Advances in glycoprotein synthesis
Lei
Liu
,
Clay S.
Bennett
and
Chi-Huey
Wong
*
Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: wong@scripps.edu; Fax: +1-858-784-2409; Tel: +1-858-784-2487
First published on 23rd November 2005
Abstract
The development of chemical and enzymatic methods for the synthesis of homogeneous glycoproteins is a fascinating challenge at the interface between chemistry and biology. Discussed here are the currently available methods for preparation of homogeneous glycoproteins. These methods include (1) glycopeptide ligation; (2) glycoprotein remodeling; and (3) in vivo suppressor tRNA technology.
 Lei Liu Lei Liu | Dr Lei Liu received his BS from the University of Science and Technology of China in 1999, and his PhD in chemistry from Columbia University with Prof. Ronald Breslow in 2004. He is now working as a postdoctoral fellow with Prof. Chi-Huey Wong. His current research is concerned with the synthesis and property studies of homogeneous glycoproteins. |
 Clay S. Bennett Clay S. Bennett | Dr Clay S. Bennett received his BA from Connecticut College in 1999, and his PhD in chemistry from the University of Pennsylvania with Prof. Amos B. Smith, III in 2005. He is now working as a postdoctoral fellow with Prof. Chi-Huey Wong. His current research is concerned with the development of new methods for glycoprotein synthesis and the study of the effect of glycosylation on protein function. |
 Chi-Huey Wong Chi-Huey Wong | Prof. Chi-Huey Wong received his BS and MS from the National Taiwan University, and his PhD in chemistry from the Massachusetts Institute of Technology. After a year as a postdoctoral fellow at Harvard University, he moved to Texas A&M University in 1983. He became Ernest W. Hahn Professor of Chemistry in 1989 at The Scripps Research Institute. His research interests encompass organic and chemoenzymatic methods in synthesis with a specific focus on the study of carbohydrate-mediated biological recognition. |
1 Introduction
The function of a gene is exhibited at the protein level through transcription, translation and, in many instances, posttranslational modifications. Among the posttranslational events that are known so far, the most complex is protein glycosylation that usually occurs at Asn, Ser, and Thr residues (Fig. 1).1 Due to the inherent complexity of oligosaccharides, glycosylation can introduce enormous structural diversity to proteins and consequently, greatly expand the information of an otherwise concise genome. It has been speculated that glycosylation may represent a level of variability that is necessary for complex processes in higher organisms. This is evidenced by the fact that more than 50% of human proteins are glycosylated, while bacteria such as E. coli do not have this modification machinery. | ||
| Fig. 1 Common O-linked serine and threonine glycosides and N-linked asparagine glycosides in protein glycosylation. | ||
Besides encoding molecular recognition information, protein glycosylation can also affect protein folding and improve the biological half-life of proteins by increasing their water solubility and inhibiting their proteolysis and thermal denaturation. Many important biological events such as cell adhesion and cell differentiation are also mediated by glycoproteins. Protein glycosylation has been implicated in diverse physiological processes ranging from receptor-mediated endocytosis to bacterial and viral infection. Furthermore, aberrant protein glycosylation has been found in various diseases such as autoimmune diseases and cancer. As a consequence, glycoproteins have potentially important clinical roles in the contexts of vaccines, diagnostics, and therapeutics.
Despite the tremendous importance of glycoproteins, progress towards understanding their structures and functions has been very slow due to the difficulty of obtaining homogeneous glycoproteins. Glycoproteins are naturally expressed as mixtures of glycoforms that possess the same peptide backbone but differ in both the nature and site of glycosylation. The isolation of homogeneous glycoproteins in significant quantity from natural sources is virtually impossible with current techniques. At present chemical and enzymatic synthesis is the only way to solve the availability problem in the study of glycoproteins. It can provide well-defined materials that were previously not attainable for the investigation of the structures and biological properties of glycoproteins. It can also allow the introduction of unnatural amino acids or carbohydrate moieties for additional glycoprotein studies and therapeutic development. Herein, we discuss the currently available tools and approaches for preparation of homogeneous glycoproteins, with a focus on (1) glycopeptide ligation; (2) glycoprotein remodeling; and (3) in vivo suppressor tRNA technology (See Fig. 2). An additional approach that is not discussed here is pathway re-engineering in yeast systems, which can produce certain human-type N-linked glycoforms.2
 | ||
| Fig. 2 Methods developed for the synthesis of homogeneous glycoproteins: (1) glycopeptide ligation; (2) glycoprotein remodeling; and (3) in vivo suppressor tRNA technology. | ||
2 Glycopeptide ligation
The general idea of the glycopeptide ligation method is shown in Fig. 2. A relatively short glycopeptide is synthesized at first, which is then coupled with longer polypeptides via a chemoselective ligation reaction to generate the full-length homogeneous glycoprotein. An important advantage of the glycopeptide ligation method is that it allows the incorporation of both natural and unnatural glycans and amino acids at single or multiple positions in the glycoprotein products. The current challenges of this method are: (1) how to attain short glycopeptides with complex oligosaccharide side chains? and (2) how to accomplish mild, chemoselective ligation between peptides and glycopeptides?2.1 Glycopeptide synthesis
In principle there are three strategies for the synthesis of a glycopeptide with an oligosaccharide side chain (See Fig. 3). Most commonly, pre-formed glycosyl amino acid building blocks are employed in the stepwise assembly of the peptide preferentially via solid-phase peptide synthesis (SPPS). It is important to note that O-glycosidic and intersaccharidic linkages are relatively labile towards acid.3 Under strongly basic conditions, β-elimination of the carbohydrate may also occur at the O-linked serine and threonine glycosides.4 Thus, the proper selection of protecting groups, which is necessary to block the functional groups on both the glycan and the peptide part, is a matter of importance for the solid-phase as well as solution-phase synthesis of glycopeptides. | ||
| Fig. 3 Three general strategies for the synthesis of relatively short glycopeptides. | ||
It has been found that the glycosidic linkages of fully acylated carbohydrates can withstand short treatment with TFA used for removal of side-chain protecting groups of amino acids. Furthermore, β-elimination of the O-linked serine and threonine glycosides is usually not promoted by relatively weak bases such as morpholine and piperidine. Thus, it is feasible to apply the well-established Fmoc SPPS chemistry to the synthesis of glycopeptides. A typical example of the current art of solid-phase synthesis of glycopeptides is shown in Scheme 1.5 In this example a fully acetylated glycosyl amino acid (Fmoc-Thr-(O-β-GlcNAc(OAc)3)-OH) was used in the peptide assembly. Removal of the acetyl groups was accomplished in the last step by short treatment with NaOMe. It is worth mentioning that the glycopeptide product shown in Scheme 1 is a model of the repeating C-terminal domain of RNA polymerase II. Our spectroscopic study of this glycopeptide suggested that glycosylation of the native, randomly coiled peptide with a single, biologically relevant sugar can lead to the formation of a turn.
