Methyl transfer reaction from monomethyltin reagent under palladium(0) catalysis: a versatile method for labelling with carbon-11†
Mickaël Huibana, Aline Huetb, Louisa Barréa, Franck Sobrio*a, Eric Fouquet*b and Cécile Perrio*ac
aGroupe de Développements Méthodologiques en Tomographie par Emission de Positons, DRM-DSV, UMR CEA 2 FRE CNRS 2698, Université de Caen-Basse Normandie, Centre Cyceron, 15 Boulevard Becquerel, BP 5229, F-14074, Caen Cedex, France. E-mail: sobrio@cyceron.fr; e.fouquet@lcoo.u-bordeaux1.fr; perrio@cyceron.fr
bLaboratoire de Chimie Organique et Organométallique, UMR CNRS 3802, Université Bordeaux I, 351 Cours de la Libération, F-33405, Talence Cedex, France
cLaboratoire de Chimie Moléculaire et Thioorganique, UMR CNRS 6507, ENSICAEN, Université de Caen-Basse Normandie, 6 Boulevard Maréchal Juin, 14050, Caen Cedex, France
First published on 11th November 2005
Abstract
The 11C-monomethylstannate prepared from [11C]-methyl iodide and Lappert's stannylene, was subject to a palladium-mediated cross-coupling reaction with an aryl halide under ligand-free conditions, to afford easily purified 11C-methyl(hetero)arenes in high radiochemical yields.
In recent years, the palladium mediated Stille reaction has proved to be an important route for the synthesis of radiotracers for Positron Emission Tomography.1–3 It usually involves the reaction of an aryltriorganostannane with easily synthesized [11C]-methyliodide as the electrophilic partner, leading to a 11C-carbon–carbon coupled product. However, preparation of organostannane precursors is not always straightforward, and in the case of functionalized organostannanes, nucleophilic groups have to be protected in order to prevent methylation as a side-reaction.2,4 Moreover, the use of trimethylstannyl derivatives as substrates would result in low specific activities, due to the competitive transfer of an unlabelled methyl group.3 Finally, difficulties might be encountered in separating the tracer from triorganotin residues. For these reasons, the use of an original [11C]-methyl labelled version of Vedejs' 1-aza-5-stannabicyclo[3,3,3]undecane was attempted,5 and the Suzuki reaction6 was also proposed as an alternative approach. However, no real improvement in terms of efficiency or in the scope of the reaction has been demonstrated so far.
Monoorganotins, activated by a fluoride source, have been shown to be very reactive in the Stille coupling reaction,7 allowing the transfer of vinyl, allyl, alkynyl, aryl, benzyl and even alkyl groups onto aryl or vinyl halides8,9 or triflates10 in good to excellent yields. They are far less toxic than di-, tri- and tetra-alkylstannanes11 and inorganic tin by-products are easily removed.12 Since such reagents are readily prepared from Lappert's stannylene13 (Sn[N(TMS)2]2) and an alkyl or aryl halide,8,9 we anticipated the possibility of synthesising the 11C-monomethyltin reagent [11C]-1 from [11C]-iodomethane, and transferring the 11C-methyl group onto an aromatic or heteroaromatic structure by reaction with an electrophile 2 (Scheme 1).
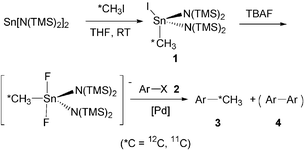 | ||
| Scheme 1 Methyl transfer reaction from stannane 1. | ||
We first evaluated the feasibility of the cross-coupling reaction under rapid conditions4 using unlabelled methyl iodide. Formation of monoorganotin 1 from iodomethane and stannylene in THF according to the previously described procedure,9 was quantitative and immediate. After in situ activation with TBAF (tetrabutylammonium fluoride, 3 equiv.), the resulting hypervalent stannate reagent was engaged in a palladium-catalyzed cross-coupling reaction with an aryl halide 2 (0.6 equiv.) in refluxing dioxane. In order to minimize reaction times and optimize the reaction, the nature and the amount of the catalytic system were varied (Table 1). Results for reactions starting from 2-bromonaphthalene 2a were found to be representative.
