Dendritic supermolecules – towards controllable nanomaterials
David K. Smith*
Department of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: dks3@york.ac.uk
First published on 1st September 2005
Abstract
Dendritic molecules constitute one of the most exciting areas of modern nanochemistry, largely as a consequence of the unique properties associated with their branched architectures. This article describes how ‘dendritic function’ can also be achieved using small, synthetically accessible branched building blocks (individual dendrons) which simply self-assemble via non-covalent interactions to generate dendritic nanoscale architectures with novel behaviour. (a) Using non-covalent interactions at the focal point of a dendron allows the self-assembly of nanometre-sized supramolecular dendrimers around an appropriate template species. Such systems have potential applications in the controlled encapsulation and release of active ingredients. (b) Employing non-covalent intermolecular dendron–dendron interactions can give rise to the hierarchical assembly of nanostructured materials. Such assemblies of dendritic molecules ultimately express their molecular scale information on a macroscopic scale, and therefore have applications in materials science, for example as gels. (c) The multiple surface groups of dendrons are capable of forming multiple interactions with large surfaces, such as those found on biomolecules or in biological systems. Employing multivalent interactions between dendron surfaces and biological molecules opens up the potential application of dendritic systems as medicinal therapies. In summary, dendritic supermolecules offer a potentially cost-effective approach to the future application of dendritic systems to a range of real-world problems.
 David K. Smith | Dave Smith completed his BA and D.Phil at Oxford University in the group of Paul Beer. He then held a Royal Society postdoctoral fellowship at ETH Zürich in the labs of Francois Diederich. In 1999, Dave returned to the UK where he took up a lectureship at the University of York, and in 2004 he was promoted to senior lecturer. His research interests lie in the areas of supramolecular chemistry, dendrimer chemistry and nanoscale science. He particularly enjoys research which is truly interdisciplinary – for example working with materials physicists and biologists. He regularly participates in scientific outreach – giving schools lectures, as well as talks to the general public discussing medicinal chemistry and the impact of nanotechnology. In 2004, he was awarded the Royal Society of Chemistry Higher Education Teaching Award. Outside of work, Dave enjoys travel, skiing, walking and cooking. |
Introduction
At present, there is increasing excitement about the way in which researchers working at the interfaces between chemistry, biology and physics are uncovering the secrets and unique properties of nanometre-sized objects – the emergent field of nanotechnology.1 Amongst the most interesting nanoscale molecular architectures developed over the past 20 years are dendrimers.2 Dendrons and dendrimers are comprised of repeating branched building blocks and their structures are monodisperse. Dendrons can be considered as individual dendritic branches, whilst dendrimers are structures in which a number of dendrons are attached to a single core unit. The relatively straightforward iterative chemical synthesis of high molecular weight dendrimers has made them ideal candidates for nanotechnological applications – indeed dendrons and dendrimers can be considered to be a unique family of nanoscopic building blocks.3 Dendrimers have a number of unique properties as a consequence of their branched architectures.4 The core of a dendrimer can be considered to be isolated from the bulk environment by the dendritic shell, and hence experiences a unique microenvironment which leads to new types of behaviour. Meanwhile, the multiple surface groups of dendritic molecules have the potential to form multiple interactions, either with bulk solvent, or with another chemical/biological species.However, although the synthesis of dendritic molecules is an apparently straightforward iterative process, it is still relatively difficult to make bulk quantities of high generation dendrimers at a low price, and with high purity. This has meant that thus far, the majority of potential applications of this class of molecule have a high added value (e.g., biomedical applications).5 Indeed, dendritic molecules are currently being exploited by Starpharma – a company involved in the development of dendrimers with multivalent surfaces which possess effective anti-viral (in particular, anti STD) activity.6
One approach to achieving ‘dendritic function’ whilst avoiding the cost of synthesising dendritic molecules is to make use of hyperbranched polymers (HBPs).7 Hyperbranched systems are imperfect branched polymers generated in a single synthetic step by polymerising a branched monomer, and as such, they have considerable potential for the development of bulk applications. For example, DSM have developed hybrane, which is used to prevent ice crystal formation and inhibit blockage of oil pipelines.8 Meanwhile, Perstorp have developed the Boltorn HBPs which have been used to modify polyurethane, with applications in (e.g.) car seating.9 However, hyperbranched polymers are not homogeneous systems, and the use of well-defined dendritic molecules may offer significant advantages.
This paper therefore outlines a different approach to achieving ‘dendritic function’, which avoids much of the time-consuming synthetic chemistry. Rather than using covalent chemistry to synthesise complex, nanoscopic perfect branched architectures, small dendritic building blocks can instead be endowed with suitable chemistries to allow them to self-assemble via well-defined, specific, non-covalent interactions. This approach, which can be considered as supramolecular dendrimer chemistry, enables the spontaneous assembly of complex nanoscale systems (Fig. 1).10 In recent years, increasing use has been made of ‘bottom-up’ self-assembly for the fabrication of interesting nanostructured architectures in solution.11 Self-assembly methods rely on the fundamental principles of supramolecular chemistry12 (i.e., the use of non-covalent interactions between molecules) and offer a straightforward approach by which complex systems can be generated using minimum synthetic input. This is of great interest in the field of ‘soft’ nanotechnology, in which the unique behaviour of nanoscale assemblies is harnessed. Molecular self-assemblies have potential applications ranging from materials chemistry through to biological intervention. The reversibility of non-covalent interactions means that such systems can ‘error-check’ their assembled structures. Furthermore, they exhibit exquisite tunabilities and, if built from biologically compatible building blocks, should not pose long-term health or environmental risks, as they can disassemble into smaller units which may be readily broken down.