 | ||
| Scheme 1 A typical example of the solid-phase synthesis of glycopeptide. | ||
Glycosyl amino acids with either monosaccharide or oligosaccharide side chains can be utilized in the solid-phase or solution-phase synthesis of glycopeptides. However, in many cases it can be a concern that coupling reactions involving rather bulky glycosyl amino acids may be low-yielding. Furthermore, there is an availability problem for the glycosyl amino acid building blocks that carry complex oligosaccharide side chains. In order to avoid these problems, it is sometimes more strategic to use a glycosyl amino acid with a relatively simple saccharide side chain in the peptide assembly and then elaborate the glycopeptide chemically or chemoenzymatically to expand the glycan. As a demonstration of this strategy, we previously accomplished the synthesis of an N-terminal fragment of PSGL-1 (P-selectin glycoprotein ligand 1), which is an important cell surface glycoprotein counter-receptor (See Scheme 2).6 A threonine residue with a disaccharide side chain was incorporated into a glycopeptide via the solid-phase peptide synthesis, which then underwent a series of chemoenzymatic manipulations to provide the final product as a sulfated glycopeptide carrying a pentasaccharide side chain.
 | ||
| Scheme 2 Chemoenzymatic syntheses of a sulfated PSGL-1 glycopeptide. The synthetic glycopeptide was sequentially treated with the appropriate sugar donors (UDP-Gal, CMP-Sia, and GDP-Fuc) and glycosyltransferases (GalT: galactosyl transferase; SiaT: sialyltransferase; and FucT: fucosyltransferase). | ||
An additional method for the synthesis of glycopeptides is the direct peptide glycosylation, which does not require the use of any glycosyl amino acids. This approach attempts to reach the maximum convergence by building a complex glycodomain first and then incorporating it directly into a peptide setting. For example, in 1990 Lansbury and coworkers reported an HBTU-mediated coupling of GlcNAc glycosamine with the side chain aspartate carboxylate in a pentapeptide which allowed the formation of an Asn-N-linked GlcNAc-containing glycopeptide.7 Using an improved version of this approach Danishefsky and coworkers very recently accomplished the first chemical synthesis of gp120 glycopeptide fragments.8 As shown in Scheme 3, the complex high-mannose-type glycans were conjugated with the gp120 peptide segments through direct aspartylation. This impressive work demonstrates that the direct peptide glycosylation method promises high convergence in the synthesis of glycopeptides. Nonetheless, it should be noted that although N-linked glycopeptides can be successfully synthesized through the direct peptide glycosylation approach, the convergent O-glycosylation of oligopeptides of any significant length to create O-linked (Ser, Thr, or Tyr) glycopeptides has never been achieved.
 | ||
| Scheme 3 Convergent synthesis of an N-linked glycopeptide (a gp120 glycopeptide fragment) via direct peptide glycosylation. | ||
2.2 Native chemical ligation
The methods described above permit access to structurally diverse but relatively short glycopeptides consisting of about 10–50 residues. During the synthesis of longer glycopeptides by the linear solid-phase assembly method, uncoupled sequences, side products, and epimers accumulate resulting in dramatically decreased yields and purities of the final products. To surpass the size limits inherent in linear SPPS, the convergent condensation of unprotected or partially protected glycopeptide segments has emerged as an approach to total synthesis of glycoproteins. Using the peptide ligation methods, the intermediates can be purified, and the isolation of the condensation product, which differs markedly in molecular weight from the individual fragments, is carried out easily.In order to accomplish the convergent coupling of glycopeptides, the C terminus of the acceptor fragment must be selectively activated in the presence of a variety of proteinogenic functional groups, while at the same time a donor fragment with compatible reactivity must be available. The thioester has proven to be one of the acyl acceptors that satisfy all these requirements. When an unprotected peptide thioester is mixed with a second unprotected peptide containing an N-terminal cysteine residue as the donor fragment, they undergo rapid transthioesterification (See Fig. 4). The resulting thioester intermediate is then subject to a spontaneous and irreversible S → N acyl transfer. In this way the two peptide fragments are linked together via a “native” amide bond.9 This process, termed native chemical ligation, has been widely applied to the synthesis of numerous moderately sized proteins. It is important to note that native chemical ligation is effected in aqueous solution with unprotected peptides, and the reaction conditions are entirely compatible with carbohydrates and native proteins.
