Investigating the latent polymorphism of maleic acid†
Graeme M.
Day
a,
Andrew V.
Trask
a,
W. D. Samuel
Motherwell
b and
William
Jones
*a
aThe Pfizer Institute for Pharmaceutical Materials Science, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: wj10@cam.ac.uk; Fax: +44 1223 336362; Tel: +44 1223 336468
bThe Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK CB2 1EZ
First published on 21st November 2005
Abstract
The unexpected appearance of a new polymorph of maleic acid is reported and a computational study addresses the predictability of this new polymorph and future potential polymorphism.
The phenomenon of polymorphism, well-known and widely studied, is the ability of a chemical substance to adopt more than one crystal structure. Different crystal structures of the same chemical material offer distinct physicochemical properties; as a result, polymorphism has attracted interest from a variety of industries, including pharmaceuticals, agrochemicals, pigments, dyestuffs, foods and explosives. A great deal of research has illustrated the intricacies of polymorphic behaviour in general,1 but, despite decades of investigation, a real understanding continues to elude researchers intent on reliably controlling and predicting the emergence of different forms. Indicative of the complexity of the matter is the continual emergence of experimental variables that may affect the polymorphic outcome of a crystallisation.2 Screening for polymorphs may require complementing the creative use of crystallisation variables with high-throughput methods that attempt to counter the stochastic nature of crystallisation with thousands of parallel experiments.3 Furthermore, developments in computational methods of predicting crystal structures add a new tool to the understanding and anticipation of polymorphism.
In light of the current research interest in polymorphism, McCrone's assertion that “the number of forms known for a given compound is proportional to the time and money spent in research on that compound”4 increasingly appears to have been prophetic. McCrone cited as supporting evidence the fact that all chemicals which may be described as “common and important” were in fact known to be polymorphic; the list of chemicals to which this descriptor may be applied is surely subjective, and indeed will have evolved over the course of forty years. Despite the apparent sagacity of McCrone's statement, a great many chemicals today remain monomorphic, belying the belief that “every compound has different polymorphic forms”.4 The discrepancy was noted by Dunitz and Bernstein,5 who observed that fewer than 5% of the compounds in the Cambridge Structural Database (CSD) are reported to be polymorphic.‡ Furthermore, they stated that “some very widely studied compounds have shown no evidence of polymorphic behaviour, even though they have been crystallised and handled for many years under a far-ranging variety of conditions.”5
Maleic acid is an example of one such compound. Crystals of maleic acid were studied as early as 1881, at which time crystal data were reported by Bodewig.6 X-ray analysis was performed in 1925 to assist in space group determination,7 and additional determinations followed in 1939, 1952 and 1974:8 each of these described the same crystal form.§ Today, maleic acid is a bulk product of the chemical industry, produced in quantities in excess of 1000 tonnes per year,¶ and is an extensively used salt-forming agent in the pharmaceutical industry.||
It was thus to our surprise that a second polymorph was obtained in our lab, 124 years after evidence of the first crystal form was reported. In the course of crystallisation experiments of a series of cocrystals of pharmaceutical molecules,10 a cocrystal of caffeine and maleic acid (2 ∶ 1), which had been prepared by solid-state grinding, was dissolved in chloroform with gentle heating and allowed to evaporate at ambient temperature and pressure. Colourless plate-like single crystals were observed in the mother liquor after one day and submitted for XRD structure determination, leading to the structure of maleic acid form II (space group Pc).** For comparison, the previously reported form I (P21/c) was grown by vapour diffusion of n-hexane into a chloroform solution of maleic acid and the structure was solved at the same temperature (T = 180 K).††
That polymorphism has been observed in maleic acid after such a long era of monomorphism is remarkable, so we sought to understand whether the existence of form II may have been predictable, and indeed whether any additional polymorphs may potentially emerge in the future. A computational study was undertaken to determine the most probable polymorphic forms of this molecule by searching packing space for the structures with lowest lattice energy, following essentially the same methodology as applied previously.11,12 Such computational studies now sometimes play a role in, or inspire, polymorph screens, occasionally leading to new forms.13
Considering conformational flexibility in computational searches for crystal structures is a major challenge, because of the difficulties involved with simultaneously treating inter- and intramolecular degrees of freedom.14 We therefore examined the CSD for guidance in choosing a molecular model that we could treat as rigid during our calculations. We describe the molecular conformation in reported crystal structures of n = 4 α,ω unsaturated cis-dicarboxylic acids by the torsion angles about the three C–C bonds (Scheme 1) and the presence of the intramolecular hydrogen bond forming an S11(7) ring (Table 1). The search reveals that, ignoring cocrystals and solvates, reported crystal structures of n = 4 α,ω unsaturated cis-dicarboxylic acids almost exclusively adopt a nearly planar intramolecularly hydrogen bonded conformation (all τ ≈ 0°).