 | ||
| Fig. 1 Schematic overview of the types of self-assembly using simple dendritic molecules described in this article. | ||
This article will provide selected references to the work of other groups who are actively working on nanochemistry using the self-assembly of simple, synthetically accessible dendritic building blocks, but will primarily focus on our own work in this area. For a more complete overview, interested readers are directed to a major review we have recently published which deals with this exciting topic in a comprehensive manner.13
Assembling discrete supramolecular dendrimers
This combination of supramolecular methodology and dendrimer chemistry has been of interest for some time,10 however perhaps the first example in which non-covalent interactions were used to assemble individual dendrons into a supramolecular dendrimer was reported by Zimmerman and co-workers in 1996.14 They reported dendrons which were able to self-assemble as a consequence of hydrogen bond mediated carboxylic acid dimerisation to form a hexameric rosette. Although relatively difficult to synthesise, these dendrons illustrated the way in which multiple dendritic building blocks could assemble in a controlled manner to yield a well-defined nanoscale architecture. Indeed, the dendritic branching exerted direct control over the self-assembly process – if the dendrons were too small, an ill-defined supramolecular polymer resulted, but if they were sufficiently large they acted as a steric buttress and encouraged the formation of the desired hexameric rosette-type architecture. Subsequently, the research groups of both Reinhoudt and Fréchet also reported supramolecular dendrimers based on hydrogen bonded hexameric rosettes.15Interestingly, Zimmerman and co-workers went on to use an organic template to direct the dendritic self-assembly process via anthyridine–amidinium interactions (Fig. 2).16 In this way, they assembled a supramolecular dendrimer with a mass of over 10,000. The template cation was chosen because it is active against Pneumocystis carinii pneumonia, and this example therefore provides insight into the ways in which supramolecular dendrimers can be assembled around active cores using individual dendron building blocks.
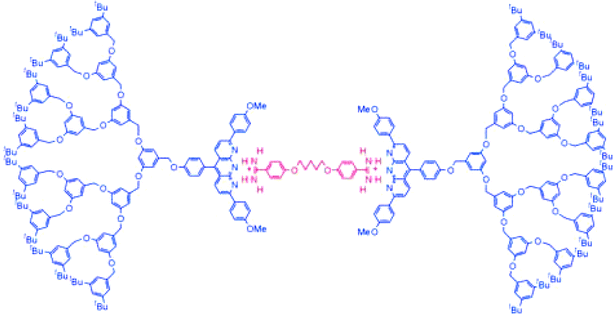 | ||
| Fig. 2 Self-assembled dendrimer reported by Zimmerman and co-workers based on the interaction between anthyridine units and amidinium cations. | ||
We realised that the self-assembly of dendritic building blocks around a templating species could effectively place the templating species in a unique microenvironment, something that had only previously been achieved using covalent dendritic encapsulation.17 With this goal in mind, we investigated the interactions between a polybasic hydrophilic dye and a dendron with an acidic group at the focal point18 (and vice versa – a polyacidic hydrophilic biologically active dye and dendrons with a basic group at the focal point – Fig. 3).19 In both cases, the hydrophilic dyes were solubilised into apolar solvents, in which they otherwise had no solubility. Furthermore, increasing the dendritic generation led to enhanced levels of solubilisation. Careful control experiments with focal-point protected dendrons showed that the acid–base interactions played a key role in the solubilisation process. It was proposed that the acid–base interactions generated a complex in which the hydrophilic dye was effectively encapsulated within a non-covalently generated dendritic microenvironment. Most interestingly, the optical properties of the dye were modified by the process – indeed the observed λmax values depended on the dendritic generation – once again in agreement with the formation of a non-covalent dendritic microenvironment.
 | ||
| Fig. 3 Solubilisation of a hydrophilic dye (aurin tricarboxylic acid) into apolar solvents as a consequence of acid–base interactions with the focal point of an appropriate dendron – two interactions shown. | ||
We also reported electron spin resonance (ESR) investigations using a spin-active probe which could bind to the focal point of our dendrons via acid–base interactions.20 These investigations demonstrated that complexation at the focal point of the dendron was able to slow down the motion of the spin-active template. Notably, this effect was most pronounced with the largest dendron investigated (third generation). We demonstrated that the presence of an appropriate functional group at the focal point of the dendron, capable of interacting non-covalently with the spin-active probe, was essential for this ESR effect to be observed.
With our previous results in mind, we went on to modify the non-covalent interactions responsible for the construction of our supramolecular dendrimers. By functionalising the focal point of L-lysine dendrons21 with a crown-ether derivative, it became possible to employ the relatively strong interaction between the crown ether and a protonated amine in order to assemble supramolecular dendrimers.22 This interaction is well-defined and can even survive in relatively competitive solvents such as methanol. Using NMR methods, we demonstrated that a bis-ammonium cation was bound by two dendritic crown ethers (Fig. 4) and its NMR spectrum reported that it experienced an encapsulated microenvironment.
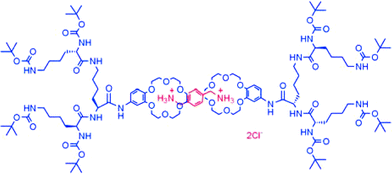 | ||
| Fig. 4 Supramolecular dendrimer formed by interactions between crown ethers and protonated amines. | ||
Interestingly, we were able to disassemble the supramolecular dendrimer either by adding K+ ions, or base.23 Potassium ions bind two orders of magnitude more strongly to 18-crown-6 than ammonium cations, hence triggering the release of the encapsulated organic template. Base simply deprotonates the encapsulated ammonium cation, hence preventing binding. This study therefore demonstrated: (i) the ability of supramolecular chemistry to assemble well-defined nanoscale architectures using simple biocompatible dendritic building blocks, and (ii) the reversibility of non-covalent assembly methods (one of the key advantages of taking a supramolecular approach to dendritic materials). In future work, we intend to demonstrate the ability of this system to modify the function of different encapsulated templates – for example, redox active or catalytic templates. It is worth noting that Gibson and co-workers have also taken a crown ether based approach to dendritic supermolecules,24 using the interaction between a protonated secondary amine and dendritic dibenzo-24-crown-8 derivatives to yield a non-covalent dendrimer via ‘rotaxane-like’ complexation.