 | ||
| Fig. 4 The mechanism of native chemical ligation. | ||
Using the native chemical ligation method, Bertozzi and coworkers recently finished the total synthesis of a chemically defined version of diptericin, an 82-residue antimicrobial glycoprotein from insects (Scheme 4).10 Because diptericin entirely lacks cysteine, Gly25 was strategically changed to the cysteine residue required for the native chemical ligation. Thus the whole glycoprotein was split into two fragments, a 58-mer C-terminal glycopeptide and a 24-mer N-terminal glycopeptide thioester. The former was synthesized readily by a standard SPPS protocol. However, the N-terminal glycopeptide thioester was difficult to prepare by the standard Fmoc strategy because thioesters would not withstand the conditions required for the deprotection of the Fmoc groups. To overcome this obstacle, Fmoc-based SPPS was performed on a sulfonamide safety-catch resin developed by Ellman and coworkers that allowed for a post-assembly formation of the glycopeptide thioester under mild conditions via nucleophilic thiolysis. The ligation of the 24-mer thioester and the 58-mer cysteinyl glycopeptide was performed in the presence of thiophenol which is known to accelerate native chemical ligation.9 Finally, O-deacetylation under mild conditions afforded the fully deprotected 82-mer glycoprotein.
 | ||
| Scheme 4 Native chemical ligation of a glycopeptide thioester to an N-terminal cysteinyl glycopeptide provides an 82-mer glycoprotein, diptericin. | ||
Recognizing the difficulty of synthesizing glycopeptide thioesters, Danishefsky and coworkers recently developed a novel solution to native chemical ligation. They discovered that a peptide phenolic ester equipped with an ortho disulfide moiety can be ligated to a peptide containing an N-terminal cysteine residue after the disulfide bond is reductively cleaved (See Fig. 5).11 The mechanism of this interesting ligation reaction probably involves a dynamic O → S acyl transfer in the reduced phenolic ester intermediate equipped with an ortho mercaptal group. This means that an ester equipped with an adjacent mercaptal group is possibly present in an equilibrium with a thioester, which can be interdicted by the machinery appropriate to native chemical ligation. It is worth noting that unlike a glycopeptide thioester, a glycopeptide ester can tolerate the Fmoc SPPS chemistry and therefore, can be more readily synthesized. Thus the new ligation method is not only mechanistically interesting, but also advantageous for practice. Using this new method, Danishefsky and coworkers have successfully synthesized several model glycopeptides containing two glycosylation sites (See Fig. 5).
 | ||
| Fig. 5 The ligation between a glycopeptide phenolic ester equipped with an ortho disulfide moiety and a glycopeptide containing an N-terminal cysteine group. | ||
A limitation of native chemical ligation is its intrinsic reliance on having a cysteine residue at the ligation juncture. Cysteine is uncommon, comprising only 1.7% of all residues in proteins. It is often difficult to find a cysteine residue located within a reasonable distance from the glycosylation site in a glycoprotein. Hence, a number of glycoproteins cannot be readily prepared by any ligation method that allows for glycopeptides to be coupled only at cysteine residues. To solve this problem an emerging strategy in protein synthesis has been the use of removable auxiliaries that act as cysteine surrogates to mediate the chemical ligation of peptide fragments.12 After the amide bond between the two peptide fragments is formed, the auxiliary group can be removed chemically under acidic conditions or via photochemical irradiation (See Fig. 6). Up to now very few of these removable auxiliary methods have been tested for the ligation of glycopeptides. Macmillan and coworkers recently reported synthetic routes to two classes of TFA-cleavable auxiliaries for cysteine-free glycopeptide ligation. It was demonstrated that the auxiliary introduction and cleavage were compatible with the presence of glycosidic linkages and to function in cysteine-free ligations across Gly-Gly junctions. Unfortunately, the auxiliaries failed to deliver ligation products at more general junctions such as Leu-Gly and Ser-Gly due to the steric problems, which seriously limited their use in glycoprotein synthesis.13
 | ||
| Fig. 6 Removable auxiliary mediated native chemical ligation. Note that a secondary amine participates in the acyl transfer, which is strongly affected by the steric hindrance. | ||
2.3 Expressed protein ligation
In many instances, one or more of the peptide segments used to assemble a protein is produced recombinantly and the assembly process is referred to as expressed protein ligation.14 The expressed protein ligation method has the decided advantage that much larger proteins can be tackled than are accessible through SPPS alone. Furthermore, facile production of recombinant fragments also renders expressed protein ligation a cost efficient method.In the context of native chemical ligation, either the peptide thioester fragment or the peptide fragment containing an N-terminal cysteine can be produced recombinantly. This premise was put to the test first by Verdine and coworkers, who developed a mutagenesis/proteolysis strategy to introduce an N-terminal cysteine into a recombinant version of the transcription factor protein, AP-1.15 Their work confirmed that native chemical ligation could be used in a semisynthetic fashion, and provided the strategy by which to attach a synthetic peptide fragment to the N terminus of a recombinant polypeptide. This strategy is obviously applicable to the preparation of homogeneous glycoproteins in which the glycosylation site is close to the N terminus.
Currently the most common protease to be used in the mutagenesis/proteolysis strategy to generate N-terminal cysteines is Factor Xa. However, this protease is not completely sequence-specific, sometimes cleaving peptide bonds after positively charged residues in the absence of its primary recognition sequence. For many protein applications a more selective protease than Factor Xa with a lower probability of cleaving internally during affinity tag removal would be advantageous. In this context we recently investigated the use of tobacco etch virus protease (TEV protease) to remove N-terminal affinity tags from fusion proteins to produce proteins with N-terminal cysteines.16 This protease is a highly selective cysteine protease with a seven amino acid recognition site, recognizing six amino acids N-terminal to the scissile bond (See Fig. 7). A major advantage of using TEV protease to generate N-terminal cysteines is the high selectivity of the protease which makes it generally applicable to the cleavage of a wide range of fusion proteins. Furthermore, TEV protease can be expressed in active form in E. coli and yeast without affecting cell viability, making production of TEV protease for large-scale applications inexpensive.
 | ||
| Fig. 7 Two general approaches to glycoprotein synthesis via expressed protein ligation. (1) N-terminal engineering via mutagenesis/proteolysis; (2) C-terminal engineering via intein technique. | ||
Using the TEV protease method, we recently accomplished the semisynthesis of a chemically defined version of a glycoprotein, human interleukin-2, which is a T-cell growth factor that is used therapeutically to treat renal cell carcinoma and metastatic melanoma (See Scheme 5).17 Bacterial expression was utilized to produce a fusion protein His-tagged interleukin-2 (amino acids 6–133), which was readily purified and then cleaved by the TEV protease to generate a truncated version of interleukin-2 containing an N-terminal cysteine residue. The N-terminal cysteine containing truncated interleukin-2 was then successfully joined to a synthetic glycopeptide thioester utilizing native chemical ligation under denaturing conditions to afford the desired glycoprotein in high yields.
 | ||
| Scheme 5 Expressed protein ligation of a glycopeptide thioester to a recombinant N-terminal cysteinyl polypeptide provides a chemically defined version of human interleukin-2. | ||
The TEV protease method provides a strategy by which to synthesize glycoproteins whose glycosylation sites are close to the N terminus (See Fig. 7). This leaves the question of how to prepare glycoproteins whose glycosylation sites are close to the C terminus. To accomplish this goal a polypeptide thioester needs to be prepared recombinantly and then ligated with an N-terminal cysteinyl glycopeptide. A currently available solution to this problem is the intein technique developed by Evans et al. and Muir and colleagues.18 Briefly speaking, an intein is a natural splicing element that is analogous to the intron of nucleic acids. An intein mediates its own excision from a peptide sequence through a series of acyl transfer reactions that ultimately results in the splicing of its flanking peptides. An important intermediate of the intein excision process is a thioester between the N-terminal flanking peptide and the cysteinyl SH group of the intein moiety. Thus, by fusion of a target polypeptide to a modified intein, it is possible to install a thioester at the C terminus of the target polypeptide as the intein excision intermediate (See Fig. 7).
Using the intein technique, we and other groups have successfully incorporated glycosylation onto the C-terminus of biologically expressed polypeptides.19 For example, a 392-amino acid maltose binding protein (MBP) was expressed in E. coli as a fusion to the N terminus of an intein from Saccharomyces cerevisiae, which bears a chitin-binding domain at its C terminus (See Scheme 6). The N-terminal cysteine residue of the intein was then triggered to form a thioester with the C terminus of the MBP, which could be readily purified on chitin beads before being transthioesterified to give a soluble thioester of MBP. Next the soluble MBP thioester underwent native chemical ligation with an N-terminal cysteinyl glycopeptide such as H-Cys-Asn(GlcNAc)-OH to give a homogeneous glycoprotein that carried a monosaccharide. Further enzymatic elaboration of the synthetic monosaccharide-containing glycoprotein to expand its glycan was also demonstrated to be feasible.
 | ||
| Scheme 6 Expressed protein ligation of an MBP thioester to a synthetic N-terminal cysteinyl glycopeptide provides a homogeneous glycoprotein. | ||
2.4 Protease-catalyzed peptide ligation
Proteases have long been known to be useful catalysts for the coupling of peptide fragments under kinetically controlled conditions.20 In this method, one peptide, the acyl donor, is activated as an ester and the condensation reaction is conducted in a mixture of water and an organic cosolvent, such as DMF, DMSO, or MeOH. The reaction is a competition between hydrolysis of the acyl donor with water and aminolysis of the acyl donor with the amine of an acyl acceptor peptide to form the desired product. The amount of hydrolysis can be minimized by decreasing the concentration of water in the reaction by increasing the amount of cosolvent, increasing the aminolysis rate by increasing the concentration of the acyl acceptor peptide, and use of mutated forms of subtilisin that have been optimized for peptide bond formation or stability in organic solvents. Self-condensation reactions of peptides can be prevented by blocking the N-terminus of the acyl donor peptide with an amine-protecting group and blocking the C-terminus of the acyl acceptor peptide as an amide.The protease-catalyzed condensation method is highly stereoselective and racemization-free. It does not require protection of amino acid side chain functions due to the high regioselectivity of most proteases under common reaction conditions. Furthermore, it is important to note that the protease-catalyzed ligation does not suffer from the cysteine limitation problem. Many types of amino acid–amino acid junctions can be successfully coupled together by this approach. Previous studies have shown that proteins such as native and mutated RNase A can be synthesized from smaller peptide fragments via protease-catalyzed peptide ligations.21 Our own interest is the application of proteases to the synthesis of glycoproteins. To accomplish this goal we have focused on suppressing the hydrolytic activity of proteases under kinetically controlled conditions. In addition, we have systematically studied how the position and type of the glycosidic linkage affect the protease-catalyzed peptide bond formation.
The primary proteases used in our studies are the serine protease subtilisin BPN′, its stable variant 8397, and a thiosubtilisin derived from 8397. The 8397 variant was designed for synthesis in anhydrous or high concentrations of DMF (half life = 14 days at 25 °C in anhydrous DMF compared to 30 min for the wild-type enzyme).22 The thiosubtilisin variant was developed for peptide synthesis in aqueous solution.23 As shown in Fig. 8, the acyl thiosubtilisin favors aminolysis over hydrolysis in aqueous solution by a factor of ∼2 kcal/mol compared to acyl subtilisin BPN′. This enhancement of aminolysis has been attributed to both enzymatic and chemical reasons as the acyl thiosubtilisin has a higher affinity for and is also more reactive toward the amine nucleophile versus water.
 | ||
| Fig. 8 Top: free energy diagrams for subtilisin BPN′ catalyzed reactions. Dotted line indicates the difference in free energies observed with thiosubtilisin BPN′. Bottom: mechanisms of hydrolysis and aminolysis for subtilisin and thiosubtilisin. | ||
The scope of subtilisin-catalyzed glycopeptide coupling reactions has been extensively studied, and examples of subtilisin condensation of glycopeptides containing O-linked and N-linked glycans, as well as acylated and unprotected sugars have been reported.24 The active site of subtilisin has been carefully mapped out to determine which residues of substrate peptides can be glycosylated. It was found that many areas of the subtilisin active site, including the S4, S3, S2′, S3′, and S4′ sites, would accept glycosylated amino acid residues, whereas areas proximal to the cleavage/ligation junction, the S1, S2, and S1′ sites, would not accept glycosylation.