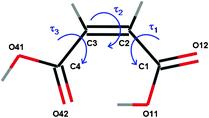 | ||
| Scheme 1 The molecular structure used in the search and numbering of the non-hydrogen atoms. | ||
| Refcode | S11(7) | τ 1 (deg.)a | τ 2 (deg.)b | τ 3 (deg.)c | |
|---|---|---|---|---|---|
| a τ 1 = O11–C1–C2–C3. b τ 2 = C1–C2–C3–C4. c τ 3 = O42–C4–C3–C2. See Scheme 1 for atom labels. | |||||
| BCOCDC | yes | −3.02 | 8.65 | −6.69 | Z′ = 2 |
| BCOCDC | yes | −19.77 | 0.01 | 21.14 | |
| BICYER | yes | 0.96 | 5.42 | 6.58 | Z′ = 2 |
| BICYER | yes | −3.45 | 0.94 | 2.04 | |
| CBUDCX | yes | −3.21 | 0.51 | 1.00 | |
| MALIAC | yes | 2.78 | 5.88 | 6.72 | |
| MALIAC02 | yes | 1.73 | 0.58 | 1.75 | |
| MALIAC11 | yes | 2.10 | 0.56 | 0.05 | |
| MCINDE | yes | 4.56 | 3.32 | −4.06 | |
| NEDNOZ | no | 5.22 | 1.34 | −98.30 | |
| QAPTUW | yes | −1.33 | 2.18 | 3.28 | |
| TELZOZ | yes | 5.93 | 2.50 | −7.14 | |
This observation simplified our computational study; we could confidently restrict our crystal structure search to this planar molecular geometry, optimised using density functional theory (using the program DMol315) and kept rigid throughout the calculations. The phase space in the 20 most common space groups for organic molecular crystals was explored using the simulated annealing algorithm implemented in the Cerius2 Polymorph Predictor module.16 During this initial generation of crystal structures, lattice energies were calculated using the W99 exp-6 repulsion-dispersion parameters17 and atomic charges fitted to the calculated molecular electrostatic potential. As it is well established12,18 that an atomic multipole description of electrostatic interactions leads to more reliable modelling of organic molecular crystals, the lowest energy predicted crystal structures were re-minimised in the program DMAREL19 with the atomic charges replaced by a distributed multipole analysis20 of the calculated charge density.
To aid comparison with our predicted structures, the two observed polymorphs were lattice energy minimised using the same model potential, after replacing the X-ray determined molecular structure with that of the optimised molecule. These energy minimised experimental structures (Fig. 1), represented by open circles on an energy vs. density plot of the lowest energy predictions (Fig. 2), are the closest to the two known forms that a lattice energy search with this model potential could generate. Indeed, these were found as the two lowest energy structures in the computational search – form II as the global minimum and form I 0.15 kJ/mol above. The predicted structures match the energy minimised versions of the observed structures within numerical accuracy (< 0.01 Å in lattice parameters).
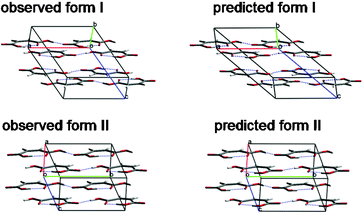 | ||
| Fig. 1 Observed and predicted polymorphs of maleic acid. | ||
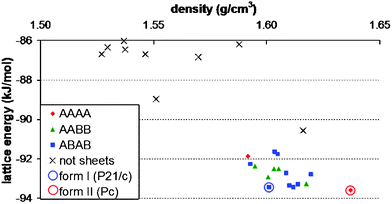 | ||
| Fig. 2 Energies and densities of the lowest energy predicted crystal structures of maleic acid (solid symbols and crosses) and energy minimised known polymorphs (open circles). | ||
Both polymorphs are built up from molecular sheets (Fig. 3), within which molecules interact via OH…O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds, forming 1D chains, which alternate in direction and interact through close contacts between anti-parallel aligned hydroxyl groups. The geometry of the layers in the observed structures is reproduced very well in these predicted structures and the lattice parameters of the global minimum are all within a few percent of form II's observed values. Form I is predicted less well – there is a slipping of the sheets between the observed structure and lattice energy minimum (Fig. 1), reflected in the 15° increase of β and associated 25% lengthening of the c-axis to maintain the 3 Å inter-planar distance. This distortion reflects the soft nature of the inter-planar interactions and shortcomings of the modelling method – limitations of the model potential, as well as the static, temperatureless nature of the energy minimisation approach.