In addition to using organic templates, there has been significant interest in the use of metal ions as templates for dendritic assembly.25 In a nice example, Kawa and Fréchet assembled three dendrons with carboxylate groups at the focal point around trivalent lanthanide cations, as a consequence of electrostatic carboxylate–Ln3+ interactions (Fig. 5).26 They showed that the optical properties of the lanthanide ion were controlled by the presence of the self-assembled dendritic shell. We took a similar approach to the encapsulation of Ln3+ cations using carboxylate anion focussed L-lysine dendritic branches.27 In this case, there was little difference in the optical properties of the lanthanide, but we showed that the Lewis acidic lanthanide ion could be considered to be effectively encapsulated within the dendritic shell – an observation which had implications for the catalytic proficiency of the metal ion. The lessons learned from this system may, in the future, be useful in the design of new encapsulated asymmetric catalysts.
 | ||
| Fig. 5 Self-assembled dendritic lanthanide complex reported by Fréchet and co-workers. | ||
As well as encapsulating individual metal ions, it is possible to use dendritic ligands as stabilisers for the growth of metal clusters and nanoparticles.28 Sulfur containing ligands are frequently used to stabilise Au(0) surfaces, and a number of groups have pioneered the use of dendritic ligands with an appropriate thiol stabilising group at the focal point with the goal of controlling nanoparticle synthesis and enhancing the stability of the resulting assembly.29 In collaboration with Chechik, we have developed gold nanoparticles in which the stabilising unit was a disulfide with dendritic L-lysine groups attached (Fig. 6).30 When disulfides stabilise gold nanoparticle surfaces, they are known to cleave, and the ligand binds to Au(0) in the thiolate form. This approach enabled us to generate gold nanoparticles with a biocompatible stabilising shell based on L-lysine – a goal of considerable interest. Interestingly, we found that the dendritic generation of the stabilising ligand controlled the size of the resultant gold nanoparticles (G1 – 2.9 nm, G2 – 2.4 nm, G3 – 1.8 nm). Furthermore, the assembly of a more extensive, higher generation dendritic shell around the nanosized gold core appeared to enhance the thermal stability of the particles. This indicates how molecular scale information programmed into dendritic branches can directly influence the size and stability of nanoscale assemblies.
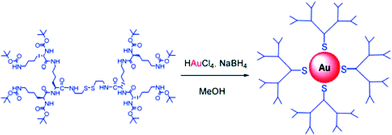 | ||
| Fig. 6 Second generation dendritic disulfide used for the stabilisation of self-assembled gold(0) nanoparticles. | ||
The discussion above has focussed attention on the way in which supramolecular dendrimers can be assembled using well-designed interactions between the focal point of individual dendrons and an appropriate template species. By assembling simple, synthetically accessible dendritic building blocks in this way, they are effectively ‘multiplied-up’, and this offers an easy approach to achieving control over the behaviour of the encapsulated template. In essence, such supramolecular dendrimers are of particular interest when they behave as more than the sum of their individual parts.
Hierarchical assembly of supramolecular dendrimers
In 2000, we made the serendipitous discovery of a supramolecular dendrimer which exhibited particularly interesting synergistic behaviour. In an extension of our research investigating acid–base interactions at the focal point of L-lysine dendrons, we performed an experiment to form a complex between this type of dendron and diaminododecane (Fig. 7).31 The dendron was soluble in organic solvents (e.g., toluene) whilst the diamine was not – however, on mixing (with sonication) the diamine became fully solubilised. We argued that the acid group at the focal point of the dendron formed a supramolecular complex with the amine groups, leading to effective encapsulation of the diamine within the dendritic shell, thus ensuring that it became compatible with the bulk solvent. However, before we could characterise the complex which had formed, we realised that the solution had immobilised the bulk solvent, forming a gel-phase material. | ||
| Fig. 7 Formation of gel-phase materials using a two-component approach based on acid–base interactions to form a gelator complex. | ||
Individual small molecules capable of assembling into soft gel-phase materials at very low weight percentages (e.g., 1% gelator, 99% solvent) are referred to as low molecular weight gelators, and they constitute a research field of intense current interest.32 In this case, we realised we had uncovered a two-component gel – such systems are relatively rare.33 It is generally accepted that the formation of supramolecular gels of this type takes place via a hierarchical self-assembly process. In this process, individual molecules assemble to form a complex. The complex subsequently assembles into one-dimensional strands or fibrils. Increasing the concentration further results in the formation of bundles of fibrils, which ultimately form a sample spanning network of fibres. Hierarchical self-assembly across extended length scales can be determined using a variety of experimental techniques, including small angle X-ray scattering (SAXS), electron microscopy and rheometry. A cryo-scanning electron microscopy (SEM) image of the entangled, gel-phase network formed using the two-component gelator complex (Fig. 7) is illustrated in Fig. 8. It is this fibrous network that ultimately gives rise to macroscopic gelation. The gel can be visualised with the naked eye, as the bulk solvent does not flow on tube inversion – a consequence of capillary forces which hold the bulk solvent within the pores of the material (individual solvent molecules retain their mobility within these pores).
 | ||
| Fig. 8 Cryo SEM image of gel-phase material formed using the building blocks shown in Fig. 7, only using bromobenzene as solvent. | ||
Only a limited number of dendritic gel-phase materials have been reported in the literature.34 Indeed, dendritic molecules are of particular interest for the formation of gels as the repeating functional groups inherent within the branched framework of the molecule have the potential to form multiple non-covalent interactions. From the reports in the literature it is also clear that simple dendron-type architectures have a much greater predisposition for gelation than fully formed spherical dendrimers. It is probable that this is, at least in part, a consequence of the need for anisotropic (i.e., directional) self-assembly underpinning the microstructure of the gel.
In a series of papers following our initial publication, we have rigorously investigated in detail the features which control the hierarchical self-assembly of dendritic molecular building blocks. All of the gels have thermal properties which respond to increasing concentration of the dendron:diamine complex (Fig. 9). We measure the thermal stability by finding the temperature at which the gel begins its transition to a sol (using a tube inversion methodology), and we denote this gel boundary temperature as Tgel. In each case, two concentration regimes can be identified. Initially increasing the concentration of the gelator complex increases the thermal stability. We refer to this as the ‘gel-building’ concentration regime. Ultimately, however, at a certain concentration (often about 1% wt/vol), the gel-phase network is essentially complete and the Tgel value becomes invariant with concentration – we refer to this as the plateau region.