| Entry | Ar–X | Precatalyst/mol% | Ligand/mol% | Time/min | Ar–CH3 | Yieldb (%) | Ar–Ar | Yieldc (%) |
|---|---|---|---|---|---|---|---|---|
| a 1 (1 mmol), 2 (0.6 mmol), TBAF (3 mmol).b Isolated yield.c Estimated by GC. | ||||||||
| 1 | 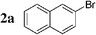 | Pd(PPh3)2Cl2 (1) | — | 30 | 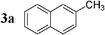 | 91 | 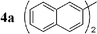 | 2 |
| 2 | Pd2dba3 (1) | PPh3 (4) | 240 | 60 | 25 | |||
| 3 | Pd2dba3 (1) | P(OMe)3 (4) | 30 | 91 | 2 | |||
| 4 | Pd2dba3 (1) | P(OiPr)3 (4) | 20 | 54 | 1 | |||
| 5 | Pd2dba3 (1) | — | 120 | 70 | — | |||
| 6 | Pd2dba3 (2) | — | 90 | 60 | 30 | |||
| 7 | Pd2dba3 (5) | — | 5 | 90 | <1 | |||
| 8 | 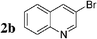 | Pd(PPh3)2Cl2 (1) | — | 15 | 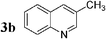 | 82 | 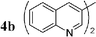 | <1 |
| 9 | Pd2dba3 (1) | P(OiPr)3 (4) | 5 | 85 | 11 | |||
| 10 | Pd2dba3 (5) | — | 2 | 90 | <1 | |||
| 11 | 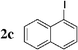 | Pd2dba3 (1) | — | 120 | 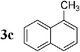 | 60 | 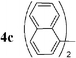 | — |
| 12 | Pd2dba3 (2) | — | 60 | 60 | — | |||
| 13 | Pd2dba3 (5) | — | 2 | 100 | — | |||
| 14 | 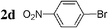 | Pd2dba3 (1) | — | 30 | 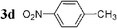 | 95 | 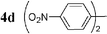 | — |
| 15 | 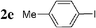 | Pd2dba3 (1) | — | 5 | 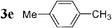 | 86 | 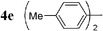 | — |
Initially, bis(triphenylphosphine)palladium(II) chloride (1 mol%), which was found to accelerate the transfer of n-alkyl groups more than tetrakis(triphenylphosphine)palladium,9 was used as precatalyst (entry 1). The anticipated 2-methylnaphthalene 3a was obtained in 91% yield after 30 min. Only traces of substrate–substrate homocoupling product 4a were detected as side-products. The catalyst was also generated in situ from Pd2dba3 (tris(dibenzilideneacetone)dipalladium, 1 mol%), used in conjunction with a ligand (4 mol%, 2 equiv. for each Pd atom). We observed that the steric and electronic properties of the ligand significantly influenced the course of the reaction. Using triphenylphosphine, the formation of the cross-coupling product 3a was greatly reduced, whereas that of dinaphthalene 4a was significantly enhanced (entry 2). Less donating trialkylphosphites, and especially less sterically hindered trimethylphosphite, facilitated the cross-coupling reaction, resulting in the recovery of product 3a in high yields after a short time (entries 3 and 4). Finally, the reaction was performed under ligand-free conditions (entries 5–7). The cross-coupling reaction took place, and was strongly accelerated by increasing the amount of catalyst up to 5 mol%. Under the latter conditions, the yield of 3a reached 90% after 5 min reaction time. The methyl transfer was found to be efficient either starting from 3-bromoquinoline 2b (entry 10), 1-iodonaphthalene 2c (entry 11–13), 4-bromonitrobenzene 2d (entry 14) or 4-iodotoluene 2e (entry 13).