C hydrogen bonds, forming 1D chains, which alternate in direction and interact through close contacts between anti-parallel aligned hydroxyl groups. The geometry of the layers in the observed structures is reproduced very well in these predicted structures and the lattice parameters of the global minimum are all within a few percent of form II's observed values. Form I is predicted less well – there is a slipping of the sheets between the observed structure and lattice energy minimum (Fig. 1), reflected in the 15° increase of β and associated 25% lengthening of the c-axis to maintain the 3 Å inter-planar distance. This distortion reflects the soft nature of the inter-planar interactions and shortcomings of the modelling method – limitations of the model potential, as well as the static, temperatureless nature of the energy minimisation approach.
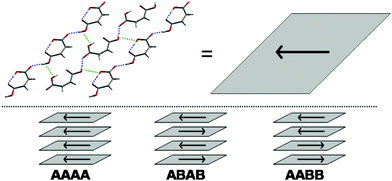 | ||
| Fig. 3 Stacking of the molecular sheets in the observed and predicted crystal structures, with arrows representing the dipole direction. Dashed lines indicate close intermolecular contacts. | ||
In fact, the 16 lowest energy structures all form the same molecular sheets and differ only in the stacking of these polar layers – both in their relative orientation and horizontal offset. These structures group into 3 classes based on the alternation of layers (Fig. 3): AAAA, where the layers have the same orientation in a polar structure; ABAB, where layers alternate direction; and AABB structures, where the direction swaps at every second layer. Within each group, the structures differ by slipping of the sheets over each other. We expect that the minima within each group are related by low energy barriers and, while they are distinct structures on the T = 0 K energy surface, they may easily interconvert at finite temperatures.‡‡ The barrier to transformation between these groups of structures, on the other hand, must be considerable, so stress-induced interconversion of structures with different stacking patterns is unlikely and the observed pattern of stacking must be determined early in the nucleation or growth regimes. Assuming that the molecules are then able to find the most favourable offset between layers, the lowest energy structure in each class is most likely to be observed. Indeed, forms I and II are the lowest in energy of the ABAB and AAAA groups, respectively. Of the other low energy structures, eight have the ABAB stacking of layers, one has an AAAA arrangement and five AABB; the most stable of these is a Z′ = 2 Pna21 crystal (Fig. 4), intermediate in density between the energy minimised forms I & II, only 0.18 kJ/mol higher in lattice energy than form I. Should form III ever be observed, this seems a likely candidate.
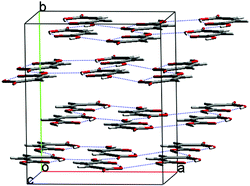 | ||
| Fig. 4 Packing diagram of the lowest energy AABB putative crystal structure of maleic acid. | ||
The appearance of maleic acid form II should serve as a warning against assuming that consistent production of one crystal form rules out the appearance of new polymorphs. Here, the presence of caffeine may have played a structure directing role in the growth of this latent crystal form—polymorphs of other common chemicals may be discovered with increasing interest in cocrystallisation and exploration of new solvent/cosolute combinations.21 Furthermore, computational modelling has allowed for a rationalisation of the emergence of the second polymorph as well as insight into other possible polymorphs that may emerge in the future.
We thank the Pfizer Institute for Pharmaceutical Materials Science for funding and Dr J. E. Davies for X-ray data collection and structure determination.
Notes and references
- J. Bernstein, Polymorphism in Molecular Crystals; Clarendon Press: Oxford, 2002 Search PubMed; B. Rodríguez-Spong, C. P. Price, A. Jayasankar, A. J. Matzger and N. Rodríguez-Hornedo, Adv. Drug Delivery Rev., 2004, 56, 241–274 Search PubMed.
- B. A. Garetz, J. Matic and A. S. Myerson, Phys. Rev. Lett., 2002, 89, 175501 CrossRef; J. M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382–3383 CrossRef CAS; A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter, Chem. Commun., 2005, 880–882 RSC.
- S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef.
- W. C. McCrone, in Physics and Chemistry of the Organic Solid State; D. Fox, M. M. Labes and A. Weissberger, Eds.; Interscience Publishers: New York, 1965; Vol. II, pp. 725–767 Search PubMed.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- C. Bodewig, Z. Kristallogr., 1881, 5, 559–560 Search PubMed.
- K. Yardley, J. Chem. Soc., Trans., 1925, 2207–2219 RSC.
- K. Lonsdale, Proc. R. Soc. London, Ser. A, 1939, 171, 541–568 Search PubMed; M. Shahat, Acta Crystallogr., 1952, 5, 763–768 CrossRef CAS; M. N. G. James and G. J. B. Williams, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1249–1257 CrossRef CAS.