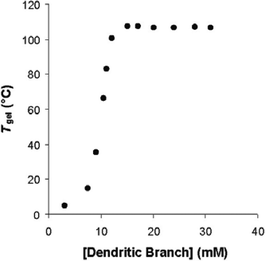 | ||
| Fig. 9 Concentration dependence of the gel-phase material formed using the building blocks illustrated in Fig. 7. Solvent – toluene. | ||
We have found that our system is one of the most highly tunable gelation systems in the literature, with the possibility to control a range of different structural parameters and obtain gels with a variety of properties. This indicates the power of this kind of system in the ‘bottom-up’ fabrication of controlled nanostructures. In particular, we have shown the following structural features all play a key role in the self-assembly process:
(a) Acid–base interactions. It is essential that there is an interaction between the two components. When the carboxylic acid at the focal point of the dendron was protected as a methyl ester, no gelation was observed.28 However, this acid–base interaction, which underpins the formation of the complex responsible for gelation, could be replaced by other appropriate interactions to generate an analogous complex. Therefore, when crown ether dendrons were assembled around a long-chain protonated aliphatic diamine, a gel-phase material still resulted (Fig. 10).23 Furthermore, when the two components were permanently connected via covalent bond formation (Fig. 11), these systems formed one-component gels.35
 | ||
| Fig. 10 Dendritic gel-phase material in which the two components interact to form a complex as a consequence of interactions between crown ethers and protonated amines. | ||
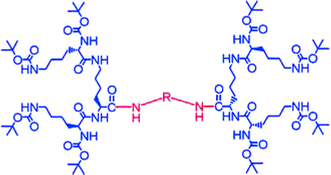 | ||
| Fig. 11 Single component dendritic gelator with a molecular structure which is directly analogous to the two-component system. | ||
(b) Spacer chain.36 The spacer chain had a profound effect on the thermal properties of the gels. Indeed decreasing the spacer chain in length from C12 to C6 reduced the Tgel value in the plateau region from 105 °C to 4 °C – a remarkable macroscopic change for a very subtle molecular scale change. In many gels, packing of aliphatic chains is important in controlling the self-assembly process, and in such cases an odd–even effect of spacer chain on gelation is often observed. In this case, however, there was no such effect. It is also very difficult to ascribe such a large change in thermal properties to an incremental increase in van der Waals interactions between spacer chains. Circular dichroism (CD) and NMR spectroscopic evidence indicated that increasing the length of the spacer chain enables more efficient hydrogen bonding between the chiral dendritic head groups, yielding an effective overall chiral nanostructure. We argued that the flexibility of the spacer chain provides sufficient room for the head groups to effectively interact and organise their mutual hydrogen bonding array, hence increasing the thermal stability of the gel.
(c) Dendritic generation.37 For these two-component gels, we have demonstrated that using the second generation dendron gave rise to the optimal materials in terms of Tgel values and the formation of an entangled network as visualised by SEM (Fig. 12). We argued that the first generation system did not contain sufficient hydrogen bonding N-H groups to enable the formation of an effective fibrous assembly – indeed an unusual ‘sausage-like’ morphology was observed. The third generation system, however, although containing lots of potential hydrogen bonding sites, was too sterically demanding to generate a perfectly organised anisotropic self-assembled fibre and gave rise instead to flattened fibres.
 | ||
| Fig. 12 SEM images of two-component gel-phase materials assembled using (A) first, (B) second and (C) third generation dendritic branches. Scale bars are 100 nm. | ||
(d) Solvent.38 By performing a thorough investigation of the effect of solvent on the thermal properties of the gels, we determined that gel behaviour could be correlated with specific solvent parameters. In particular, there was a general correlation with the Hildebrand polar solubility parameter (δa), as might be expected for a gel held together by hydrogen bond interactions. It was also necessary to consider whether the solvent was hydrogen bond donating or accepting using the Kamlet–Taft parameters (α and β respectively) – it was clear that whilst hydrogen bond acceptor solvents had little effect on the gelation process, hydrogen bond donors were easily able to disrupt gelation. This type of physical organic study of supramolecular gels is relatively rare, yet yields powerful predictive information.
(e) Chirality of the dendritic building blocks.39 Our initial gelators were synthesised using the naturally occurring L-lysine building blocks. Circular dichroism (CD) studies clearly indicated that whilst the dendron had minimal CD signal in its own right, in the presence of the diamine a large CD signal was observed – indicative of nanoscale chiral organisation. In this case, the CD signal is ascribed to the chiral organisation of the CONH groups (λmax = ca. 220 nm) within the branched structure. This can be best ascribed to a helical stacked organisation of the gelator complex. When using dendrons based on D-lysine, the resultant materials have identical thermal properties (e.g., Tgel values), but equal and opposite CD spectra, indicating the formation of identical fibrous assemblies but with opposite helicities. On mixing a small amount of the ‘wrong’ enantiomer into the gel, the nanoscale chiral organisation was rapidly destroyed, and the thermal stability of the macroscopic gel was significantly reduced. Most interestingly, SEM imaging indicated that the racemic gel had a new nanoscale morphology – flattened woven ribbons (Fig. 13). This study was reinforced by investigating the behaviour of the materials based on L, D, D or D, L, L dendrons, which also exhibited depressed Tgel values. This indicates that the effective organisation of the chiral centres in the gel is an important pre-requisite for anisotropic self-assembly – demonstrating that molecular scale features have a direct impact on both nanoscale self-assembly and macroscopic behaviour.
 | ||
| Fig. 13 SEM images of gel-phase materials formed from the system illustrated in Fig. 7 using (A) L, L, L, (B) L, L, L ∶ D, D, D (50 ∶ 50) and (C) D, D, D dendritic peptides. | ||
(f) Molar ratio of the two components.40 Perhaps the most interesting way of controlling the self-assembly process was to modify the ratio of the two components. This led to the formation of completely new nanoscale morphologies. In the presence of excess diamine, instead of a fibrillar morphology being formed, a morphological transition to discrete structures was observed which could be imaged by SEM (Fig. 14). At relatively high mole fractions of diamine, we argue that the dendron acts simply to stabilise the surface of organized discrete regions of aliphatic diamine, and a controlled ‘crystallization’ process begins to control the aggregate morphology. Depending on the length of the spacer chain, different discrete morphologies, with different materials properties were generated. For the C12 spacer chain, a suspension of micron sized platelets was visualised. With the C10 spacer chain, micron sized platelets were once again observed. These platelets formed a sample spanning ‘honeycomb’ network which led to the formation of a transparent gel-phase material at ambient temperatures (Fig. 15A). Gel-phase materials which are not based on fibrillar morphologies are extremely rare.41 When using the C9 and C8 spacer chains, even more interesting morphologies were observed – nanoscale squares and rosettes respectively (Figs. 15B/C). Fascinatingly, and unusually, these discrete aggregates were again still capable of forming extended networks underpinning gel-phase materials.
 | ||
| Fig. 14 SEM images of gel-phase materials formed from the system illustrated in Fig. 7, only formed using (A) 2 ∶ 1 and (B) 1 ∶ 4.5 dendron ∶ diamine molar ratios. | ||
 | ||
| Fig. 15 SEM images of the morphologies formed by gel-phase system in the presence of excess diamine when using diamines with different spacer chain lengths: (A) C10, scale bar: 200 nm; (B) C9, scale bar: 200 nm; (C) C8, scale bar: 1 μm. | ||
These observations are consistent with the proposal that when excess diamine is present, its dendritically controlled nano-‘crystallization’ becomes the dominant feature of macroscopic gelation, with the length of the spacer chain controlling the preferred morphology of the observed nanocrystals. Organic nanocrystals are of considerable interest, as they have previously proven very difficult to fabricate and have applications in pharmaceutical formulation.42 These results therefore indicate how simple building blocks can be combined to yield nanoscale assemblies with interesting materials properties and unique gel-phase morphologies.
(g) Gelator surface groups. We have also modified the surfaces of the gelators. The surfaces of dendritic molecules are of particular interest because their large surface area means they interact very effectively with the bulk surroundings (i.e., solvent in this case). By modifying the periphery of our dendrons with long hydrophobic chains (Fig. 16) we have significantly enhanced the solvent range which can be gelated by these systems.43 Interestingly, the mode of self-assembly was also modulated – indeed, the individual dendrons themselves were now capable of gelation either on their own (i.e., as single component gelators), or as a two-component mixture with a diamine. This ability to form single component gels is presumably a consequence of the multiple van der Waals interactions which are now possible between the surface groups giving rise to an enhanced self-assembly pathway. Interestingly, the second generation system formed a more effective one-component gel (presumably due to the multiple van der Waals interactions) whilst the first generation dendritic system yielded a more effective two-component gel. This system could therefore be controlled either dendritically, or by the addition of a second component.
 | ||
| Fig. 16 Dendrons functionalised on the surface with long hydrophobic chains act as gelators in their own right, as well as in the presence of an appropriate aliphatic diamine. They form gels in a wide range of organic solvents. | ||
We are interested in extending this kind of research to yield novel switchable and controllable gel-phase materials. Furthermore, we are using this kind of hierarchical self-assembly approach to generate gels in biocompatible solvents, and intend to investigate their potential as controlled release formulation media. It is worth noting that all of the gels we have developed are based on L-lysine building blocks. Currently, L-lysine based dendrimers are the only dendritic systems to have been applied in vivo in humans.44
Since developing our basic understanding of the hierarchical self-assembly of supramolecular dendrimers into extended nanoscale morphologies, we have also begun to apply these gels for the fabrication of inorganic structures in collaboration with Chechik. There has been considerable interest in the self-assembly and synthesis of organic–inorganic hybrid materials, in which a self-assembled nanostructured organic system is able to control the subsequent synthesis or behaviour of an inorganic component.45 With this in mind, we used one of our dendritic gel-phase materials46 as a matrix for the photochemical synthesis of gold nanoparticles.47
In order to ‘load’ these organogels with an effective precursor for the synthesis of gold nanostructures, a diffusion method was used. HAuCl4 was transferred into toluene using tetraoctylammonium bromide (TOAB) and then the solution was allowed to stand above a sample of gel. The yellow coloration was observed to diffuse slowly (days) into the gel until an even distribution was obtained. On UV irradiation, the colour of the gel rapidly changed from orange/yellow to colourless (ca. 30 min) as Au(III) was converted to Au(I), and subsequently the gel changed from colourless to intense purple (Fig. 17) over a period of about 6 hours as Au(I) was reduced to Au(0). The final sample appeared as a coloured purple band running through the gel – which did not diffuse, even over extended periods of time (weeks). TEM indicated that gold nanoparticles with average diameters of 13 nm had been formed – although no additional nanostructuring of the individual particles (other than some aggregation) was observed in this case. It was shown that the nanoparticles were both stabilised by, and embedded within, the gel-phase network. We postulate that this approach may, in the future, generate materials with interesting optoelectronic applications, such as immobilised addressable nanoparticles, or gels with potential uses in sensor technology.
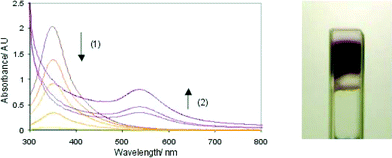 | ||
| Fig. 17 UV-Visible spectra on irradiation of HAuCl4 and TOAB within a gel-phase material. The spectra illustrate (1) the loss of colour as Au(III) is reduced to Au(I) and (2) the increase in the surface plasmon band as Au(0) nanoparticles are formed. The photo illustrates the purple band formed on irradiation. | ||
Interactions between dendrimers and biological targets
Thus far, we have considered using controlled self-assembly either at the focal point of dendrons, to yield well-defined supramolecular dendrimers, or through the branching to yield hierarchical arrays of supramolecular dendrimers. It is, however, also possible to employ the surface groups of simple, synthetically accessible dendrons to achieve self-assembly. As stated above, the surface groups of a dendritic structure have high surface areas and are highly accessible to the bulk medium. Furthermore, the fact that there are multiple copies of the surface groups can enhance weak binding events as a consequence of multivalency.48 Multivalent arrays of ligands can exhibit enhanced binding to biological surfaces as a consequence of the lesser entropic cost of organising them at the binding sites. This makes simple dendritic molecules with tailored surfaces of particular interest in biological ‘soft nanotechnology’ with potential applications in medicinal chemistry.Work from the groups of Roy and Stoddart developed the use of multivalent dendritic saccharides to enhance weak sugar binding processes with biological systems such as lectins which are of importance on cell surfaces.49 Control of this class of interactions is of importance in generating new therapeutics. It was shown that the precise structure of the dendritic scaffold played a key role in organising the surface saccharide units in order to maximise binding.50
There has also been enormous interest in the binding of DNA using dendritic systems. DNA binding is of potential therapeutic relevance as DNA–dendrimer complexes can in principle act as vectors for the delivery of therapeutic DNA into cells – so called gene therapy.51 It has been proposed that gene therapy may provide an approach for treating diseases as diverse as cystic fibrosis, arthritis, sickle cell anemia and cancer. The groups of Tomalia and Szoka pioneered the use of high generation poly(amidoamine) (PAMAM) dendrimers to bind and transfect DNA52 – indeed such molecules form the basis of the commercial ‘Superfect™’ kits. In recent years, a wide range of different dendritic DNA binding systems have been developed.13 All of these rely on interactions between protonated amines and polyanionic DNA for the formation of dendrimer–DNA complexes. In the majority of cases, large dendritic scaffolds are required.