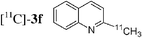
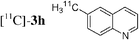
Having in hand conditions allowing a few minutes' reaction time compatible with the half-life of carbon-11,4 studies using [11C]-CH3I were undertaken. To do so [11C]-CH3I obtained from cyclotron produced [11C]-CO2, was distilled into THF containing the stannylene for the formation of [11C]-1. TBAF was added at room temperature, and THF was removed by heating at 120 °C under nitrogen. After addition of the catalyst and the electrophile 2a,b,f–i in dioxane, the mixture was heated at 120 °C for 5 min, then quenched with water. Analyses of the crude products were performed by radio TLC and HPLC, by comparison with authentic stable samples. Due to the small amount of [11C]-CH3I,4 and consequently of [11C]-1, it has to be kept in mind that the coupling reactions were performed with an excess of palladium catalyst. Furthermore, the presence of remaining stannylene associated with the large excess of the electrophile might favour the non-radioactive homocoupling reaction over the radioactive methyl transfer. Thus, the development of the cross-coupling reaction in radioactive chemistry required some adjustments and the influence of the amount of each reagent was carefully examined. The reaction using 3-bromoquinoline 2b as substrate was taken as model (Table 2). In all cases, formation of the expected radioactive 3-methylquinoline [11C]-3b was detected. Homocoupling product 4b was obtained as a non-radioactive side-product which was easily separated from [11C]-3b by preparative HPLC. Whatever the amount of stannylene used (7–20 µmol), the trapping of [11C]-CH3I leading to [11C]-1, was quantitative and immediate, and the radiochemical yield in [11C]-3b was not changed (entries 1–3). As anticipated, the amount of TBAF additive was shown to be crucial for an optimum 11C-methyl transfer. The incorporation rate of carbon-11 increased from 4 to 75% by using 1 to 3 equiv. of TBAF compared to the stannylene (entries 5, 9–10). The need for 3 equiv. of TBAF was explained as resulting from the side-reaction occurring between the stannylene and the substrate 2b, leading to the corresponding pentavalent tin species.9 The radiochemical yield of [11C]-3b was not altered by decreasing the quantity of substrate 2b from 110 to 37 µmol, since it was always used in excess with respect to the stannylene (entry 2 versus entry 4, and entry 5 versus entry 11). On the other hand, the catalyst amount could not be lowered below 3 µmol, otherwise formation of [11C]-3b was significantly reduced (entries 4–5, 8). Taking into account all results we identified as optimum conditions, the use of 15 µmol of stannylene, 45 µmol of TBAF, 50–70 µmol of 3-bromoquinoline 2b, and 5 µmol of Pd2dba3. Under these conditions, a radiochemical yield of [11C]-3b up to 78% was obtained. We noted that the cross-coupling reaction did not continue beyond 5 min (entries 5–7). In the course of further studies aiming to simplify the procedure, dioxane was found to be necessary for the coupling reaction. We observed that the yield of [11C]-3b decreased significantly if evaporation of THF was not complete (data not presented).
| Entry | Sn[N(TMS)2]2/µmol | TBAF/µmol | 2b/µmol | Pd2dba3/µmol | Time/min | [11C]-3b (%)a |
|---|---|---|---|---|---|---|
| a Radiochemical yield decay corrected, based on [11C]-CH3I (n = 2 or 3). | ||||||
| 1 | 20 | 60 | 110 | 5 | 5 | 74 ± 5 |
| 2 | 15 | 40 | 110 | 5 | 5 | 77 ± 7 |
| 3 | 7 | 21 | 110 | 5 | 5 | 70 ± 8 |
| 4 | 15 | 45 | 75 | 5 | 5 | 78 ± 2 |
| 5 | 15 | 45 | 75 | 3 | 5 | 75 ± 4 |
| 6 | 15 | 45 | 75 | 3 | 10 | 74 ± 6 |
| 7 | 15 | 45 | 75 | 3 | 15 | 72 ± 3 |
| 8 | 15 | 45 | 75 | 1.5 | 5 | 52 ± 23 |
| 9 | 15 | 30 | 75 | 3 | 5 | 38 ± 2 |
| 10 | 15 | 15 | 75 | 3 | 5 | 4 ± 2 |
| 11 | 15 | 45 | 37 | 3 | 5 | 75 ± 1 |
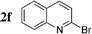
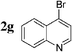
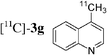
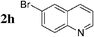
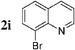
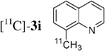
The radiosynthesis was extended to 2-bromonaphthalene 2a and quinolines 2f–i bearing a bromine atom at different positions (Table 3). Radiochemical yields of corresponding [11C]-3 were reproducible, in the range of 63–78% except for 2-methylquinoline [11C]-3f (around 40%, entry 3).
In summary, we have developed the palladium-catalyzed methyl transfer reaction from monomethylstannane 1 or [11C]-1 to various aryl halide 2. The quantitative and immediate conversion of [11C]-methyl iodide into methyltin reagent [11C]-1, the efficiency of the rapid coupling reaction under ligand-free and rapid conditions, the use of easily available electrophiles as substrates, and the ease of purification, make this methodology highly suitable for radiosynthesis of PET tracers. We are currently further investigating the scope of this methodology for the radiosynthesis of complex biologically active compounds.
