Stabilisation of a paramagnetic BH4−-bridged dinickel(II) complex by a macrodinucleating hexaaza-dithiophenolate ligand†
Yves
Journaux
*a,
Vasile
Lozan
b,
Julia
Klingele‡
b and
Berthold
Kersting
*b
aLaboratoire de Chimie Inorganique et Matériaux Moléculaires Bat F UMR CNRS 7071, case courrier 42, Université Pierre et Marie Curie, 4 place Jussieu, F75252, Paris cedex 05, France. E-mail: jour@ccr.jussieu.fr; Tel: +33 14427 5562
bInstitut für Anorganische Chemie, Johannisallee 29, Universität Leipzig, D-04013 Leipzig, Germany. E-mail: b.kersting@uni-leipzig.de; Fax: +49-341-9736199; Tel: +49-341-9736143
First published on 23rd November 2005
Abstract
The first paramagnetic borohydrido-bridged dinuclear nickel(II) complex, [(L)NiII2(μ1,3-BH4)]+, stabilised by a sterically demanding hexaaza-dithiophenolate macrocycle, has been obtained by the reaction of [(L)NiII2(μ-ClO4)]+ with NnBu4BH4.
The search for dinuclear dithiolato-bridged complexes which model key features of the active site of hydrogenase enzymes is an active research area.1,2 Two main strategies exist to access such compounds. One involves the addition of an electrophilic metal–carbonyl fragment to a nucleophilic metal complex with cis-oriented thiolate functions.3 The resulting [NiFeS2], or [Fe2S2], assemblies are co-ligated with CO and CN− and represent good structural analogues of the proposed active site structures, as demonstrated recently by a number of research groups.4–7 In the other strategy, macrocyclic dinucleating polyaza-dithiolate ligands are used for the cluster assembly.8 Until now this strategy has only allowed for the production of homodinuclear nickel complexes, and it is unclear at present whether these more classical Werner-type coordination compounds will ever be able to bind the biologically relevant co-ligands CO, CN− and H−. Herein we provide the first evidence for nickel–hydrogen interactions in such compounds.
Our study was initiated by the recent discovery of Desrochers et al.9 who demonstrated that the sterically encumbered hydrotris(3,5-dimethyl-pyrazolyl)borate ligand (Tp*−) can stabilize a hydrogen-rich nickel environment in [Tp*NiII(μ1,3-BH4)]. In order to test whether similar dinickel(II) complexes with a bridging borohydride co-ligand are supported by the dinucleating hexaaza-dithiophenolate ligand (L)2− (Scheme 1),10 an acetonitrile solution of the chloro-bridged complex [(L)NiII2(μ-Cl)]ClO4 (1·ClO4) was treated with NnBu4BH4 under an argon atmosphere at ambient temperature. Unlike [Tp*NiIICl],9 however, no reaction occurred. In a second approach, the reaction was carried out using the dark-green perchlorato-bridged complex [(L)NiII2(μ-ClO4)]ClO4 (2·ClO4) which was prepared by Cl− abstraction from 1·ClO4 with Pb(ClO4)2.† This gave a pale-green solution of the desired borohydrido-bridged complex 3, which was isolated as its BPh4− salt in ca. 75% yield. In the absence of air and protic reagents this compound is stable for weeks, both in the solid state and in solution. This stability is quite remarkable given that nickel(II) complexes of sterically less demanding ligands are readily reduced to nickel boride.11
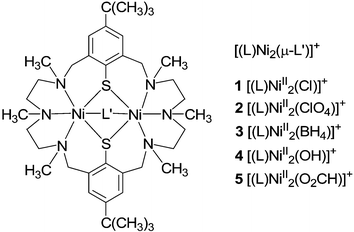 | ||
| Scheme 1 Complexes 1–5. | ||
IR measurements of solid 3·BPh4 showed intense absorption bands at 2390, 2360, 2153 and 2071 cm−1 indicative of terminal B–H and bridging B–H⋯Ni functions.12 The UV-Vis spectrum recorded in acetonitrile suggested the presence of octahedral Ni(II) ions [λ = 650 (ν2) and 1074 nm (ν1)].13 Final confirmation came from an X-ray crystal structure determination of 3·BPh4·2MeCN (Fig. 1).§ As can be seen, the BH4− ion bridges the two Ni(II) centres in a symmetrical fashion to generate a bioctahedral N3NiII(μ-S)2(μ-BH4)NiIIN3 core structure that has never been observed before in nickel–thiolate chemistry. There are no interactions between the MeCN of solvent of crystallization and the [(L)NiII2(μ-BH4)]+ cations. The average Ni–H distance at 1.89(4) Å compares well with that in the mononuclear NiS4H2 complex [NiII(bmp)2] (bmp = bis(2-mercapto-1-methylimidazolyl)borate),14 the only other sulfur-rich Ni(II) complex with B–H⋯Ni interactions that has been structurally characterised.
 | ||
| Fig. 1 ORTEP representation of the structure of complex 3 at 50% probability ellipsoids. Hydrogen atoms, except those of the BH4− co-ligand, have been omitted for clarity. Selected bond lengths (Å) and angles (°): Ni1–N1 2.219(4), Ni1–N2 2.152(4), Ni1–N3 2.284(4), Ni1–S1 2.437(2), Ni1–S2 2.459(1), Ni1–H1 1.93(4), Ni2–N4 2.271(4), Ni2–N5 2.135(4), Ni2–N6 2.260(4), Ni2–S1 2.439(2), Ni2–S2 2.457(2), Ni2–H2 1.84(4), B1–H1 1.25(4), B1–H2 1.24(4), B1–H3 1.12(4), B1–H4 1.12(4); Ni1⋯Ni2 3.458; Ni1–S1–Ni2 90.33(5), Ni2–S2–Ni1 89.41(5). | ||
Magnetic susceptibility measurements have been carried out to see whether magnetic exchange interactions are present in 3·BPh4. As can be seen from Fig. 2, the product χmT (per dinuclear complex) gradually increases from 2.69 cm3 K mol−1 at 295 K to a maximum of 3.29 cm3 K mol−1 at 28 K, and then decreases rapidly to 2.74 μB at 2 K. This behaviour indicates an intramolecular ferromagnetic exchange interaction between the two Ni2+ ions in 3.15
 | ||
| Fig. 2 Plot of χmT against T for 3·BPh4. The solid line represents the best theoretical fit of the magnetic susceptibility data by full-matrix diagonalization of the spin Hamiltonian H = −2JS1·S2 + D(Sz12 + Sz22 − 4/3) + gβ(S1 + S2)B. Experimental and calculated data are provided as ESI.† | ||
Least-squares fit of the magnetic susceptibility data by full-matrix diagonalization of the appropriate spin Hamiltonian H = −2JS1·S2 + D(Sz12 + Sz22 − 4/3) + gβ(S1 + S2)B gave J = 27 cm−1, D = +4.3 cm−1, and g = 2.09 (or alternatively J = 27.5 cm−1, D = −3.9 cm−1, and g = 2.09). Interestingly, the J value of 27 cm−1 is by far the largest value that has ever been observed in dinuclear nickel(II) complexes supported by (L)2−.16 It indicates that the ferromagnetic magnetic exchange interactions are not only propagated via the bridging thiolate functions but also through the μ1,3-bridging borohydride ion. To the best of our knowledge, this property of the BH4− ion has not been documented previously in the literature. This suggests its use as a building block in the construction of molecular based magnetic materials.
Preliminary results show that 3 reacts with protic reagents HA, such as HCl, H2O, or HCO2H with liberation of H2 and formation of the respective [(L)NiII2(A)]+ species (A = Cl−1, OH−4 and HCO2−5). Complexes 1 and 4 have been reported earlier. The new complex 5 is also readily produced by the reaction of 3 with CO2. IR measurements of 5·BPh4 showed two absorption bands at 1602 and 1424 cm−1, attributable to the symmetric and antisymmetric stretching frequencies of a μ1,3-bridging formate ion. This was also confirmed by an X-ray crystal structure determination of 5·BPh4 (see ESI†).
In conclusion, we have prepared the first stable dinuclear nickel(II) borohydrido-bridged complex of a macrodinucleating hexaaza-dithiophenolate ligand. Work in progress is directed towards the synthesis of related compounds with bridging hydride ligands by taking advantage of the steric protection offered by the supporting ligand. Such compounds may also aid in understanding the electronic structures and the reactivities of the dinuclear active sites of the hydrogenase enzymes.
This work was supported by the Deutsche Forschungsgemeinschaft (Project KE 585/4-1,2).
Notes and references
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268 RSC.
- (a) A. C. Marr, D. J. E. Spencer and M. Schröder, Coord. Chem. Rev., 2001, 219–221, 1055 CrossRef CAS; (b) S. Brooker, Coord. Chem. Rev., 2001, 222, 33–56 CrossRef CAS.
- C. A. Grapperhaus and M. Y. Darensbourg, Acc. Chem. Res., 1998, 31, 451 CrossRef CAS.
- C.-H. Lai, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed. Engl., 1996, 35, 2390 CrossRef CAS.
- S. C. Davies, D. J. Evans, D. L. Hughes, S. Longhurst and J. R. Sanders, Chem. Commun., 1999, 1935 RSC.
- D. Sellmann, F. Geipel, F. Lauderbach and F. W. Heinemann, Angew. Chem., Int. Ed., 2002, 41, 632 CrossRef CAS.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726 CrossRef CAS.
- A. J. Amoroso, S. S. M. Chung, D. J. E. Spencer, J. P. Danks, M. W. Glenny, A. J. Blake, P. A. Cooke, C. Wilson and M. Schröder, Chem. Commun., 2003, 2020 RSC.
- P. J. Desrochers, S. LeLievre, R. J. Johnson, B. T. Lamb, A. L. Phelps, A. W. Cordes, W. Gu and S. P. Cramer, Inorg. Chem., 2003, 42, 7945 CrossRef CAS.
- G. Steinfeld and B. Kersting, Chem. Commun., 2001, 1376 RSC.
- N. F. Curtis, J. Chem. Soc., 1965, 924 RSC.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263 CrossRef CAS.
- B. Kersting, Angew. Chem., Int. Ed., 2001, 40, 3988.
- H. M. Alvarez, M. Krawiec, B. T. Donovan-Merkert, M. Fouzi and D. Rabinovich, Inorg. Chem., 2001, 40, 5736 CrossRef CAS.
- In the solid state the dinuclear nickel complexes are well separated by the bulky tetraphenylborate anions. The abrupt decrease in χmT below 28 K is therefore most likely due to zero field splitting of NiII and not to intermolecular exchange interactions.
- J. Hausmann, M. H. Klingele, V. Lozan, G. Steinfeld, D. Siebert, Y. Journaux, J. J. Girerd and B. Kersting, Chem. Eur. J., 2004, 10, 1716 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation and characterisation data for complexes 2–5. See DOI: 10.1039/b512744k |
| ‡ Present address: Institut für Anorganische und Analytische Chemie, Universität Freiburg, Albertstrasse 21, D-79104 Freiburg, Germany. |
§ Crystal data for [(L)NiII2(μ1,3-BH4)](BPh4)·2MeCN (3·BPh4·2MeCN): C66H94B2N8Ni2S2, M = 1202.65. Triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , (no. 2), a = 15.992(3), b = 16.080(3), c = 16.515(3) Å, α = 63.82(3), β = 70.97(3), γ = 66.47(3)°, V = 3436(1) Å3, Z = 2, Dcalcd. = 1.162 g cm−3, μ(Mo-Kα) = 0.651 mm−1, T = 210 K. Using Mo-Kα radiation (0.71073 Å), a total of 31051 reflections were collected of which 16023 were independent. Refinement converged to R1 = 0.0749, wR2 = 0.2053 [I > 2σ(I)]. Hydrogen atoms for the η2-BH4 ligand were located from the final difference map and were refined isotropically with Ueq(H) 1.2 times that of the boron to which they are attached. The MeCN solvate molecules were found to be disordered over two positions. CCDC 280004. Crystal data for [(L)NiII2(μ1,3-O2CH)](BPh4) (5·BPh4): C63H85BN6Ni2O2S2, M = 1150.72. Monoclinic, space group P21/n, a = 18.452(4), b = 34.531(7), c = 18.452(4) Å, β = 92.22(3)°, V = 11748(4) Å3, Z = 8, Dc = 1.301 g cm−3, μ(Mo-Kα) = 0.761 mm−1, T = 210 K. Using Mo-Kα radiation (0.71073 Å), a total of 74914 reflections were collected of which 28479 were independent. Refinement converged to R1 = 0.0778, wR2 = 0.1651 [I > 2σ(I)]. CCDC 280005. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512744k , (no. 2), a = 15.992(3), b = 16.080(3), c = 16.515(3) Å, α = 63.82(3), β = 70.97(3), γ = 66.47(3)°, V = 3436(1) Å3, Z = 2, Dcalcd. = 1.162 g cm−3, μ(Mo-Kα) = 0.651 mm−1, T = 210 K. Using Mo-Kα radiation (0.71073 Å), a total of 31051 reflections were collected of which 16023 were independent. Refinement converged to R1 = 0.0749, wR2 = 0.2053 [I > 2σ(I)]. Hydrogen atoms for the η2-BH4 ligand were located from the final difference map and were refined isotropically with Ueq(H) 1.2 times that of the boron to which they are attached. The MeCN solvate molecules were found to be disordered over two positions. CCDC 280004. Crystal data for [(L)NiII2(μ1,3-O2CH)](BPh4) (5·BPh4): C63H85BN6Ni2O2S2, M = 1150.72. Monoclinic, space group P21/n, a = 18.452(4), b = 34.531(7), c = 18.452(4) Å, β = 92.22(3)°, V = 11748(4) Å3, Z = 8, Dc = 1.301 g cm−3, μ(Mo-Kα) = 0.761 mm−1, T = 210 K. Using Mo-Kα radiation (0.71073 Å), a total of 74914 reflections were collected of which 28479 were independent. Refinement converged to R1 = 0.0778, wR2 = 0.1651 [I > 2σ(I)]. CCDC 280005. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512744k |
| This journal is © The Royal Society of Chemistry 2006 |