In a recent example, however, Diederich and co-workers reported the ability of a small dendron (Fig. 18) to self-assemble and bind to DNA, yielding efficient transfection.53 This dendron once again employed protonated amine surface groups to bind to the anionic DNA, but also possessed hydrophobic tails to encourage additional self-assembly of the dendron. As such, this system is a cross between polymeric DNA binders such as Superfect and lipidic systems such as lipofectamine.54 Remarkable transfection efficiencies were observed – the first time that a ‘small’ dendron had been shown to have potential for this kind of application.
 | ||
| Fig. 18 Dendron reported by Diederich and co-workers to self-assemble with DNA. | ||
Around this time, we started the development of dendrons in which we hoped to enhance the strength of the DNA binding event. In particular we wanted to take a biomimetic approach to the development of DNA binders. With this goal in mind, we functionalised a dendritic surface with multiple spermine groups (Fig. 19). Spermine is present in human cells, where it plays a key role in the regulation of DNA.55 However, because the strength of spermine–DNA binding is relatively modest, its presence is required at millimolar concentrations. It is known that spermine binds to DNA in the minor groove and interestingly, that it finds it relatively difficult to compete against inorganic cations such as Na+, which are present in cells at high concentrations (> 100 mM).56 By placing multiple spermine groups onto the surface of our dendron, we hoped to generate a system which had multiple points of binding to the DNA helix and would consequently exhibit a multivalency effect. In addition, we also hoped that by using a naturally occurring DNA binder, and placing it on a relatively small dendron, the toxicity of the construct would be limited.
 | ||
| Fig. 19 Spermine derivatives investigated for ability to bind DNA in a multivalent manner – regions with affinity for DNA are shown in blue. | ||
We determined the ability of the first and second generation dendritic systems (and appropriate models and controls, Fig. 19) to bind to DNA using a variety of different methods.57 Ethidium bromide displacement assays allowed a clear comparison of the different DNA binders under equivalent conditions. Interestingly, whilst a single spermine unit (G0) only showed effective ethidium bromide displacement (> 50%) at concentrations well above 1 µM, the first (G1) and second (G2) generation systems showed binding at very low concentrations (76 and 30 nM, respectively). Furthermore, under biological salt conditions (150 mM NaCl), the binding of G0 was very adversely affected by two orders of magnitude (> 200 µM). However, the binding of the first and second generation dendrons to DNA still occurred at low concentrations (300 and 28 nM, respectively) – indeed the binding of the second generation system was salt independent. This is evidence of a clear multivalency effect of the spermine units on the dendritic surface, and indicates that effective DNA binding is possible even with small quantities of the dendron. Gel electrophoresis studies were supportive of the data described above. Furthermore, TEM studies indicated that the dendrons were capable of compacting the DNA into approximately spherical nanoscale structures (unlike simple spermine or G0 where the strength of binding is too low).
We have begun to investigate the potential of these dendrons as gene transfection agents in collaboration with Pack.58 Pleasingly, the structures exhibited low toxicities, and in the presence of chloroquine, were capable of transfecting cancer cell lines with genetic material coding for luciferase. However, the levels of transfection achieved with these initial structures were relatively modest. Nonetheless, there is plenty of potential for further synthetic modification to enhance the transfection process and develop effective simple vectors based on ‘small’ dendritic structures. Furthermore, we are interested in applying these very high affinity DNA binders for the encapsulation and protection of genetic material (e.g., from enzymatic degradation)59 and work to investigate this is currently in progress.
Conclusions and outlook
It is clear that the self-assembly of dendritic architectures can be exploited in a number of different ways to generate genuine nanostructured systems.(a) By using non-covalent interactions at the focal point of a dendron it is possible to achieve the effective self-assembly of nanometre-sized supramolecular dendrimers. Such systems have potential applications in controlled encapsulation and release processes.
(b) Employing non-covalent interactions between the branches of adjacent dendritic systems can give rise to hierarchical self-assembly of nanostructured materials, which ultimately expresses their molecular scale structural information on a macroscopic scale. The soft gel-phase materials we have assembled in this way have potential applications as biocompatible materials. Furthermore, this approach can in principle be used as a versatile approach for the ‘bottom-up’ nano-construction of a wide range of different morphologies.
(c) Finally, employing the surface of dendritic systems for self-assembly purposes allows the exploitation of multiple non-covalent interactions and consequent multivalency-type effects. This is of particular interest in enabling interactions between simple dendritic building blocks and complex biological macromolecules which possess large surface areas. We have demonstrated that a biomimetic approach, using multiple spermine groups on a dendritic surface, can enhance DNA binding – an area of research with potential future therapeutic implications.
In all cases, the general approach employed within our research, i.e., using small dendritic molecules which self-assemble to achieve their function, offers a potentially cost-effective approach to the future application of dendritic molecules to a wide range of real-world problems.
Acknowledgements
Most importantly, I am deeply indebted to the talented researchers working in my group over the past six years – their individual names can be found in the reference list. It is their enthusiasm and tenacity which has enabled all of the work discussed in this article. I am also indebted to our collaborators: Victor Chechik (University of York, UK), Martin Feiters (University of Nijmegen, NL), Ian Hamley (University of Leeds, UK) and Dan Pack (University of Illinois at Urbana Champaign, USA). Furthermore, I would like to thank EPSRC and The Leverhulme Trust for funding this research, as well as ICI Technology and Syngenta for their ongoing support of our research in this area. I also acknowledge Worldwide Universities Network and The Royal Society for providing travel expenses to visit collaborators. Finally, I would like to thank the University of York for some financial support, and more importantly for giving me the freedom to follow my research interests into new and fruitful areas.Notes and references
- (a) M. De Wild, S. Berner, H. Suzuki, L. Ramoino, A. Baratoff and T. A. Jung, Ann. N. Y. Acad. Sci., 2003, 1006, 291–305 CrossRef; (b) I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS; (c) G. M. Whitesides, Small, 2005, 1, 172–179 Search PubMed.
- (a) G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendrimers and Dendrons: Concepts, Syntheses, Applications, VCH, Weinheim, 2001 Search PubMed; (b) J. M. J. Fréchet and D. A. Tomalia (Eds.), Dendrimers and Other Dendritic Polymers, John Wiley and Sons, New York, 2002 Search PubMed.
- (a) D. A. Tomalia, Aldrichim. Acta, 2004, 37, 39–57 CAS; (b) J. M. J. Fréchet, J. Polym. Sci., Part A, 2003, 41, 3713–3725 CrossRef CAS.
- D. K. Smith and F. Diederich, Chem. Eur. J., 1998, 4, 1353–1361 CrossRef CAS.
- S. E. Stiriba, H. Frey and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 1329–1334 CrossRef CAS.
- (a) Y. H. Jiang, P. Emau, J. S. Cairns, L. Flanary, W. R. Morton, T. D. McCarthy and C. C. Tsai, AIDS Res. Hum. Retroviruses, 2005, 21, 207–213 CrossRef CAS; (b) M. Witvrouw, V. Fikkert, W. Pluymers, B. Matthews, K. Mardel, D. Schols, J. Raff, Z. Debyser, E. De Clercq, G. Holan and C. Pannecouque, Mol. Pharmacol., 2000, 58, 1100–1108 CAS.
- (a) B. I. Voit, Comp. Rev. Chim., 2003, 6, 821–832 Search PubMed; (b) C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS.
- P. Froehling, J. Polym. Sci., Part A, 2004, 42, 3110–3115 CrossRef CAS.
- (a) A. Hult, M. Johansson and E. Malmstrom, Macromol. Symp., 1995, 98, 1159–1161 CAS; (b) http://www.perstorp.com/upload/boltorn.pdf (accessed 24/06/2005).
- (a) D. K. Smith and F. Diederich, Top. Curr. Chem., 2000, 210, 183–227 CAS; (b) S. C. Zimmerman and L. J. Lawless, Top. Curr. Chem., 2001, 217, 95–120 CAS; (c) J. M. J. Fréchet, Proc. Natl. Acad. Sci. USA, 2002, 99, 4782–4787 CrossRef CAS; (d) F. Diederich and B. Felber, Proc. Natl. Acad. Sci. USA, 2002, 99, 4778–4781 CrossRef CAS; (e) P. J. Gittins and L. J. Twyman, Supramol. Chem., 2003, 15, 5–23 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- (a) P. D. Beer, P. A. Gale and D. K. Smith, Supramolecular Chemistry, Oxford University Press, Oxford, UK, 1999 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley and Sons, Chichester, UK, 2000 Search PubMed.
- D. K. Smith, A. R. Hirst, C. S. Love, J. G. Hardy, S. V. Brignell and B. Huang, Prog. Polym. Sci., 2005, 30, 220–293 CrossRef CAS.
- (a) S. C. Zimmerman, F. Zeng, D. E. C. Reichert and S. V. Kolotuchin, Science, 1996, 271, 1095–1098 CrossRef CAS; (b) F. Zeng, S. C. Zimmerman, S. V. Kolotuchin, D. E. C. Reichert and Y. G. Ma, Tetrahedron, 2002, 58, 825–843 CrossRef CAS.
- (a) W. T. S. Huck, R. Hulst, P. Timmerman, F. C. J. M. van Veggel and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1997, 36, 1006–1008 CrossRef CAS; (b) A. W. Freeman, R. Vreekamp and J. M. J. Fréchet, Abstr. Am. Chem. Soc. (PMSE), 1997, 214, 128 Search PubMed.
- Y. Wang, F. W. Zeng and S. C. Zimmerman, Tetrahedron Lett., 1997, 38, 5459–5462 CrossRef CAS.
- (a) C. J. Hawker, K. L. Wooley and J. M. J. Fréchet, J. Am. Chem. Soc., 1993, 115, 4375–4376 CrossRef CAS; (b) D. L. Stone, D. K. Smith and P. T. McGrail, J. Am. Chem. Soc., 2002, 124, 856–864 CrossRef CAS.
- D. K. Smith, Chem. Commun., 1999, 1685–1686 RSC.
- G. M. Dykes, L. J. Brierley, D. K. Smith, P. T. McGrail and G. J. Seeley, Chem. Eur. J., 2001, 7, 4730–4739 CrossRef CAS.
- G. M. Dykes, D. K. Smith and A. Caragheorgheopol, Org. Biomol. Chem., 2004, 2, 922–926 RSC.
- (a) R. G. Denkewalter, J. Kolc and W. J. Lukasavage (Allied Corp), US 4289872, 1981 [Chem. Abstr., 1985, 102, 79324q] Search PubMed; (b) M. Driffield, D. M. Goodall and D. K. Smith, Org. Biomol. Chem., 2003, 1, 2612–2620 RSC.
- G. M. Dykes, D. K. Smith and G. J. Seeley, Angew. Chem., Int. Ed., 2002, 41, 3254–3257 CrossRef CAS.
- G. M. Dykes and D. K. Smith, Tetrahedron, 2003, 59, 3999–4009 CrossRef CAS.
- (a) N. Yamaguchi, L. M. Hamilton and H. W. Gibson, Angew. Chem., Int. Ed., 1998, 37, 3275–3279 CrossRef CAS; (b) H. W. Gibson, N. Yamaguchi, L. Hamilton and J. W. Jones, J. Am. Chem. Soc., 2002, 124, 4653–4665 CrossRef CAS.
- P. A. Chase, R. J. M. K. Gebbink and G. van Koten, J. Organomet. Chem., 2004, 689, 4016–4054 CrossRef CAS.
- M. Kawa and J. M. J. Fréchet, Chem. Mater., 1998, 10, 286–296 CrossRef CAS.
- D. L. Stone, G. M. Dykes and D. K. Smith, Dalton Trans., 2003, 3902–3906 RSC.
- For a review see: Z. Y. Zhong, K. B. Male and J. H. T. Luong, Anal. Lett., 2003, 36, 3097–3118 Search PubMed.
- (a) M. K. Kim, Y. M. Jeon, W. S. Jeon, H. J. Kim, S. G. Hong, C. G. Park and K. Kim, Chem. Commun., 2001, 667–668 RSC; (b) S. Nakao, K. Torigoe, K. Non-No and T. Yonezawa, J. Phys. Chem. B, 2002, 106, 12097–12100 CrossRef CAS.
- C. S. Love, V. Chechik, D. K. Smith and C. Brennan, J. Mater. Chem., 2004, 14, 919–923 RSC.
- K. S. Partridge, D. K. Smith, G. M. Dykes and P. T. McGrail, Chem. Commun., 2001, 319–320 RSC.
- For reviews see: (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef; (b) O. Gronwald, E. Snip and S. Shinkai, Curr. Opin. Colloid Interface Sci., 2002, 7, 148–156 Search PubMed.
- For a review see: A. R. Hirst and D. K. Smith, Chem. Eur. J., 2005 Search PubMed DOI: 10.1002/chem.200500241.
- For a review see: A. R. Hirst and D. K. Smith, Top. Curr. Chem., 2005, 256, 237–273 Search PubMed.
- B. Huang, A. R. Hirst, D. K. Smith, V. Castelletto and I. W. Hamley, J. Am. Chem. Soc., 2005, 127, 7130–7139 CrossRef CAS.
- A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Langmuir, 2004, 20, 7070–7077 CrossRef CAS.
- A. R. Hirst and D. K. Smith, Org. Biomol. Chem., 2004, 2, 2965–2971 RSC.
- A. R. Hirst and D. K. Smith, Langmuir, 2004, 20, 10851–10857 CrossRef CAS.
- A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Chem. Eur. J., 2004, 10, 5901–5910 CrossRef CAS.
- (a) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef CAS; (b) A. R. Hirst, D. K. Smith and J. P. Harrington, Chem. Eur. J., 2005 Search PubMed DOI: 10.1002/chem.200500501.
- D. J. Abdallah, S. A. Sirchio and R. G. Weiss, Langmuir, 2000, 16, 7558–7561 CrossRef CAS.
- R. H. Muller and C. M. Keck, J. Biotechnol., 2004, 113, 151–170 CrossRef CAS.
- J. G. Hardy, A. R. Hirst, D. K. Smith, I. Ashworth and C. Brennan, Chem. Commun., 2005, 385–387 RSC.
- D. K. Smith, Spec. Chem., 2004, 24(Dec/Jan), 42–44 Search PubMed.
- C. Sanchez, G. J. D. A. Soler-Illia, F. Ribot and D. Grosso, Compt. Rend. Chim., 2003, 6, 1131–1151 Search PubMed.
- C. S. Love, A. R. Hirst, V. Chechik, D. K. Smith, I. Ashworth and C. Brennan, Langmuir, 2004, 20, 6580–6585 CrossRef CAS.
- C. S. Love, V. Chechik, D. K. Smith, K. Wilson, C. Brennan and I. Ashworth, Chem. Commun., 2005, 1971–1973 RSC.
- For reviews of multivalency see: (a) S.-K. Choi, Synthetic Multivalent Molecules: Concepts and Biomedical Applications, Wiley Interscience, New York, 2004 Search PubMed; (b) A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 RSC; (c) For a thermodynamic model of multivalency see: P. I. Kitov and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 16271–16284 Search PubMed.
- For reviews see: (a) R. J. Pieters, Trends Glycosci. Glycotechnol., 2004, 16, 243–254 Search PubMed; (b) T. K. Lindhorst, Top. Curr. Chem., 2002, 218, 201–235 CAS.
- D. Zanini and R. Roy, J. Am. Chem. Soc., 1997, 119, 2088–2095 CrossRef CAS.
- (a) I. M. Verma and N. Somia, Nature, 1997, 389, 239–242 CrossRef CAS; (b) D. Luo and M. Saltzmann, Nat. Biotechnol., 2000, 18, 33–37 CrossRef CAS; (c) T. Merdan, J. Kopecek and T. Kissel, Adv. Drug Deliv. Rev., 2002, 54, 715–758 CrossRef CAS.
- (a) J. Haensler and F. C. Szoka, Bioconjugate Chem., 1993, 4, 372–379 CrossRef CAS; (b) J. F. Kukowska-Latallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. USA, 1996, 93, 4897–4902 CrossRef CAS. For a review see: (c) J. Dennig, Top. Curr. Chem., 2003, 228, 227–236 CAS.
- D. Joester, M. Losson, R. Pugin, H. Heinzelmann, E. Walter, H. P. Merkle and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1486–1490 CrossRef CAS.
- B. Martin, M. Sainlos, A. Aissaoui, N. Oudrhiri, M. Hauchecorne, J. P. Vigneron, J. M. Lehn and P. Lehn, Curr. Pharm. Des., 2005, 11, 375–394 Search PubMed.
- (a) C. W. Tabor and H. Tabor, Annu. Rev. Biochem., 1984, 740–790; (b) V. Vijayanathan, T. Thomas, A. Shirahata and T. J. Thomas, Biochemistry, 2001, 40, 13644–13651 CrossRef CAS.
- (a) N. Korolev, A. P. Lyubartsev, L. Nordenskiöld and A. Laaksonen, J. Mol. Biol., 2001, 308, 907–917 CrossRef CAS; (b) Y. Burak, G. Ariel and D. Andelman, Curr. Opin. Colloid Interface Sci., 2004, 9, 53–58 Search PubMed.
- M. A. Kostiainen, J. G. Hardy and D. K. Smith, Angew. Chem., Int. Ed., 2005, 44, 2556–2559 CrossRef CAS.
- J. G. Hardy, M. A. Kostiainen, D. K. Smith, N. P. Gabrielson and D. W. Pack, unpublished results.
- H. G. Abdelhady, S. Allen, M. C. Davies, C. J. Roberts, S. J. B. Tendler and P. M. Williams, Nucleic Acids Res., 2003, 31, 4001–4005 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |