A highly selective charge transfer fluoroionophore for Cu2+†
Zhen-Chang
Wen
a,
Rui
Yang
ab,
Hui
He
a and
Yun-Bao
Jiang
*a
aDepartment of Chemistry and the MOE Key Laboratory of Analytical Sciences, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
bCollege of Chemistry and Chemical Engineering, Southwest University, Chongqing 410075, China. E-mail: ybjiang@xmu.edu.cn; Fax: +86 592 2185662; Tel: +86 592 2185662
First published on 17th November 2005
Abstract
A dual fluorescent charge transfer fluoroionophore (1) with its ionophore incorporated in the electron acceptor was developed and was found to show a highly selective fluorescent response to Cu2+ with a dramatic enhancement in its CT emission.
Because of its biological and environmental importance, detection and monitoring of Cu2+, especially via methods that allow selective and sensitive assays, are highly demanding. Fluorescence signaling is one of the first choices due to its high detection sensitivity and intrinsic operation simplicity. Designing fluoroionophores for Cu2+ has drawn much recent attention.1 These fluoroionophores consist in principle of a metal ionophore and a signaling fluorophore that are either conjugated or connected by a flexible spacer.2 As a transition metal ion known for its efficient fluorescence quenching character, detection of Cu2+ by fluoroionophores hitherto reported operates mostly in fluorescence quenching mode under an electron or energy transfer mechanism.3,4 Due to sensitivity reasons, fluoroionophores showing fluorescence enhancement as a result of metal-ion binding are to be favored over those exhibiting fluorescence quenching. A number of fluoroionophores have been reported for Cu2+ that show fluorescence enhancement.5–7 Among those reported, Cu2+ binding resulting in the inhibition of photo-induced electron transfer (PET) in the fluoroionophores has been an important mechanism,6 with the metal ionophore located in the electron donor. Obviously, the metal binding selectivity is subject to the choice of a sophisticated ionophore that is selective for this metal ion. Intramolecular charge transfer (CT) fluoroionophores for Cu2+ were also reported that showed an enhancement in the short-wavelength emission of the locally excited state (LE state), at the expense of the long-wavelength CT emission.7 This is in principle similar to PET inhibition, with the metal ionophore incorporated in both the electron donor7a,c and acceptor.7b Cu2+ fluoroionophores showing enhanced excimer emission were recently reported.8 Herein is described a new CT dual fluorescent fluoroionophore 1 (Scheme 1)9 that shows a highly selective and sensitive fluorescent response toward Cu2+.
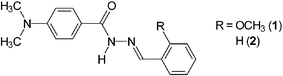 | ||
| Scheme 1 Structure of 1 and its OCH3-free control molecule 2. | ||
1 was designed on the basis of the dual fluorescent CT fluorophore 4-(N,N-dimethylamino)benzamide10 whose electron acceptor is derived into 2-methoxybenzaldehyde hydrazone, the metal ionophore.11 2-Hydroxybenzaldehyde acylhydrazones are known as good ligands for transition metal ions.11 The 2-OH group in the salicylaldehyde hydrazone moiety, however, may open an ESIPT (excited-state intramolecular proton transfer) channel which would decrease the fluorescence quantum yield and complicate the photophysics.12 The 2-OCH3 group instead of 2-OH was therefore chosen in 1, in which the N′-acylhydrazone moiety as metal ionophore is located in the electron acceptor.
The absorption spectrum of 1 in ACN exhibits three bands peaked at 227, 272, and 334 nm, respectively (Fig. 1a). In the presence of Cu2+, the band at 334 nm was attenuated while a new peak appeared at longer wavelength (388 nm). Two clear isosbestic points at 363 and 293nm were observed during the spectral titration, indicating the formation of a well-defined 1–Cu2+ complex. Similar spectral variations were observed with the other tested transition metal ions such as Zn2+, Hg2+ and Pb2+ (Fig. S1 in ESI†), which, after non-linear fitting assuming a 1 ∶ 1 stoichiometry, afforded binding constants of comparable value at 104 M−1 orders of magnitude (Table S1 in ESI†). These observations indicate that Cu2+, Zn2+, Hg2+ and Pb2+ bind to a similar extent to 1 in the ground state.
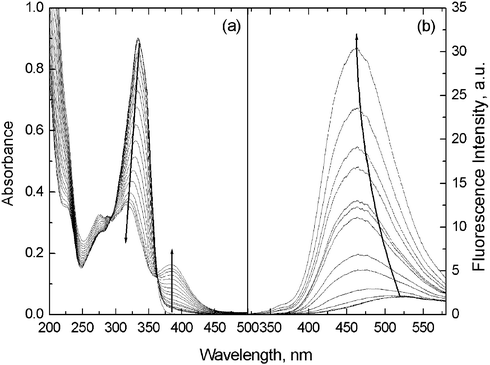 | ||
| Fig. 1 Absorption (a) and fluorescence (b) spectra of 1 (2.0 × 10−5 M) in ACN in the presence of increasing concentration of Cu2+ (0–6.0 × 10−5 M in (a) and 0–5.0 × 10−5 M in (b)). The excitation wavelength for acquiring fluorescence spectra was 293 nm, an isosbestic wavelength observed in the absorption spectral titration. Cu2+ and other metal ions were used as their perchlorate salts. | ||
1 in ACN emits dual fluorescence, with a total fluorescence quantum yield Φ of 0.0082 (Fig. 1b). This is indicative of the occurrence of the excited-state CT known for the parent CT fluorophore 4-(N,N-dimethylamino)benzamide.10 The short-wavelength emission at ca. 350 nm assigned to the LE state of 1 showed very weak dependence on solvent polarity, whereas a continuous red-shift was observed in the long-wavelength emission from 410 nm in diethyl ether to 525 nm in ACN of increasing polarity, confirming the CT nature of its emissive state. A dipole moment of 24 D was deduced from the solvatochromic data for the CT state of 1 following a reported procedure,13 see details and Fig. S2 in ESI.†
The response of the CT dual fluorescence of 1 toward a variety of metal ions was examined first in ACN. Upon addition of Li+, Na+, K+, Mg2+, Ca2+, Ba2+, Al3+, Fe3+, Co2+, Ni2+, Cu2+, Ag+, Zn2+, Cd2+, Hg2+, and Pb2+ to the ACN solution of 1, a prominent fluorescence enhancement was only observed for Cu2+ (30 fold) with a substantial blue shift in its CT emission from 525 nm to 460 nm, Fig. 1b. Other transition metal ions, as expected, quenched the fluorescence to differing extents while the CT emission of 1 blue shifted too, whereas alkali and alkaline earth metal ions had hardly any effect on the emission of 1 (see Fig. S3 in the ESI† and Fig. 2). Different from the previously reported CT fluoroionophores for Cu2+ with which the LE emission was greatly enhanced upon Cu2+ binding whereas the CT emission was quenched,71 showed only minor changes in its LE emission in both the position and intensity, while its CT emission was dramatically enhanced and blue-shifted. The total fluorescence of 1 was enhanced 30 fold when 2.5 equivalent of Cu2+ was present and further increase in Cu2+ concentration led to slight fluorescence quenching (Fig. S3 in ESI†).
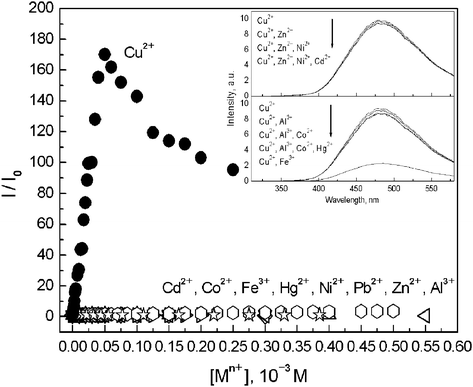 | ||
| Fig. 2 Plot of the fluorescence enhancement (I/I0 or Φ/Φ0) versus concentrations of Cu2+ and other metal ions in a mixture of ACN and tris-HCl (0.005 M, pH = 7.2) aqueous buffer solution (9 ∶ 1, v/v). Inset shows the fluorescence spectra of 1 (2.0 × 10−5 M) in the presence of 2 equiv of Cu2+ and 2 equiv of Cu2+ plus 10 equiv of other metal ions, respectively. | ||
Preliminary examination of the response of its dual fluorescence toward Cu2+ in ACN–H2O buffer solutions promised practical applications of 1 as a highly selective and sensitive fluoroionophore for Cu2+ (Fig. 2). Compared to that in pure ACN, the dual fluorescence of 1 was quenched by 75% with a total Φ of 0.002 in ACN–H2O (9 ∶ 1, v/v). However, an unexpectedly dramatic enhancement of the CT emission was observed when Cu2+ was added to the solution of 1 in ACN–H2O (9 ∶ 1, v/v) buffered by 0.005M tris-HCl (pH = 7.2). A substantial blue shift was also observed in the long-wavelength CT emission, now from 530 nm to 482 nm. The fluorescence of 1 was enhanced 170 fold when 2.5 equivalents of Cu2+ were introduced (Fig. 2), together with a substantial increase in the CT to LE intensity ratio (Fig. S4 in ESI†). The fluorescence enhancement factor decreased with increasing water content in ACN–H2O, but could still be more than 40 fold when the water content was 90% by volume (Table S2 in ESI†). With increasing water content in ACN, a continuous red-shift from 460 nm to 526 nm was observed in the long-wavelength emission of the 1–Cu2+ complex (Table S2 in ESI†), establishing its CT nature of the emissive state. The dipole moment of the CT state of the 1–Cu2+ complex was similarly deduced as 23 D by assuming an unchanged ground-state dipole moment of the fluoroionophore upon binding to Cu2+, see details and Fig. S5 in ESI.†
The total fluorescence intensity of 1 in aqueous ACN solution was found to be independent of the aqueous phase pH over 3–12 (Fig. S6 in ESI†), all of the experiments were hence carried out in the 9 ∶ 1 (v/v) mixture of ACN and 0.005 M tris-HCl (pH = 7.2). The fluorescence of 1 was only slightly altered by Li+, Na+, K+, Mg2+, Ca2+, Ba2+, Al3+, Co2+, Ni2+, Zn2+, Cd2+, Hg2+, and Pb2+, whereas Fe3+ at high concentration led to substantial quenching. Meanwhile, the fluorescence of 1 in the presence of 2 equivalents of Cu2+ was hardly changed by the co-existence of 10 equivalents of other metal ions except Fe3+; in the latter case half of the fluorescence of 1–Cu2+ was quenched, Fig. 2 inset. These results indicated that 1 was highly selective for Cu2+via substantial enhancements in its CT fluorescence. Preliminary data indicated that 1 can be employed to detect Cu2+ down to 1.8 × 10−8 M when 1 was used at 2.5 × 10−6 M.
Absorption titration and ESI-MS data (Table S1 and Fig. S7 in ESI†) indicated that 1 bound with the tested transition metal ions such as Cu2+, Pb2+ and Zn2+etc. in a 1 ∶ 1 stoichiometry. According to the reported coordination mode of 2-hydroxybenzaldehyde acylhydrazones with transition metals including Cu2+,11c1 was assumed to chelate Cu2+ in a tridentate mode via its carbonyl O, imino N and 2-methoxy O atoms. This tridentate binding mode was supported by IR data. In the IR spectrum of 1 (Fig. S8 in ESI†) the peaks at 1624, 1609 and 1253 cm−1 ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN and C–O stretching, respectively, were found, upon Cu2+ binding, to shift to lower wavenumbers of 1609, 1570 and 1245 cm−1 respectively.14
O, CN and C–O stretching, respectively, were found, upon Cu2+ binding, to shift to lower wavenumbers of 1609, 1570 and 1245 cm−1 respectively.14
Several factors can be listed for rationalizing the observed emission enhancement. (i) The low fluorescence quantum yield of 1 in ACN in the absence of Cu2+ (0.0082) might be attributed to radiationless channels from the nπ* state.10a Indeed, we observed in the absorption spectrum of 1 in cyclohexane a weak shoulder at 369 nm due to the nπ* transition.15 In the presence of Cu2+ that coordinates with the lone pair of the carbonyl oxygen, the energy of the nπ* state would be raised so that the ππ* state becomes the lowest excited state, leading to a substantial increase in the fluorescence quantum yield.7b,16 (ii) The blue shifted CT emission of 1 in the presence of Cu2+ actually pointed to an enlarged energy gap between the emissive CT state and its corresponding ground state and hence a decreased radiationless rate constant17 and an increased quantum yield. (iii) Cu2+ binding to 1 could also induce a conformation restriction, resulting in increased fluorescence quantum yield18 and preventing Cu2+ from quenching the fluorescence of the fluorophore. In supporting this assignment, it was observed that the OCH3-free control molecule of 1, 2 (Scheme 1) that chelates Cu2+ in a bidentate manner, showed only 10 or 70 fold fluorescence enhancement when fully complexed with Cu2+ in ACN or ACN–H2O (9 ∶ 1, v/v) solution, respectively (Fig. S9 in ESI†). It is of significance to indicate that, although all the tested transition metal ions such as Cu2+, Pb2+ and Hg2+ bind to a similar extent to 1 in the ground state and the afore-listed factors should exist with all of them, only Cu2+ dramatically enhances the CT emission of 1. This means that the coordination of 1 with Cu2+ in either the ground or the excited state is not exactly the same as that of 1 with other metal ions, although the detailed mechanism for this highly selective fluorescent response of 1 toward Cu2+ remains to be clarified. It was found that the blue-shifted CT emission in ACN of the 1–Cu2+ complex appeared at more or less the same position as that of N′-isopropyl 4-(N,N-dimethylamino)benzamide at ca. 460 nm (Fig. S10 in ESI†). This suggests that the electron acceptor in 1 becomes smaller upon Cu2+ binding and might be a useful structural hint for clarifying the high selectivity observed for Cu2+. We are currently working on a series of 1 derivatives with 4-substituents other than the electron donating N(CH3)2 in which the excited-state CT channel in 1 is removed.
In summary, we reported a simple yet highly selective and sensitive CT fluoroionophore for Cu2+, in which the metal ionophore was incorporated in the electron acceptor of the CT fluorophore. A blue shift and dramatic enhancement in the CT fluorescence of 1 in ACN and aqueous ACN solutions were observed in the presence of Cu2+, a transition metal ion known as an efficient fluorescence quencher. A noteworthy character of this CT fluoroionophore is that its highly selective fluorescent response toward Cu2+ is not due to the binding preference in the ground state. As both the electron donor (4-N(CH3)2) and the ionophore in the electron acceptor in 1 can be structurally modified or replaced, the results reported here provide a new strategy for constructing turn-on CT fluorophores for transition metal ions.
We thank the NSF of China (Grant 20425518), the Ministry of Education (MOE) of China, NSF of Fujian Province (D0220001) and VolkswagenStiftung (I/77 072, with K. A. Zachariasse, Max-Planck-Institut für biophysikalische Chemie, Göttingen, Germany) for financial support.
Notes and references
- (a) R. Krämer, Angew. Chem., Int. Ed., 1998, 37, 772–773 CrossRef CAS; (b) K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 2161–2195 CrossRef CAS.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef; (b) A. P. de Silva, D. B. Fox, A. J. M. Huxley and T. S. Moody, Coord. Chem. Rev., 2000, 205, 41–57 CrossRef CAS.
- (a) L. Fabrizzi, M. Licchelli, P. Pallavicini, A. Perotti and D. Sacchi, Angew. Chem., Int. Ed. Engl., 1994, 33, 1975–1977 CrossRef; (b) A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609–610 CrossRef CAS; (c) Y. Li and C. M. Yang, Chem. Commun., 2003, 2884–2885 RSC; (d) E. Brasola, F. Mancin, E. Rampazzo, P. Tecilla and U. Tonellato, Chem. Commun., 2003, 3026–3027 RSC; (e) Y. Zheng, X. Cao, J. Orbulescu, V. Konka, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, Anal. Chem., 2003, 75, 1706–1712 CrossRef CAS.
- (a) A. W. Varnes, R. B. Dodson and E. L. Wehry, J. Am. Chem. Soc., 1972, 94, 946–950 CrossRef CAS; (b) L. Prodi, F. Bolletta, M. Montaldi and N. Zaccheroni, Eur. J. Inorg. Chem., 1999, 455–460 CrossRef CAS; (c) A. Mokhir, A. Kiel, D.-P. Herten and R. Kraemer, Inorg. Chem., 2005, 44, 5661–5666 CrossRef CAS.
- (a) V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386–7387 CrossRef CAS; (b) Q. Y. Wu and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 14682–14683 CrossRef CAS; (c) Z. Xu, X. Qian and J. Cui, Org. Lett., 2005, 7, 3029–3032 CrossRef CAS.
- (a) P. Ghosh, P. K. Bharadwaj, J. Roy and S. Ghosh, J. Am. Chem. Soc., 1997, 119, 11903–1111909 CrossRef CAS; (b) B. Ramachandram and A. Samanta, Chem. Commun., 1997, 1037–1038 RSC; (c) B. Ramachandram and A. Samanta, J. Phys. Chem. A, 1998, 102, 10579–10587 CrossRef CAS; (d) K. A. Mitchell, R. G. Brown, D. Yuan, S.-C. Chang, R. E. Utecht and D. E. Lewis, J. Photochem. Photobiol., A, 1998, 115, 157–161 CrossRef CAS; (e) B. Ramachandram, G. Saroja, N. B. Sankaran and A. Samanta, J. Phys. Chem. B, 2000, 104, 11824–11832 CrossRef CAS; (f) S. Kaur and S. Kumar, Chem. Commun., 2002, 2840–2841 RSC.
- (a) K. Rurack, M. Kollmannsberger, U. Resch-Genger and J. Daub, J. Am. Chem. Soc., 2000, 122, 968–969 CrossRef CAS; (b) J. P. Malval and R. Lapouyade, Helv. Chim. Acta, 2001, 84, 2439–2451 CrossRef CAS; (c) Z. Xu, Y. Xiao, X. Qian, J. Cui and D. Cui, Org. Lett., 2005, 7, 889–892 CrossRef CAS.
- (a) J.-S. Yang, C.-S. Lin and C.-Y. Hwang, Org. Lett., 2001, 3, 889–892 CrossRef CAS; (b) G. Hennrich, W. Walther, U. Resch-Genger and H. Sonnenschein, Inorg. Chem., 2001, 40, 641–644 CrossRef CAS.
- The syntheses of 1 and its control molecule 2 are described in the Electronic Supplementary Information (ESI†).
- (a) D. Braun, W. Rettig, S. Delmond, J. F. Létard and R. Lapouyade, J. Phys. Chem. A, 1997, 101, 6836–6841 CrossRef CAS; (b) F.-Y. Wu and Y.-B. Jiang, Chem. Phys. Lett., 2002, 355, 438–444 CrossRef CAS.
- (a) S. S. Katiyar and S. N. Tandon, Talanta, 1964, 11, 892–894 CrossRef; (b) D. K. Johnson, T. B. Murphy, N. J. Rose, W. H. Goodwin and L. Pickart, Inorg. Chim. Acta, 1982, 67, 159–165 CrossRef CAS; (c) P. F. Lee, C.-T. Yang, D. Fan, J. J. Vittal and J. D. Ranford, Polyhedron, 2003, 22, 2781–2786 CrossRef CAS.
- E. Bardez, I. Devol, B. Larrey and B. Valeur, J. Phys. Chem. B, 1997, 101, 7786–7793 CrossRef CAS.
- K. A. Zachariasse, M. Grobys and E. Tauer, Chem. Phys. Lett., 1997, 274, 372–382 CrossRef CAS.
- E. W. Ainscough, A. W. Brodie, A. J. Dobbs, J. D. Ranford and J. M. Waters, Inorg. Chim. Acta, 1998, 267, 27–38 CrossRef CAS.
- (a) S. Dähne, W. Freyer, K. Teuchner, J. Dobkowski and Z. R. Grabowski, J. Lumin., 1980, 22, 37–49 CrossRef; (b) J. Dobkowski, E. Kirkor-Kamińska, J. Koput and A. Siemiarczuk, J. Lumin., 1982, 27, 339–355.
- (a) I. Leray, F. O' Reilly, J.-L. Habib-Jiwan, J.-Ph. Soumillion and B. Valeur, Chem. Commun., 1999, 795–796 RSC; (b) H. Zhang, L.-F. Han, K. A. Zachariasse and Y.-B. Jiang, Org. Lett., 2005, 7, 4217–4220 CrossRef CAS.
- R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635–3641 CrossRef CAS.
- (a) S. A. McFarland and N. S. Finney, J. Am. Chem. Soc., 2002, 124, 1178–1179 CrossRef CAS; (b) J. V. Mello and N. S. Finney, Angew. Chem., Int. Ed., 2001, 40, 1536–1538 CrossRef CAS; (c) G. Hennrich, H. Sonnenschein and U. Resch-Genger, Tetrahedron Lett., 2001, 42, 2805–2808 CrossRef CAS; (d) S. Watanabe, O. Onogawa, Y. Komatsu and K. Yoshida, J. Am. Chem. Soc., 1998, 120, 229–230 CrossRef CAS; (e) K. Sandanayake, K. Nakashima and S. Shinkai, J. Chem. Soc., Chem. Commun., 1994, 1621–1622 RSC; (f) D. H. Lee, J. H. Im, J.-H. Lee and J.-I. Hong, Tetrahedron Lett., 2002, 43, 9637–9640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full characterization of 1 and 2; absorption and fluorescence spectra of 1 in the presence of metal ions; solvatochromic data, IR and ESI-MS of 1 and 1–Cu2+ complex. See DOI: 10.1039/b510546c |
| This journal is © The Royal Society of Chemistry 2006 |