From Pd nanoparticles to single crystals: 1,3-butadiene hydrogenation on well-defined model catalysts†
Joaquin
Silvestre-Albero
,
Günther
Rupprechter
* and
Hans-Joachim
Freund
Fritz-Haber-Institut, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: rupprechter@fhi-berlin.mpg.de
First published on 18th November 2005
Abstract
Although 1,3-butadiene hydrogenation is known to be a structure-sensitive reaction, correlation of the catalytic activity with the exact Pd particle surface structure shows that the reaction is in fact particle size independent.
The hydrogenation of unsaturated hydrocarbons on Pd catalysts is among the most frequently applied reactions of heterogeneous catalysis.1 We have previously examined the adsorption, decomposition and reaction (hydrogenation) of olefins such as ethene or pentene with clean and well-defined alumina supported Pd nanoparticles, employing a number of surface-spectroscopic techniques under ultrahigh vacuum (UHV).2–6 A general observation was that the properties of disordered small Pd particles may substantially deviate from those of Pd single crystals (due to the higher fraction of low-coordinated sites on small Pd particles), while larger Pd particles start to approach the behaviour of a Pd(111) single crystal. However, a quantitative comparison in terms of turnover frequencies (TOFs; molecules reacted per surface site and second) has not been carried out so far, since this requires reaction studies at ambient pressure, similar to those of technical heterogeneous catalysis.
Here we report atmospheric pressure kinetic studies of 1,3-butadiene hydrogenation on Pd–Al2O3 catalysts for a wide range of mean particle size, from 2 to 8 nm. By employing well-defined model catalysts, i.e. Pd nanoparticles grown in UHV on thin Al2O3 films,7–9 and by determining their absolute activity for butadiene hydrogenation, we are able to conclusively prove that the behaviour of large Pd particles, in fact, is identical to that of Pd(111), while for Pd particles below 4 nm it is not. This is only feasible because we correlate the microscopic structural information on the Pd nanoparticles with their macroscopic catalytic properties. We also show that the selective hydrogenation of 1,3-butadiene—although being structure-sensitive10—is particle size independent if the observed reaction rates are normalized taking into account the exact size and surface structure of the Pd nanoparticles.
The Pd catalysts were grown and characterized in UHV and subsequently transferred (under UHV) to a high-pressure reaction cell,11 where reaction kinetics were determined in a recirculated batch mode by on-line gas chromatography. When compared to previous studies,1,10,12,13 our model catalysts enable full control of particle size and cleanliness. This is crucial because recent studies on wet-chemically prepared Pd–Al2O3 catalysts have revealed a strong dependence of the particle adsorption properties on the synthesis procedure.14 Furthermore, we are also able to distinguish between the different butene isomers. Most importantly, only an exact characterization of the Pd nanoparticle morphology and surface structure by scanning tunnelling microscopy (STM) and a comparison of the absolute reaction rates of Pd nanoparticles with reference measurements on Pd(111) and Pd(110) single crystals allow us to propose a model which explains the observed apparent particle size dependence.
Although butadiene hydrogenation is an important and well-optimized technological process to remove undesired diene impurities from alkene streams,1 a complete microscopic understanding has not been obtained yet. Apart from this, butadiene hydrogenation is also an interesting reaction for model catalysis because it has four different products (1-butene, trans-2-butene, cis-2-butene, n-butane) and therefore allows the study of both activity and selectivity issues. To start with, we first discuss the reaction kinetics on small Pd nanoparticles (mean size ∼2 nm) on Al2O3 at 373 K (Fig. 1a). Fig. 1b and 1c show corresponding measurements on Pd particles with a mean size of 8 nm, and on Pd(111), respectively. All four hydrogenation products were observed but the kinetic behaviour and the selectivity are remarkably different for the three model catalysts. As evident from the consumption of 1,3-butadiene (100% relates to 5 mbar butadiene), the hydrogenation rate increases with decreasing butadiene pressure for 2 nm Pd nanoparticles (Fig. 1a), while the 8 nm particles and the (111) single crystal surface exhibit nearly zero order reaction kinetics (Fig. 1b, c; i.e. the reaction rate is (nearly) independent of the actual butadiene pressure). This indicates that for the 2 nm Pd nanoparticles the rate-limiting step(s) change with the butadiene surface concentration. The observed kinetics on small particles somewhat resemble the transition from diffusion-control to a kinetic regime. At high butadiene pressure (coverage) the reaction rate is limited by adsorption/desorption processes, as indicated by the high (apparent) reaction activation energy of 75 kJ mol−1. With increasing butadiene consumption the reaction itself becomes rate-limiting. Similar high activation barriers were reported for highly-dispersed Pd–Al2O3 catalysts,13,15 which indicates that our model catalyst mimics real catalysts quite well. The similarity of the kinetic behaviour of the larger Pd nanoparticles (mean size = 8 nm) with that of Pd(111) can be rationalized considering the STM characterization of large Pd particles. High-resolution STM images2,16 indicated that larger particles have a truncated cubo–octahedral shape with sometimes incomplete (111) facets (comprising ∼80% of the surface) and with (100) side facets (cf.Fig. 1b, 2b).