The authors thank Professor M.-C. Lasne (ENSICAEN), O. Tirel and M. Ibazizene (Centre Cyceron). The present work was supported by the “Ministère de la Recherche et des Nouvelles Technologies” (ACI program), CEA (Commissariat à l'Energie Atomique), CNRS (Centre National de la Recherche Scientifique), the «PUNCH-Orga» Network (Pôle Universitaire Normand de Chimie Organique), the “Région Basse-Normandie” and the European Union (FEDER funding).
Notes and references
- L. Samuelsson and B. Långström, J. Labelled Compd. Radiopharm., 2003, 46, 263 CrossRef CAS; F. Karimi and B. Långström, J. Labelled Compd. Radiopharm., 2002, 45, 423 CrossRef CAS; J. Tarkiainen, J. Vercouillie, P. Emond, J. Sandell, J. Hiltunen, Y. Frangin, D. Guilloteau and C. Halldin, J. Labelled Compd. Radiopharm., 2001, 44, 1013 CrossRef; M. Björkman, H. Doi, B. Resul, M. Suzuki, R. Noyori, Y. Watanabe and B. Långström, J. Labelled Compd. Radiopharm., 2000, 43, 1327 CrossRef CAS; M. Suzuki, H. Doi, M. Björkman, Y. Andersson, B. Långström, Y. Watanabe and R. Noyori, Chem.-Eur. J., 1997, 3, 2039 CrossRef CAS; Y. Andersson, A. Cheng and B. Långström, Act. Chem. Scand., 1995, 49, 683 Search PubMed.
- O. Langer, T. Forngren, J. Sandell, F. Dollé, B. Långström, K. Någren and C. Halldin, J. Labelled Compd. Radiopharm., 2003, 46, 55 CrossRef CAS; J. Sandell, M. Yu, P. Emond, L. Garreau, S. Chalon, K. Någren, D. Guilloteau and C. Halldin, Bioorg. Med. Chem. Lett., 2002, 12, 3611 CrossRef CAS.
- J. Madsen, P. Merachtsaki, P. Davoodpour, M. Bergström, B. Långström, K. Andersen, C. Thomsen, L. Martiny and G. M. Knudsen, Bioorg. Med. Chem., 2003, 11, 3447 CrossRef CAS.
- Chemistry using carbon-11 is linked to the short half-life of the radioisotope (t1/2 = 20.4 min) and to the use of submicromolar quantities of labelled reactant. Radiosyntheses have to be carried out within a minimum of steps, and reactions have to be rapid, efficient, selective and preferably without purification in between reaction steps, while leading to high chemical and radiochemical purities, and high specific radioactivities. Radiosynthesis strategies avoiding deprotection steps are welcome. R. A. Ferrieri, G. Antoni, T. Khilberg and B. Långström, in Handbook of Radiopharmaceuticals. Radiochemistry and Applications, ed. M. J. Welch and C. S. Redvanly, Wiley, Chichester, 2003, pp. 229–282 Search PubMed.
- T. Forngren, L. Samuelsson and B. Långström, J. Labelled Compd. Radiopharm., 2004, 47, 71 CrossRef CAS; E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114, 6556 CrossRef CAS.
- E. D. Hostetler and H. D. Burns, J. Labelled Compd. Radiopharm., 2003, 46, S1–S403 CrossRef.
- For a recent review see: A. M. Echavarren, Angew. Chem., Int. Ed., 2005, 44, 3962 Search PubMed.
- E. Fouquet, M. Pereyre and A. L. Rodriguez, J. Org. Chem., 1997, 62, 5242 CrossRef CAS.
- A. Herve, A. L. Rodriguez and E. Fouquet, J. Org. Chem., 2005, 70, 1953 CrossRef CAS.
- E. Fouquet and A. L. Rodriguez, Synlett, 1998, 1323 CrossRef CAS.
- P. J. Smith, Health and safety aspect of tin chemicals, in Chemistry of Tin, ed. P. J. Smith, 1998, ch. 11 Search PubMed.
- E. Fouquet, M. Pereyre, A. L. Rodriguez and T. Roulet, Bull. Soc. Chim. Fr., 1997, 134, 959 CAS.
- D. H. Harris and M. F. J. Lappert, J. Chem. Soc., Chem. Commun., 1974, 895 RSC; C. D. Schaeffer and J. J. Zuckerman, J. Am. Chem. Soc., 1974, 96, 7160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures and characterization data. See DOI: 10.1039/b510286c |
| This journal is © The Royal Society of Chemistry 2006 |