A typical example of subtilisin catalyzed glycopeptide ligation is shown in Scheme 7, which describes the synthesis of a partial sequence of the C-terminal region of ribonuclease B.25 The Rink amide resin loaded with the Fmoc-Ala-Pam linker was used as the initial amino acid and solid support. Standard Fmoc solid-phase peptide synthesis was employed to produce the full length, fully protected, resin-bound peptide. Standard TFA deprotection of the peptide side chains and concomitant release from the resin gave the N-terminal protected, activated peptide ester in excellent yield. Subtilisin-catalyzed peptide ligation was then performed to couple the activated peptide ester with a glycopeptide acyl acceptor. In a 7 ∶ 3 ratio of DMF/buffer, although the aminolysis product was formed, a significant portion of hydrolysis product was also detected (aminolysis-to-hydrolysis ratio = 3.95 ∶ 1). To decrease the rate of the hydrolysis reaction, the portion of DMF was increased and conversely the available water was decreased. It was found that a 9 ∶ 1 ratio of DMF/buffer gave a significant increase in aminolysis-to-hydrolysis ratio (5.25 ∶ 1). The yield of the coupled glycopeptide product under these conditions was 84%.
 | ||
| Scheme 7 Subtilisin catalyzed condensation of glycopeptides under kinetically controlled conditions. | ||
2.5 Traceless Staudinger ligation
In addition to the native chemical ligation and protease catalyzed ligation methods, a new interesting alternate approach to convergently couple peptide is the “traceless” Staudinger ligation.26 In this method an azide and a phosphinothioester react to form an iminophosphorane, which then undergoes an intramolecular S → N acyl transfer to form an amidophosphonium salt intermediate. Hydrolysis of the amidophosphonium salt produces an amide that does not contain any residual atoms from the phosphine prosthetic group (See Fig. 9). Very recently it was shown by Raines and coworkers that this ligation occurs in high yields (80–99% for dipeptide model systems) at room temperature in aqueous or wet organic solvents and is compatible with the unprotected functional groups of proteinogenic amino acids.27 Our own recent studies have demonstrated that the traceless Staudinger ligation is also applicable to the coupling of glycopeptides. Both O-linked and N-linked glycopeptides have been examined, and the use of glycopeptides as both acyl donors and acceptors has been tested. The yields of these coupling reactions are satisfactory, except in cases where there is substantial steric hindrance.28 | ||
| Fig. 9 The mechanism of traceless Staudinger ligation. | ||
Both the acyl donor and acceptor for the Staudinger ligation approach to glycoproteins can be synthesized through SPPS. However, in order to attain a more cost-effective method for relatively large scale synthesis of glycoproteins, we shall take full advantage of recombinant DNA expression technology to obtain either the acyl donor or acceptor biologically. Conceptually, peptides with a C-terminal phosphinothioester can be produced by intein/thiol exchange technology. On the other hand, currently there is no general method to produce recombinantly expressed peptides that contain an N-terminal azido group. Thus it is an interesting challenge to selectively introduce an N-terminal azido group to a polypeptide in which the functional groups of proteinogenic amino acids are unprotected. Our recent studies have demonstrated that the subtilisin-catalyzed peptide ligation method can be used to selectively N-azidonate an unprotected polypeptide. The maximum cost efficiency can be reached by using an azidoacetyl amino acid ester of trifluoroethanol as the acyl donor (See Scheme 8). The combined approach of the subtilisin-catalyzed N-azidonation and the traceless Staudinger ligation could be used to synthesize glycoproteins from a long, expressed polypeptide and a short, synthetic glycopeptide in a cost-efficient way.
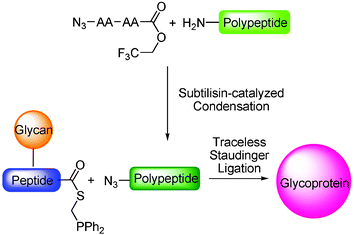 | ||
| Scheme 8 Two-step synthesis of a glycoprotein using subtilisin-catalyzed azidonation and traceless Staudinger ligation. | ||
3 Glycoprotein remodeling
An alternative to the chemical synthesis of N-linked glycans involves the use of enzymes to transform a heterogeneous population of protein glycoforms into a single homogeneous glycoprotein. In the initial step in this process endoglycosidases hydrolyze the bond between the two GlcNAc residues in the bis-N-acetylchitibiose core of N-linked glycans29 to afford a glycoprotein containing a single GlcNAc attached to an Asn residue. As different endoglycosidases recognize different glycan chains, it is possible, through proper choice of the endoglycosidase, to selectively trim high mannose (Endo F1 or Endo H)29 or hybrid and complex-type (Endo F2 and Endo F3) glycans. The resulting GlcNAc residue can then be modified using glycosyltransferases and sugar nucleotides to build up a complex glycan (See Fig. 10). Although the high cost of sugar nucleotides renders them unattractive for large-scale synthesis, the use of multiple enzyme sugar nucleotide regeneration systems allows for the in situ generation of the sugar nucleotide from simple sugars.30 | ||
| Fig. 10 Synthesis of homogeneous unnatural glycoforms of ribonuclease B possessing sialyl Lewis X epitopes that contain derivatized NeuAc residues via glycoprotein remodeling. | ||
This technique has been used by us for the synthesis of homogeneous unnatural glycoforms of ribonuclease B possessing sialyl Lewis X epitopes that contain derivatized NeuAc residues.31 The process began with treatment of commercial Ribonuclease B (present as a mixture of high-mannose glycoforms) to expose the reducing GlcNAc residue. The glycoprotein was then treated with β-1,4-galactosyltransferase in the presence of UDP-Gal (generated in situ from Gal and UDP) to afford a protein containing Galβ1,4GlcNac. Subsequent treatment of the protein with α-2,3-sialyltransferase (SialT) and α-1,3-fucosyltransferase V in the presence of the request nucleotide sugars then afforded the desired sialyl Lewis X containing protein. It is noteworthy that the SialT was tolerant of modification of the NeuAc moiety and could be used to introduce 9-MeHgS-NeuAc to the protein for crystallographic purposes. The scope of the use of glycosyltransferases in organic synthesis has been reviewed elsewhere.32
Although the use of glycosyltransferases allows for the rapid production of highly complex glycans, it suffers from a limitation in that if the reactions do not proceed to completion it is necessary to carry out highly tedious separations to isolate the desired product. An alternative approach relies on the use of the endoglycosidases Endo A and Endo M to transfer a fully elaborated glycan chain to the GlcNAc-Asn residue through a transglycosylation reaction.33,34 Under normal circumstances endoglycosidases hydrolyze the complex sugars. However, it was found that the hydrolysis pathway could be suppressed by using organic solvents and a large excess of the glycosyl acceptor.35 Through this method, Takegawa and coworkers were able to transfer (Man)6GlcNAc to a partially deglycosylated RNase B, using (Man)6GlcNAc2Asn as a donor source. More recently, it has been demonstrated that sugar oxazolines can serve as substrates for Endo A and Endo M, thereby eliminating the need to incorporate a sacrificial sugar residue into the donor.36 While this method has been shown to give excellent results with simple peptides, extension to glycoprotein synthesis has yet to be demonstrated. At present only Endo A and Endo M have been utilized in the transglycosylation reaction, and the method is currently limited to using high-mannose or hybrid and complex type glycans as donors.
4 In vivo suppressor tRNA technology
Recently, in vivo suppressor tRNA technology has been exploited for the recombinant production of neoglycoproteins and glycoproteins.37 Successful in vivo incorporation of unnatural amino acids in E. coli has been achieved systematically by (1) evolving an orthogonal tRNA synthetase–tRNA pair from Methanococcus jannaschii that is capable of accepting and charging an unnatural amino acid onto Amber-suppressing tRNACUA and (2) introducing permissible Amber stop codons (TAG) into a protein of interest that serve to site-specifically direct the incorporation of the unnatural amino acid.Using the in vivo suppressor tRNA technology, p-acetylphenylalanine was incorporated into proteins and subsequently derivatized with aminooxy saccharides to produce homogeneous neoglycoproteins (See Fig. 11).38 More recently, a naturally occurring homogeneous glycoprotein population was produced in E. coli for the first time via the direct incorporation of the core glycosylamino acids β-GlcNAc-serine and α-GalNAc-threonine. The glycoproteins were easily isolated and the incorporated monosaccharide can serve as a primary glycosylation site to which saccharides can be added sequentially with glycosyltransferases in vitro.39,40 Though the current production level is relatively low, ∼4 mg/L, this new method may eventually lead to the development of fermentation methods for the large-scale production of glycoproteins with well-defined carbohydrates at genetically controlled positions.
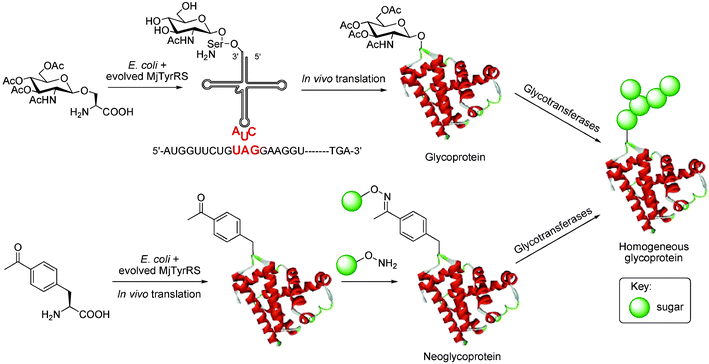 | ||
| Fig. 11 Homogeneous neoglycoproteins and glycoproteins can be produced by in vivo suppressor tRNA technology. An orthogonal tRNA synthetase and tRNA pair can be evolved to accept and to supply the unnatural amino acids N-acetylglucosamine-β-serine (a) and p-acetylphenylalanine (b) in response to the stop codon TAG during protein biosynthesis in vivo. The ketone handle can be derivatized with aminooxy saccharides, glycosyltransferases are then used to form extended glycans. Abbreviation: MjTyrRS, Methanococcus jannaschii tyrosine-tRNA synthetase. | ||
5 Conclusions
The structural and biological consequences of cellular protein modification through posttranslational glycosylation are central issues in the rapidly growing field of glycobiology. The study of natural glycoproteins and the creation of nonnatural ones require the ability to access and manipulate homogeneous glycoproteins. In this context the synthesis of glycoproteins from readily available components is an important goal in glycobiology.The currently available tools in the arsenal of glycoprotein synthesis include (1) glycopeptide ligation; (2) glycoprotein remodeling; and (3) in vivo suppressor tRNA technology. These approaches complement each other in terms of synthetic capability and cost efficiency. Armed with these tools we envisage that the ambitious goal of the total synthesis of a variety of biologically interesting glycoproteins is within reach.
Acknowledgements
We thank NIH and The Skaggs Institute for support of our research. We also thank Dr Lisa J. Whalen for proof reading the manuscript.Notes and references
- Some related reviews: (a) R. A. Dwek, Chem. Rev., 1996, 96, 683 CrossRef CAS; (b) O. Seitz, ChemBioChem, 2000, 1, 214 CrossRef CAS; (c) M. J. Grogan, M. R. Pratt, L. A. Marcaurelle and C. R. Bertozzi, Annu. Rev. Biochem., 2002, 71, 393; (d) B. G. Davis, Chem. Rev., 2002, 102, 579 CrossRef CAS; (e) S. Hanson, M. Best, M. C. Bryan and C.-H. Wong, Trends Biochem. Sci., 2004, 29, 656 CrossRef CAS; (f) M. R. Pratt and C. R. Bertozzi, Chem. Soc. Rev., 2005, 34, 58 RSC; (g) C.-H. Wong, J. Org. Chem., 2005, 70, 4219 CrossRef CAS.