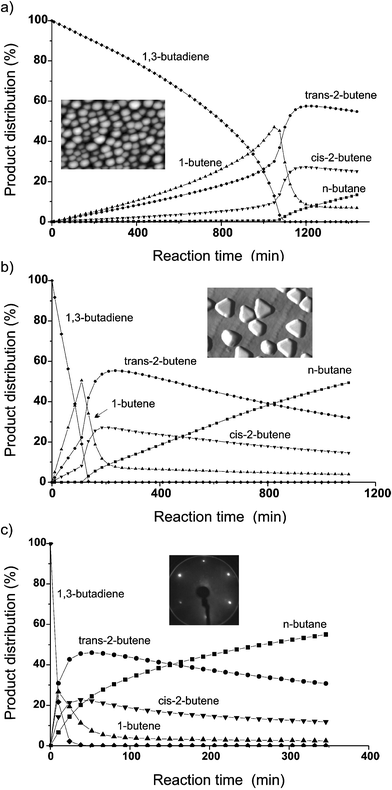 | ||
| Fig. 1 Product distribution versus reaction time for 1,3-butadiene hydrogenation at 373 K on (a, b) Pd–Al2O3/NiAl(110) catalysts (mean particle size 2 nm (a) and 8 nm (b)) and on (c) a Pd(111) single crystal. STM images 2–4 and a LEED pattern are shown as insets. | ||
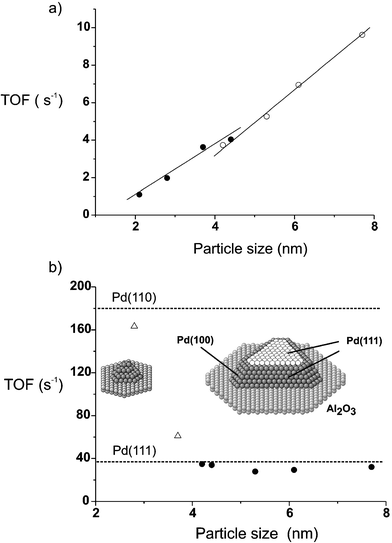 | ||
| Fig. 2 Turnover frequency (TOF) for the different Pd–Al2O3/NiAl(110) catalysts as a function of mean particle size, (a) normalized by the total number of Pd surface atoms, and (b) normalized by using the number of Pd atoms on (111) facets, using a truncated cubo-octahedron as the structural model (shown as inset). TOF values for Pd(111) and Pd(110) under identical reaction conditions are included (dashed lines). | ||
Comparing Fig. 1a with 1b and 1c suggests that butadiene is more strongly bonded to small Pd nanoparticles and hydrogen adsorption on small Pd particles is hence more efficiently hindered than on large ones. The catalytic activity of small Pd particles is thus governed by the rate of hydrogen adsorption on the hydrocarbon-covered Pd particles. Below a critical butadiene coverage (∼70% for the conditions of Fig. 1a) hydrogen adsorption is less affected and the reaction slowly switches to a kinetic regime. In fact, when the experiment whose results are shown in Fig. 1a was performed with an initial butadiene pressure of 3.5 mbar (i.e. 70% of the original concentration of Fig. 1a), the reaction rate increased from the beginning, confirming the importance of a critical hydrocarbon coverage. A stronger bonding of 1,3-butadiene with decreasing particle size (2.8–1.2 nm) was also suggested by Bertolini et al.17 on the basis of core level photoemission.
Nevertheless, even the 8 nm Pd particles are more selective to butenes than Pd(111). The adsorption of butadiene is presumably still stronger than on Pd(111), as evident from the absence of re-adsorption processes of butenes at low butadiene coverage, i.e. no butane is formed until butadiene has (nearly) completely disappeared, resulting in a 100% selectivity to butenes in the presence of butadiene.
To further explore the kinetic properties we have also measured the particle size dependence of the reaction rate. Fig. 2 displays the specific activity (turnover frequency, i.e. molecules of butadiene reacted per Pd surface atom within 1 hour) as a function of particle size. In Fig. 2a the total number of Pd surface atoms is used for normalization (assuming particles of truncated cubo-octahedral shape; filled and open circles refer to Pd deposition at 90 K and 300 K, respectively). Such a plot is common in heterogeneous catalysis and shows, for the current case, that the TOF grows linearly with particle size. This indicates that the activity does not scale with the number of Pd surface atoms and that larger Pd particles seem more active than smaller ones. Similar trends were reported for Pd particles up to ∼4 nm10,12 but no in situ structural characterization was carried out, preventing an accurate structure–activity correlation.
In contrast, our STM measurements provide exact microscopic information on the shape and surface structure of the Pd nanoparticles. This allows us to propose a more realistic model of the Pd nanoparticles including incomplete surface terraces (a ∼5 nm cubo-octahedron with incomplete (111) facets is shown in Fig. 2b as inset; for graphical reasons incomplete layers on side facets are not displayed). Taking into account that the reaction is structure sensitive, i.e. that different crystallographic orientations will have different contributions to the overall catalytic activity, and considering the aforementioned model, we are able to calculate a more accurate (“real”) TOF by dividing the total hydrogenation activity by the number of surface sites in a specific crystallographic orientation. Along these lines, the number of Pd surface atoms present in (111) and (100) terraces as well as at edges and at the metal–support border were calculated for different Pd clusters sizes. As a result we find that when the number of Pd atoms in the incomplete (111) facets is used for normalization, the TOF of butadiene hydrogenation is clearly particle sizeindependent (Fig. 2b). This suggests that the reaction takes place preferentially on the (111) facets of the Pd nanoparticles, at least for a mean particle size above 4 nm (smaller sizes will be discussed below).
To validate this model we have carried out reference measurements on Pd(111) and Pd(110) single crystals. The Pd(110) catalyst exhibited an initial TOF as high as 180 s−1 while for Pd(111) a value of 38 s−1 was obtained (values included in Fig. 2b as dashed lines). The ∼5-times lower activity of Pd(111) is probably due to the stronger bonding of 1,3-butadiene to Pd(111) as compared to Pd(110),18 which reduces the surface hydrogen concentration (see below), and/or to the higher sticking probability and adsorption energy of H2 on the more open (110) surface (0.9 eV on (111) and 1.05 eV on (110)19). A (110) (like) surface geometry may also favour the reaction because ridges and troughs allow a better co-adsorption of butadiene and hydrogen.
The specific activity of larger Pd particles and Pd(111) show excellent agreement which strongly supports that the reaction occurs on the (111) particle facets. For small Pd particles (∼2–3 nm mean size) the normalization is more difficult because these particles (with only a few atoms edge length) do no longer exhibit well-developed facets (the “facets” typically contain only 4–8 atoms; a ∼2 nm particle is shown in Fig. 2b). The TOF suggests that the small Pd particles have a catalytic activity which is more similar to Pd(110) than to Pd(111). However, as mentioned, the normalization is not straightforward for small particles (marked as △ in Fig. 2b).
The consecutive reactions on the Pd nanoparticles and single crystals, after 100% butadiene conversion was reached, are also worth noting. After nearly full 1-butene conversion mainly isomerization products (∼50% trans- and ∼35% cis-2-butene) and only ∼15% n-butane were produced on the Pd particles (Fig. 1a, b). In contrast, on Pd(111) (Fig. 1c) about 45% trans-2-butene, ∼20% cis-2-butene and ∼35% n-butane were observed. The higher selectivity of Pd particles for isomerization may be again due to the “hydrogen deficiency” as a result of site blocking by the butenes.
Summarizing, we report for the first time that butadiene hydrogenation, in spite of being structure-sensitive, is particle size independent for Pd nanoparticles supported on Al2O3 if the catalytic activity is scaled by the number of active sites, i.e. Pd atoms in incomplete (111) terraces. This is supported by comparing the catalytic activity of small and large Pd nanoparticles with Pd(111) and Pd(110) single crystals under the same reaction conditions. With this we bridge the “materials gap” between heterogeneous catalysis on metal nanoparticles and surface science studies on single crystals, by showing that here Pd particles larger than 4 nm behave very similar to Pd(111).
Spectroscopic measurements (infrared and thermal desorption2–6,11) on ethene and pentene have suggested either π-bonded or di-σ bonded species as active intermediates, depending on the hydrocarbon length. However, the complexity of butadiene allows for a large number of adsorption configurations20,21 and future spectroscopic studies on Pd nanoparticles are certainly required to further explore the reaction mechanism of diene hydrogenation.
J.S.A. acknowledges support by the Alexander von Humboldt Foundation. We thank M. Heemeier and M. Bäumer for STM sample characterization.
Notes and references
- H. Arnold, F. Dölbert and J. Gaube, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, 1997, vol. 3, p. 2165 Search PubMed; Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185 Search PubMed.
- M. Frank and M. Bäumer, Phys. Chem. Chem. Phys., 2000, 2, 3723 RSC.
- S. K. Shaikhutdinov, M. Heemeier, M. Bäumer, T. Lear, D. Lennon, R. J. Oldman, S. D. Jackson and H.-J. Freund, J. Catal., 2001, 200, 350.
- H.-J. Freund, M. Bäumer, J. Libuda, T. Risse, G. Rupprechter and S. Shaikhutdinov, J. Catal., 2003, 216, 223 CrossRef CAS.
- A. Doyle, S. Shaikhutdinov, S. D. Jackson and H.-J. Freund, Angew. Chem., Int. Ed., 2003, 42, 5240 CrossRef CAS; A. Doyle, S. Shaikhutdinov and H.-J. Freund, Angew. Chem., Int. Ed., 2005, 117, 635 CrossRef.
- M. Morkel, G. Rupprechter and H.-J. Freund, Surf. Sci. Lett., 2005, 588, L209 Search PubMed.
- M. Bäumer, J. Libuda, A. Sandell, H.-J. Freund, G. Graw, T. Bertrams and H. Neddermeyer, Ber. Bunsen-Ges. Phys. Chem., 1995, 99, 1381.
- H.-J. Freund, Angew. Chem., Int. Ed., 1997, 36, 452 CrossRef.
- H.-J. Freund, M. Bäumer and H. Kuhlenbeck, Adv. Catal., 2000, 45, 412.
- B. Tardy, C. Noupa, C. Leclercq, J.C. Bertolini, A. Hoareau, M. Treilleux, J. P. Faure and G. Nihoul, J. Catal., 1991, 129, 1 CrossRef CAS.
- G. Rupprechter, Annu. Rep. Prog. Chem., Sect. C, 2004, 100, 237 RSC.
- J. P. Boitiaux, J. Cosyns and S. Vasudevan, Appl. Catal., 1983, 6, 41 CrossRef CAS.
- B. K. Furlong, J. W. Hightower, T. Y.-L. Chan, A. Sarkany and L. Guczi, Appl. Catal., A, 1994, 117, 41 CrossRef CAS.
- T. Lear, R. Marshall, E. K. Gibson, T. Schütt, T. M. Klapötke, G. Rupprechter, H.-J. Freund, J. M. Winfield and D. Lennon, Phys. Chem. Chem. Phys., 2005, 7, 565 RSC.
- G. C. Bond, G. Webb, P. B. Wells and J. M. Winterbottom, J. Chem. Soc., 1965, 3218 RSC.
- H. K. Hansen, T. Worren, S. Stempel, E. Lægsgaard, M. Bäumer, H.-J. Freund, F. Besenbacher and I. Stensgaard, Phys. Rev. Lett., 1999, 83, 4120 CrossRef CAS.
- J. C. Bertolini, P. Delichere, B. Khanra, J. Massardier, C. Noupa and B. Tardy, Catal. Lett., 1990, 6, 215 CrossRef CAS.
- G. Tourillon, A. Cassuto, Y. Jugnet, J. Massardier and J. C. Bertolini, J. Chem. Soc., Faraday Trans., 1996, 92, 4835 RSC.
- H. Conrad, G. Ertl, J. Koch and E. E. Latta, Surf. Sci., 1974, 41, 435 CrossRef.
- F. Mittendorfer, C. Thomazeau, P. Raybaud and H. Toulhoat, J. Phys. Chem. B, 2003, 107, 12287 CrossRef CAS.
- P. Sautet and J. Paul, Catal. Lett., 1991, 9, 245 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details about experimental setup, catalysts preparation and supplementary experimental data. See DOI: 10.1039/b513030a |
| This journal is © The Royal Society of Chemistry 2006 |