Mechanism of α-oxoamine synthases: identification of the intermediate Claisen product in the 8-amino-7-oxononanoate synthase reaction†
Olivier
Kerbarh
,
Dominic J.
Campopiano
and
Robert L.
Baxter
*
School of Chemistry, Joseph Black Building, The University of Edinburgh, West Mains Road, Edinburgh, UK EH9 3JJ. E-mail: r.baxter@ed.ac.uk; Fax: +44 (0)131 6507155; Tel: +44 (0)131 6504708
First published on 14th November 2005
Abstract
The reactive β-ketoacid pyridoxal-5′-phosphate aldimine formed in the condensation step of the 8-amino-7-oxononanoate synthase reaction was ‘trapped’ in the enzyme-bound form by carrying out the reaction with L-alanine methyl ester and pimeloyl-CoA affording the more stable methyl ester of the putative intermediate, the characterisation of which provides the first definitive evidence for a β-ketoacid intermediate in an α-oxamine synthase mechanism.
The biosynthesis of biotin (vitamin H), the cofactor in most biological carboxylation and transcarboxylation reactions, follows a ubiquitous pathway in plants and microorganisms.1 8-Amino-7-oxononanoate synthase (AONS) (EC 2.3.1.47) is the pyridoxal-5′-phosphate (PLP)-dependent enzyme which catalyses the first committed step in the four step pathway, the decarboxylative condensation of L-alanine 1 and pimeloyl-CoA 2 to give (7S)-8-amino-7-oxononanoate 3 (Scheme 1).1,2
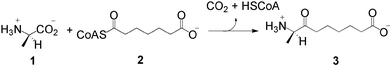 | ||
| Scheme 1 The 8-amino-7-oxononanoate synthase reaction. | ||
AONS is a member of the α-oxoamine synthase family of enzymes, a small, but widespread, group of proteins that typically catalyse Claisen condensations between amino acids and acyl-CoA thioesters with concomitant decarboxylation.3 The family includes 5-aminolevulinate synthase (ALAS), the enzyme responsible for generating the precursor of the porphyrin rings of hemes in microorganisms and animals;4 serine palmitoyltransferase (SPT), which catalyses the first step in sphingolipid biosynthesis;5 and 2-amino-3-ketobutyrate CoA ligase (KBL), which is involved in the threonine degradation pathway.6 KBL is the only member of the family the mechanism of which does not involve a decarboxylation step. Mutations in some of these enzymes have been implicated in human disease states such as hereditary sideroblastic anemia caused by an amino acid substitution in erythroid-specific ALAS (ALAS2)7 and hereditary sensory neuropathy type I caused by mutations in the SPT subunit-1 (SPTLC1).8
Our understanding of the chemical mechanisms of the α-oxoamine synthase enzymes culminates from the results of early radiolabeling studies on Rhodobacter spheroides ALAS,9 and from kinetic studies on Escherichia coli2 and Bacillus sphericus10 AONS, and murine ALAS.11 Detailed crystallographic studies of E. coli AONS and KBL and their aldimine complexes have also been carried out.2,6,12 The overall reaction catalysed by AONS can be conjectured to take place by either of two feasible routes, A or B, summarised in Scheme 2. In the first, common, step (i) formation of a substrate external aldimine intermediate (AONS–Ala) occurs via a transaldimination reaction between L-alanine and the PLP-Lys236 internal aldimine. In the second step (ii) binding of pimeloyl-CoA causes a conformational change in the enzyme dimer and this appears to allow formation of a transient quinonoid intermediate, although whether this is the species AONS-Q1, formed by decarboxylation (route A), or AONS-Q2, formed by α-proton abstraction (route B), is uncertain. The next step (iii) involves the Claisen reaction of the quinonoid (AONS-Q1 or AONS-Q2) with pimeloyl-CoA 2 liberating HSCoA and leading to the formation of either the product aldimine (AONS-AON) directly, or the β-ketoacid aldimine complex (AONS-OA) which subsequently decarboxylates to form the quinonoid of the product (AONS-Q3). If the latter route (B) is involved protonation must then occur at C5 (step iv) to give AONS-AON. Finally transaldimination of the product aldimine results in release of 3 and restoration of the PLP-Lys236 internal aldimine.
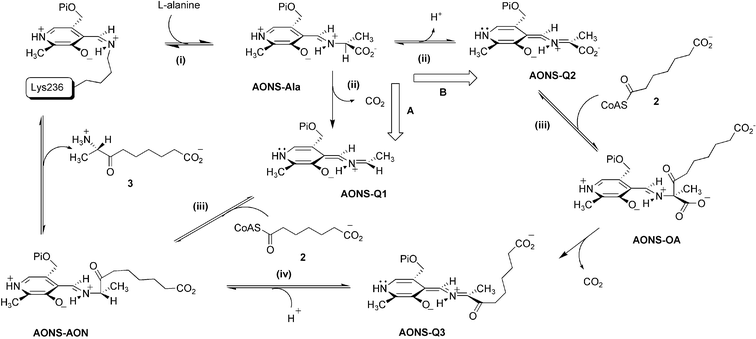 | ||
| Scheme 2 Two possible mechanisms for the AONS reaction. | ||
Evidence from kinetic isotope effects and the fact that racemic intermediate analogue, 8-amino-7-oxo-8-phosphononanoic acid, is an inhibitor of the B. sphericus enzyme provide some support for pathway B.10 However, our attempts to model the initial AONS–Ala complex in the active site of E. coli AONS were complicated by the flexibility of the enzyme and the presence of multiple catalytic residues which together did not allow us to make a distinction between decarboxylation and proton abstraction as the first committed step in the mechanism. Ascribing specific catalytic roles to individual residues on the basis of single mutations is complicated by the fact that the active site functionalities interact via a complex network of H-bonds.2 Invoking analogy by comparison with the established mechanisms of other PLP enzymes of known structure is also equivocal. To take only one relevant example—the AONS active site includes the side chains of Asn47 and Arg361 which are structurally equivalent to Gln52 and Arg406 of dialkylglycine decarboxylase (DGD). In the two different reactions catalysed by DGD, Gln52 binds the carboxylate in the course of decarboxylation and Arg406 binds it during transamination.13
To resolve this problem we examined the reaction using the methyl ester of L-alanine as a pseudo-substrate, arguing that it could not form a quinonoid species by decarboxylation but could do so if proton abstraction was involved. In the latter case this might also undergo condensation to afford the methyl ester of the β-ketoacid–aldimine complex (AONS-OA–Me), which cannot undergo decarboxylation and should therefore accumulate. There is adequate precedence in using methyl ester analogues in studies of PLP-dependent enzymes. Amino acid esters have been used in studies of histidine decarboxylase, DOPA decarboxylase and SPT and shown to give the corresponding enzyme-bound amino acid aldimine methyl esters.5c,14
In previous work we demonstrated that the first step in the reaction sequence, L-alanine external aldimine (AONS–Ala) formation, is rapid (k1 = 2 × 104 M−1 s−1) and that formation of the substrate quinonoid species, characterised by an absorbance peak at 486 nm, does not occur to any measurable extent in the absence of the second substrate but occurs rapidly (kobs = 45 s−1) upon addition of pimeloyl-CoA to the pre-formed AONS–Ala complex.2,15 In marked contrast to the behavior with L-alanine, the binding of L-alanine methyl ester to AONS in the absence of pimeloyl-CoA results in the extremely rapid (kobs = 68.25 s−1) appearance of a strong absorbance at 486 nm that can be attributed to the quinonoid intermediate, AONS-Q2 methyl ester (see Scheme 2, data not shown). This is followed by a slow decrease of the intensity of this band (kobs = 8.50 s−1) and a concomitant increase in the absorbances at 425 and 324 nm, indicative of Cα protonation of the quinonoid species to afford an external aldimine.16 The fact that reaction of the substrate methyl ester with the enzyme, in the absence of pimeloyl-CoA, can lead directly to the formation of a quinonoid intermediate may be explained, at least partly, by the different pKa of the Cα proton of the methyl ester. It has been shown that the pKa for the protonated form of the PLP aldimine of alanine is significantly lower (ca. 3 units) than that of the carboxylate form.17
The spectroscopic changes attendant on reaction of both L-alanine methyl ester and pimeloyl-CoA with the enzyme are shown in Fig. 1. Initially there is a dramatic increase in the absorption at 486 nm indicative of formation of the AONS-Q2 methyl ester species. However, here the pseudo-first order rate constant for quinonoid formation is 32.3 s−1, roughly half the rate observed for quinonoid formation in the absence of pimeloyl-CoA. The results for L-alanine and its methyl ester are quite different—pimeloyl-CoA binding is required for Cα deprotonation of AONS–Ala but it reduces the rate of deprotonation of the AONS–Ala methyl ester analog. This, coupled with the comparability of the quinonoid formation rates for L-alanine and its methyl ester, would suggest that substrate binding to AONS is not completely ordered and that some inhibition of the binding of the amino acid substrate occurs through unproductive binding of pimeloyl-CoA with the holoenzyme.
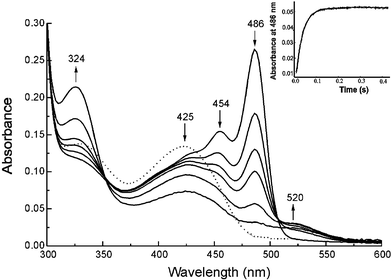 | ||
| Fig. 1 Time-course of the reaction of AONS (20 µM) with L-alanine methyl ester (10 mM) and pimeloyl-CoA (100 µM) in potassium phosphate buffer (20 mM, pH 7.5) at 25 °C. The dotted line shows the spectrum of the holoprotein before addition of substrates. Absorption spectra were recorded on a HP8453 single beam diode-array spectrophotometer as a function of time (0, 5, 10, 15, 30, 60 min). The arrows at each wavelength indicate an increase or decrease in absorbance over the course of the reaction. The insert (upper right) shows a stopped-flow absorbance time-course of the reaction of 50 µM AONS rapidly mixed with 10 mM L-alanine methyl ester and 100 µM pimeloyl-CoA under the same conditions using a Bio-Logic SFM-3 stopped-flow instrument equipped with a MOS-450 spectrometer. The change in absorption at 486 nm was followed for 400 ms. | ||
In marked contrast to the reaction with L-alanine, reaction with the methyl ester in the presence of pimeloyl-CoA gives rise to the accumulation of a new enzyme-bound species with an absorbance maximum at 454 nm. This is not due to the product quinonoid (AONS-Q3), since this absorbs at 520 nm, or to the product aldimine (AONS-AON), since this absorbs at ca. 436 nm, and thus appears likely to be due to accumulation of the β-ketoacid methyl ester aldimine complex (AONS-OA methyl ester, 4) which cannot undergo enzymatic decarboxylation. In time, there is a gradual decrease in the intensity of this latter peak and appearance of a 520 nm absorbance characteristic of the product quinonoid (AONS-Q3). The rate of appearance of this last peak correlates with the rate of HSCoA formation (kobs = 0.06 s−1) in the steady-state assay and this may be attributed to slow hydrolysis of the methyl ester of the AONS-OA–Me complex and subsequent decarboxylation. This slow hydrolysis is perhaps not unexpected since the crystal structure of the AONS product aldimine suggests that one face of the active site is relatively solvent accessible.2
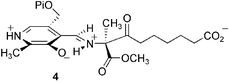
To ascertain whether the 454 nm absorption was indeed due to the putative β-ketoacid methyl ester aldimine complex 4 a sodium cyanoborohydride reduction of the incubation mixture was carried out to reduce the unstable imine bond of the complex. Electrospray mass spectrometry of the products revealed, in addition to peaks at 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 473 (±20) and 41707 (±20) amu corresponding to the apo- and the holoenzymes (predicted masses 41463 and 41690 amu), a new peak at 41957 (±21) amu corresponding to AONS monomer plus the reduced β-ketoacid methyl ester aldimine (predicted mass 41937 amu). This provides convincing evidence that the β-ketoacid intermediate (AONSOA) shown in Scheme 2 (route B) is indeed a late intermediate in the reaction sequence.
473 (±20) and 41707 (±20) amu corresponding to the apo- and the holoenzymes (predicted masses 41463 and 41690 amu), a new peak at 41957 (±21) amu corresponding to AONS monomer plus the reduced β-ketoacid methyl ester aldimine (predicted mass 41937 amu). This provides convincing evidence that the β-ketoacid intermediate (AONSOA) shown in Scheme 2 (route B) is indeed a late intermediate in the reaction sequence.
Modelling of the β-ketoacid aldimine into the active site conformation of AONS adopted in the AON aldimine (product) structure is shown in Fig. 2.2
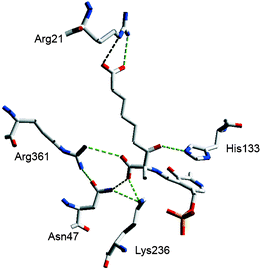 | ||
| Fig. 2 Modelling of the β-ketoacid PLP aldimine intermediate in the AONS active site showing the H-bonding network. His207 which H-bonds to Asn47 and Lys236 and the imine N of PLP is omitted for clarity It is noteworthy that mutations to Lys236, His133 and Asn47 destroy catalytic activity but that an Arg361 mutation (R361C) is marginally active (kcat = 0.002 s−1). Produced using Swiss PdB viewer. | ||
The alanine derived carboxylate can form H-bonds with the side chains of Arg361, Asn47 and Lys236 of the same protein monomer. In this structure the C8-carboxylate bond cannot adopt the Dunathan intermediate orientation (aligned orthogonal to the plane of the Schiff's base) which provides the stereoelectronic interactions required for facile decarboxylation.18 The structure suggests that the C7 ketone, protonated by His133, may play a role as an electron sink in the decarboxylation.
This study was funded by the BBSRC.
Notes and references
- M. A. Eisenberg, in Escherichia coli and Salmonella typhimurium, ed. F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter and H. E. Umbarger, American Society for Microbiology, Washington, DC, 1987, vol. 1, pp. 544–550 Search PubMed.
- S. P. Webster, D. Alexeev, D. J. Campopiano, R. M. Watt, M. Alexeeva, L. Sawyer and R. L. Baxter, Biochemistry, 2000, 39, 516 CrossRef CAS.
- A. C. Eliot and J. F. Kirsch, Ann. Rev. Biochem., 2004, 73, 383 CrossRef CAS; D. E. Metzler, Biochemistry, the chemical reactions of living cells, Harcourt/Academic Press, San Diego, CA, 2001, vol. 1, pp 737–753 Search PubMed.
- G. C. Ferreira, P. J. Neame and H. A. Dailey, Protein Sci., 1993, 2, 1959 CrossRef CAS.
- (a) K. Gable, G. Han, E. Monaghan, D. Bacikova, M. Natarajan, R. Williams and T. M. Dunn, J. Biol. Chem., 2002, 277, 10194 CrossRef CAS; (b) K. Hanada, Biochim. Biophys. Acta, 2003, 1632, 16 CAS; (c) H. Ikushiro, H. Hayashi and H. Kagamiyama, Biochemistry, 2004, 43, 1082 CrossRef CAS.
- J. Schmidt, J. Sivaraman, Y. G. Li, R. Larocque, J. Barbosa, C. Smith, A. Matte, J. D. Schrag and M. Cygler, Biochemistry, 2001, 40, 5151 CrossRef.
- J. Edgar, M. S. Losowsky, J. S. Noble and S. N. Wickramasinghe, Eur. J. Haematol., 1997, 58, 1; P. M. Shoolingin-Jordan, S. Al-Daihan, D. Alexeev, R. L. Baxter, S. S. Bottomley, I. Kahari, R. Donald, S. Ipsita, M. Sarwar, L. Sawyer and S.-W. Wang, Biochim. Biophys. Acta, 2003, 1647, 361 CAS.
- J. L. Dawkins, D. J. Hulme, S. B. Brahmbhatt, M. Auer-Grumbach and G. A. Nicholson, Nat. Genet., 2001, 27, 309 CrossRef CAS.
- Z. Zamam, P. M. Jordan and M. Akhtar, Biochem. J., 1973, 135, 257; M. M. Abboud, P. M. Jordan and M. Akhtar, J. Chem. Soc., Chem. Commun., 1974, 643 RSC.
- O. Ploux and A. Marquet, Eur. J. Biochem., 1996, 236, 301 CAS; O. Ploux, O. Breyne, S. Carrillon and A. Marquet, Eur. J. Biochem., 1999, 259, 63 CrossRef CAS.
- G. A. Hunter and G. C. Ferreira, Biochemistry, 1999, 38, 12526 CrossRef CAS; G. A. Hunter and G. C. Ferreira, J. Biol. Chem., 1999, 274, 12222 CrossRef CAS; J. S. Zhang and G. C. Ferreira, J. Biol. Chem., 2002, 277, 44660 CrossRef CAS.
- D. Alexeev, M. Alexeeva, R. L. Baxter, D. J. Campopiano, S. P. Webster and L. Sawyer, J. Mol. Biol., 1998, 284, 401 CrossRef CAS.
- M. D. Toney, E. Hohenester, J. W. Keller and J. N. Jansonius, J. Mol. Biol., 1995, 245, 151 CrossRef CAS.
- H. Hayashi, H. Mizuguchi and H. Kagamiyama, Biochemistry, 1993, 32, 812 CrossRef CAS; P. S. Moore, M. Bertoldi, P. Dominici and C. B. Voltattorni, FEBS Lett., 1997, 412, 245 CrossRef CAS; M. Olmo, F. Sanchez-Jimenez, M. Medina and H. Hayashi, J. Biochem. (Tokyo), 2002, 132, 433 Search PubMed.
- The catalytic constants for E. coli AONS have been redetermined in this study: kcat = 0.17 (0.02) s−1; Km [pimeloyl-CoA] = 142 (25) µM; Km [L-alanine] = 1.14 (0.12) mM.
- L-Alanine methyl ester is also a competitive inhibitor (Ki = 2.8 mM). It seems reasonable to suggest that, in the absence of the second substrate, protonation of the methylated quinonoid adduct occurs on the solvent accessible Si face to give the unproductive enantiomeric D-alanine aldimine.
- J. E. Dixon and T. C. Bruice, Biochemistry, 1973, 12, 4762 CrossRef CAS; S. D. Fratte, S. Iurescia, S. Angelaccio, F. Bossa and V. Schirch, Eur. J. Biochem., 1994, 225, 395 CrossRef.
- M. D. Toney, Arch. Biochem. Biophys., 2005, 433, 279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the characterisation of the intermediate AONS-OA–Me by mass spectrometry. See DOI: 10.1039/b511837a |
| This journal is © The Royal Society of Chemistry 2006 |