Int. Ed. , 1999 , 38 , 56 ) CrossRef.
- A. W. Coleman, S. G. Bott, S. D. Morley, C. M. Means, K. D. Robinson, H. Zhang and J. L. Atwood, Angew. Chem., Int. Ed. Engl., 1988, 27, 1361 CrossRef.
- J. L. Atwood, F. Hamada, K. D. Robinson, G. W. Orr and R. L. Vincent, Nature, 1991, 349, 683 CrossRef CAS; J. L. Atwood, D. L. Clark, R. K. Juneja, G. W. Orr, K. D. Robinson and R. L. Vincent, J. Am. Chem. Soc., 1992, 114, 7558 CrossRef CAS; Z. Asfari, J. Harrowfield, P. Thuéry and J. Vicens, Supramol. Chem., 2003, 15, 69 CrossRef CAS; J. L. Atwood, S. J. Dalgarno, M. J. Hardie and C. L. Raston, New J. Chem., 2004, 28, 326 RSC; S. J. Dalgarno, M. J. Hardie, J. L. Atwood and C. L. Raston, Inorg. Chem., 2004, 43, 6351 CrossRef CAS; J. L. Atwood, L. J. Barbour, S. Dalgarno, C. L. Raston and H. R. Webb, J. Chem. Soc., Dalton Trans., 2002, 23, 4351 RSC; S. G. Bott, A. W. Coleman and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 610 CrossRef CAS; A. Drljaca, M. J. Hardie, C. L. Raston and L. Spiccia, Chem.–Eur. J., 1999, 5, 2295 CrossRef CAS; A. Drljaca, M. J. Hardie and C. L. Raston, J. Chem. Soc., Dalton Trans., 1999, 3639 RSC; A. Drljaca, M. J. Hardie, J. A. Johnson, C. L. Raston and H. R. Webb, Chem. Commun., 1999, 1135 RSC; J. L. Atwood, L. J. Barbour, E. S. Dawson, P. C. Junk and J. Kienzle, Supramol. Chem., 1996, 7, 271 CrossRef CAS; L. J. Barbour, A. K. Damon, G. W. Orr and J. L. Atwood, Supramol. Chem., 1996, 7, 167 CrossRef CAS; J. L. Atwood, L. J. Barbour, M. J. Hardie, C. L. Raston, M. N. Statton and H. R. Webb, CrystEngComm, 2001, 4 Search PubMed; J. L. Atwood, L. J. Barbour, M. J. Hardie, E. Lygris, C. L. Raston and H. R. Webb, CrystEngComm, 2001, 10 Search PubMed; P. C. Leverd, P. Berthault, M. Lance and M. Nierlich, Eur. J. Org. Chem., 2000, 133 CrossRef CAS; S. Shinkai, K. Araki, T. Matsuda, N. Nishiyama, H. Ikeda, I. Takasu and M. Iwamoto, J. Am. Chem. Soc., 1990, 112, 9053 CrossRef; H. Iki, H. Tsuzuki, H. Kijima, I. Hamachi and S. Shinkai, Supramol. Chem., 1995, 4, 223; M. Selkti, A. W. Coleman, I. Nicolis, N. Douteau-Guevel, F. Villain, A. Tomas and C. de Rango, Chem. Commun., 2000, 161 RSC.
- P. J. Nichols and C. L. Raston, Dalton Trans., 2003, 2923 RSC; J. L. Atwood, T. Ness, P. J. Nichols and C. L. Raston, Cryst. Growth Des., 2002, 2, 171 CrossRef CAS; J. L. Atwood, L. J. Barbour, E. S. Dawson, P. C. Junk and J. Kienzle, Supramol. Chem., 1996, 7, 271–274 CrossRef CAS.
- P. J. Nichols, M. Makha and C. L. Raston, Dalton Trans. Search PubMed , submitted for publication.
- M. Makha, C. L. Raston, A. N. Sobolev and A. H. White, Chem. Commun., 2004, 1066 RSC.
- G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef; J. L. Atwood, L. J. Barbour, S. J. Dalgarno, M. J. Hardie, C. L. Raston and H. R. Webb, J. Am. Chem. Soc., 2004, 126, 13170–13171 CrossRef CAS.
- A. W. Coleman, S. G. Bott, S. D. Morley, C. M. Means, K. D. Robinson, H. Zhang and J. L. Atwood, Angew. Chem., 1988, 100, 1412 CrossRef CAS; J. L. Atwood, G. W. Orr, N. C. Means, F. Hamada, H. Zhang, S. G. Bott and K. D. Robinson, Inorg. Chem., 1992, 31, 603 CrossRef CAS; J. L. Atwood, A. W. Coleman, H. Zhang and S. G. Bott, J. Inclusion Phenom., 1989, 7, 203 CAS.
- J. A. Ferrell, W. K. Vencill, K. Xia and T. L. Grey, Pest Manage. Sci., 2005, 61, 40–46 Search PubMed.
- I. Dance and M. J. Scudder, Chem.–Eur. J., 1996, 2, 481–486 CrossRef CAS; I. Dance and M. J. Scudder, J. Chem. Soc., Dalton Trans., 1998, 3155–3165 RSC.
Footnotes |
| † Synthesis of compound 1, modelled as (C28H39O26S4NaYb)·(C24H20P)·5H2O: To a warm aqueous solution of sodium para-sulfonato-calix[4]arene (20 mg, 24.1 µmol)‡ and tetraphenylphosphonium chloride (10 mg, 24.1 µmol), was added ytterbium(III) chloride hexahydrate (10 mg, 24.1 µmol). The warm mixture was allowed to slowly evaporate overnight, affording large colourless crystals that were suitable for X-ray diffraction studies. Crystal data: C52H69O31NaPS4Yb, M = 1545.31, monoclinic, space group P21/c, a = 21.271(3), b = 11.375(2), c = 26.372(4) Å, β = 104.908(2)°, V = 6166(17) Å3, Dc = 1.665 g cm−3, Z = 4, µMo = 1.778 mm−1, T = 153 K, Mo-Kα radiation (λ = 0.71073 Å), θmax = 29°, Nt = 58944, R1 = 0.0297, wR2 = 0.0637, GOF = 1.03 for 12980 reflections with I > 2σ(I). CCDC 284293. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513543e |
| ‡ Sodium para-sulfonato-calix[4]arene was prepared by the reaction of concentrated sulfuric acid with para-tert-butyl-calix[4]arene, followed by precipitation on addition of sodium chloride solution. Tetraphenylphosphonium chloride and ytterbium chloride were purchased from Aldrich. |
| This journal is © The Royal Society of Chemistry 2006 |