- K. Schaum, K. Schaeling and F. Klausing, Justus Liebigs Ann. Chem., 1916, 411, 161–195 CrossRef CAS.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CrossRef CAS.
- G. M. Day, J. Chisholm, N. Shan, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2004, 4, 1327–1340 CrossRef CAS.
- G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1023–1033 CrossRef CAS.
- N. Blagden, W. I. Cross, R. J. Davey, M. Broderick, R. G. Pritchard, R. J. Roberts and R. C. Rowe, Phys. Chem. Chem. Phys., 2001, 3, 3819–3825 RSC; A. T. Hulme, S. L. Price and D. A. Tocher, J. Am. Chem. Soc., 2005, 127, 1116–1117 CrossRef CAS.
- C. Ouvrard and S. L. Price, Cryst. Growth Des., 2004, 4, 1119–1127 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- Cerius2, version 4.6, Accelrys Inc.: San Diego, 2001.
- D. E. Williams, J. Mol. Struct., 1999, 486, 321–347 CrossRef; D. E. Williams, J. Comput. Chem., 2001, 22, 1154–1166 CrossRef CAS; D. E. Williams, J. Comput. Chem., 2001, 22, 1–20 CrossRef CAS.
- D. S. Coombes, S. L. Price, D. J. Willock and M. Leslie, J. Phys. Chem., 1996, 100, 7352–7360 CrossRef CAS; G. M. Day, S. L. Price and M. L. Leslie, J. Phys. Chem. B, 2003, 107, 10919–10933 CrossRef CAS.
- DMAREL, version 3.1, S. L. Price, D. J. Willock, M. Leslie and G. M. Day, 2001.
- A. J. Stone and M. Alderton, Mol. Phys., 1985, 56, 1047–1064 CAS.
- See, for example, the reported behaviour of 1,3,5-trinitrobenzene in the presence of additives: P. K. Thallapally, R. K. R. Jetti, A. K. Katz, H. L. Carrell, K. Singh, K. Lahiri, S. Kotha, R. Boese and G. R. Desiraju, Angew. Chem., Int. Ed., 2004, 43, 1149–1155 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the computational methods and crystallisation conditions. Unit cell parameters, lattice energies and CIF files of the lowest energy predictions. See DOI: 10.1039/b513442k |
| ‡ This statistic could be biased by the fact that much crystal structure data serves the purpose of molecular structure confirmation; thus, the reporting of polymorphic structures may suffer as a result. |
| § Curiously, a report in 19169 included maleic acid among a lengthy list of polymorphic substances, but lacked any characterisation data. |
| ¶ Maleic acid is listed in the 2004 Organisation for Economic Co-operation and Development List of High Production Volume Chemicals: http://www.oecd.org/dataoecd/55/38/33883530.pdf |
| || Over 100 CSD entries represent maleate salts of organic compounds. |
| ** Interestingly, we have since then not been able to prepare the new form; this is possibly another case of disappearing polymorphism (see ref. 5) or, at least, a challenge for those investigating selective polymorph growth. |
| †† Crystal data for maleic acid form I: C4H4O4, M = 116.07, monoclinic, space group P21/c, a = 7.4749(8), b = 10.0849(12), c = 7.5465(7) Å, β = 124.423(6)°, V = 469.26(9) Å3, T = 180(2) K, Z = 4, μ(Mo-Kα) = 0.152 mm−1, Dc = 1.643 Mg/m3, λ = 0.71073 Å, 2θmax = 27.46°, 2647 reflections measured, 1061 unique (Rint = 0.0268). Final residuals for 79 parameters were R1 = 0.0387, wR2 = 0.0984 for I > 2σ(I), and R1 = 0.0510, wR2 = 0.1064 for all 1061 data. Crystal data for maleic acid form II: C4H4O4, M = 116.07, monoclinic, space group Pc, a = 3.6933(3), b = 7.4840(5), c = 8.5933(7) Å, β = 102.220(3)°, V = 232.14(3) Å3, T = 180(2) K, Z = 2, μ(Mo-Kα) = 0.154 mm−1, Dc = 1.661 Mg/m3, λ = 0.71073 Å, 2θmax = 27.46°, 1278 reflections measured, 529 unique (Rint = 0.0280). Final residuals for 80 parameters were R1 = 0.0261, wR2 = 0.0607 for I > 2σ(I), and R1 = 0.0291, wR2 = 0.0615 for all 529 data. CCDC 285153 and 285154. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513442k |
| ‡‡ Calculated shear elastic stiffness constants show facile slipping of the sheets in all of these structures (all but 3 having a direction of shear with an elastic stiffness < 1 GPa). |
| This journal is © The Royal Society of Chemistry 2006 |