- S. R. Hamilton, P. Bobrowicz, B. Bobrowicz, R. C. Davidson, H. Li, T. Mitchell, J. H. Nett, S. Rausch, T. A. Stadheim, H. Wischnewski, S. Wildt and T. U. Gerngross, Science, 2003, 301, 1244 CrossRef CAS.
- H. Kunz and C. Unverzagt, Angew. Chem., Int. Ed. Engl., 1988, 27, 1697 CrossRef.
- P. Sjolin, M. Elofsson and J. Kihlberg, J. Org. Chem., 1996, 61, 560 CrossRef.
- E. E. Simanek, D.-H. Huang, L. Pasternack, T. D. Machajewski, O. Seitz, D. S. Millar, H. J. Dyson and C.-H. Wong, J. Am. Chem. Soc., 1998, 120, 11567 CrossRef CAS.
- (a) K. M. Koeller, M. E. Smith and C.-H. Wong, Bioorg. Med. Chem., 2000, 8, 1017 CrossRef CAS; (b) K. M. Koeller, M. E. Smith, R.-F. Huang and C.-H. Wong, J. Am. Chem. Soc., 2000, 122, 4241 CrossRef CAS; (c) For a related study, see: A. Leppanen, S. P. White, J. Helin, R. P. McEver and P. D. Cummings, J. Biol. Chem., 2000, 275, 39569 Search PubMed.
- S. T. Ansfield and P. T. Lansbury, J. Org. Chem., 1990, 55, 5560 CrossRef CAS.
- (a) M. Mandal, V. Y. Dudkin, X. Geng and S. J. Danishefsky, Angew. Chem., Int. Ed., 2004, 43, 2557 CrossRef CAS; (b) X. Geng, V. Y. Dudkin, M. Mandal and S. J. Danishefsky, Angew. Chem., Int. Ed., 2004, 43, 2562 CrossRef CAS.
- P. E. Dawson and S. B. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS.
- (a) Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 11684 CrossRef CAS. For other native chemical ligation approaches to glycoproteins, see: (b) L. A. Marcauelle, L. S. Mizoue, J. Wilken, L. Oldham, S. B. Kent, T. M. Handel and C. R. Bertozzi, Chem. Eur. J., 2001, 7, 1129 CrossRef CAS; (c) S. Mezzato, M. Schaffrath and C. Unverzagt, Angew. Chem., Int. Ed., 2005, 41, 1650 CrossRef CAS.
- J. D. Warren, J. S. Miller, S. J. Keding and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6576 CrossRef CAS.
- (a) J. Offer, C. N. C. Boddy and P. E. Dawson, J. Am. Chem. Soc., 2002, 124, 4642 CrossRef CAS; (b) C. Marinzi, J. Offer, R. Longhi and P. E. Dawson, Bioorg. Med. Chem., 2002, 12, 2749 CrossRef.
- D. Macmillan and D. W. Anderson, Org. Lett., 2004, 6, 4659 CrossRef CAS.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249 CrossRef.
- D. A. Erlanson, M. Chytil and G. L. Verdine, Chem. Biol., 1996, 3, 981 CrossRef CAS.
- (a) T. J. Tolbert and C.-H. Wong, Angew. Chem., Int. Ed., 2002, 41, 2171 CrossRef CAS; (b) For a related study see: D. Macmillan and L. Arham, J. Am. Chem. Soc., 2004, 126, 9530 Search PubMed.
- T. J. Tolbert, D. Franke and C.-H. Wong, Bioorg. Med. Chem., 2005, 13, 909 CrossRef CAS.
- (a) T. C. Evans, J. Benner and M. Q. Xu, Protein Sci., 1998, 7, 2256 CrossRef CAS; (b) T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. USA, 1998, 95, 6705 CrossRef CAS.
- (a) T. J. Tolbert and C.-H. Wong, J. Am. Chem. Soc., 2000, 122, 5421 CrossRef CAS. For other expressed protein ligation approaches to glycoproteins, see: (b) D. Macmillan and C. R. Bertozzi, Tetrahedron, 2000, 56, 9515 CrossRef CAS; (c) D. Macmillan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2004, 43, 1355 CrossRef CAS; (d) C. P. R. Hackenberger, C. T. Friel, S. E. Radford and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 12882 CrossRef CAS.
- F. Bordusa, Chem. Rev., 2002, 102, 4817 CrossRef CAS.
- D. Y. Jackson, J. Burnier, C. Quan, M. Stanley, J. Tom and J. A. Wells, Science, 1994, 117, 819.
- (a) Z. Zhong, J. L.-C. Liu, L. M. Dinterman, M. A. J. Finkelman, W. T. Mueller, M. L. Rollence, M. Whitlow and C.-H. Wong, J. Am. Chem. Soc., 1991, 113, 6336 CrossRef; (b) P. Sears, M. Schuster, P. Wang, K. Witte and C.-H. Wong, J. Am. Chem. Soc., 1994, 116, 6521 CrossRef CAS; (c) R. D. Kidd, H. P. Yennawar, P. Sears, C.-H. Wong and G. K. Faber, J. Am. Chem. Soc., 1996, 118, 1645 CrossRef CAS.
- Z. Zhong, J. Bibbs, W. Yuan and C.-H. Wong, J. Am. Chem. Soc., 1991, 113, 2259 CrossRef CAS.
- (a) C.-H. Wong, M. Schuster, P. Wang and P. Sears, J. Am. Chem. Soc., 1993, 115, 5893 CrossRef CAS; (b) K. Witte, O. Seitz and C.-H. Wong, J. Am. Chem. Soc., 1998, 120, 1979 CrossRef CAS.
- T. J. Tolbert and C.-H. Wong, Methods Mol. Biol., 2004, 283, 267 Search PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141 CrossRef CAS.
- (a) B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2001, 3, 9 CrossRef CAS; (b) B. L. Nilsson, R. J. Hondal, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 5268 CrossRef CAS; (c) B. L. Nilsson, M. B. Soellner and R. T. Raines, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 91 CrossRef CAS.
- L. Liu, Z.-Y. Hong and C.-H. Wong, ChemBioChem, 2005 Search PubMed (DOI: 10.1002/cbic.200500437).
- (a) R. B. Trimble and A. L. Tarentino, J. Biol. Chem., 1991, 266, 1646 CAS; (b) P. W. Robbins, R. B. Trimble, D. F. Wirth, C. Hering, F. Maley, G. F. Maley, B. W. Das, N. Royal and K. Biemann, J. Biol. Chem., 1984, 259, 7577 CAS.
- (a) X. Chen, J. Fang, J. Zhang, Z. Liu, J. Shao, P. Kowal, P. Andreana and P. G. Wang, J. Am. Chem. Soc., 2001, 123, 2081 CrossRef CAS; (b) M. Gilbert, R. Bayer, A. Cunningham, S. DeFrees, Y. Gao, D. C. Watson, N. M. Young and W. W. Wakarchuk, Nat. Biotechnol., 1998, 16, 769 CAS; (c) T. Noguchi and T. Shiba, Biosci., Biotechnol., Biochem., 1998, 62, 1594 CrossRef CAS; (d) A. Zervosen and L. Elling, J. Am. Chem. Soc., 1996, 118, 1836 CrossRef CAS; (e) G. C. Look, Y. Ichikawa, G.-J. Shen, G.-J. Cheng and C.-H. Wong, J. Org. Chem., 1993, 58, 4326 CrossRef CAS; (f) P. Wang, G.-J. Shen, Y.-F. Wang, Y. Ichikawa and C.-H. Wong, J. Org. Chem., 1993, 58, 3985 CrossRef CAS; (g) Y. Ichikawa, Y.-C. Lin, D. P. Dumas, G.-J. Shen, E. Garcia-Junceda, M. A. Williams, R. Bayer, C. Ketcham, L. E. Walker, J. C. Paulson and C.-H. Wong, J. Am. Chem. Soc., 1992, 114, 9283 CrossRef CAS; (h) C.-H. Wong, R. Wang and Y. J. Ichikawa, J. Org. Chem., 1992, 57, 4343 CrossRef CAS; (i) D. Gygax, P. Spies, T. Winkler and U. Pfarr, Tetrahedron, 1991, 28, 5119 CrossRef; (j) Y. Ichikawa, G.-J. Shen and C.-H. Wong, J. Am. Chem. Soc., 1991, 113, 4698 CrossRef CAS; (k) C.-H. Wong, S. L. Haynie and G. M. Whitesides, J. Org. Chem., 1982, 47, 5418 CrossRef.
- (a) R. Martin, K. L. Witte and C.-H. Wong, Bioorg. Med. Chem., 1998, 6, 1283 CrossRef CAS; (b) K. Witte, P. Sears, R. Martin and C.-H. Wong, J. Am. Chem. Soc., 1997, 119, 2114 CrossRef CAS.
- (a) C.-H. Wong, R. L. Halcomb, Y. Ichikawa and T. Kajimoto, Angew. Chem., Int. Ed. Engl., 1995, 34, 521 CrossRef CAS; (b) C.-H. Wong, R. L. Halcomb, Y. Ichikawa and T. Kajimoto, Angew. Chem., Int. Ed. Engl., 1995, 34, 412 CrossRef CAS.
- (a) K. Haneda, T. Inazu, K. Yamamoto, Y. Nakahara and A. Kobata, Carbohydr. Res., 1996, 292, 61 CAS; (b) K. Yamamoto, K. Fujimori, K. Haneda, M. Mizuno, T. Inazu and H. Kumagai, Carbohydr. Res., 1998, 305, 415 CrossRef CAS.
- (a) J.-Q. Fan, K. Takegawa, S. Iwahara, A. Kondo, K. Ikunoshin, C. Abeygunawardana and Y. C. Lee, J. Biol. Chem., 1995, 270, 17723 CrossRef CAS; (b) K. Takegawa, M. Tabuchi, S. Yamaguchi, A. Kondo, I. Kato and S. Iwahara, J. Biol. Chem., 1995, 270, 3094 CrossRef CAS.
- E. Akaie, M. Tsutsumida, K. Osumi, M. Fujita, T. Yamanoi, K. Yamamoto and K. Fujita, Carbohydr. Res., 2004, 339, 719 CrossRef.
- (a) B. Li, Y. Zeng, S. Hauser, H. Song and L.-X. Wang, J. Am. Chem. Soc., 2005, 127, 9692 CrossRef CAS; (b) M. Fujita, S. Shoda, K. Haneda, T. Inazu, K. Takegawa and K. Yamamoto, Biochem. Biophys. Acta, 2001, 270, 17723; (c) K. Yamamoto, S. Kadowaki, M. Fujisaki, H. Kumagai and T. Tochikura, Biosci., Biotechnol., Biochem., 1994, 58, 72 CrossRef CAS.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34 CrossRef CAS.
- H. Liu, L. Wang, A. Brock, C.-H. Wong and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 1702 CrossRef CAS.
- Z. Zhang, J. Gildersleeve, Y.-Y. Yang, R. Xu, J. A. Loo, S. Urya, C.-H. Wong and P. G. Schultz, Science, 2004, 303, 371 CrossRef CAS.
- R. Xu, S. R. Hanson, Z. Zhang, Y.-Y. Yang, P. G. Schultz and C.-H. Wong, J. Am. Chem. Soc., 2004, 126, 15654 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |