Synthesis of a pH dependent covalent imprinted polymer able to recognize organotin species†
Mercedes
Gallego-Gallegos
a,
Riansares
Muñoz-Olivas
*a,
C.
Cámara
a,
María J.
Mancheño
*b and
Miguel A.
Sierra
b
aDepartamento de Química Analítica, Facultad de Química, Universidad Complutense, 28040 Madrid, Spain
bDepartamento de Química Orgánica, Facultad de Química, Universidad Complutense, 28040 Madrid, Spain
First published on 22nd November 2005
Abstract
The covalent imprinting approach has for the first time been successfully applied for the synthesis of an imprinted polymer able to recognize organotin species. The synthesis has been accomplished by co-polymerization of the complex Bu2SnO-m-vinylbenzoin as the imprinting template plus co-monomer sodium methacrylate, and ethylene glycol dimethacrylate as cross-linker. The imprinting effect has been evidenced within the narrow pH range 2.5 < pH < 3.5. At lower pH values, the imprinting effect is prevented by the exclusive existence of non-specific interactions, whereas pH > 3.5 provokes a strong rebind of the template in both imprinted and non-imprinted polymers. This pH dependency can be explained as a selective chemical modification which reduces bind diversity following a model based on enolization by protonation of the specific cavities. Characterization of the adsorption isotherms showed good agreement with the Langmuir–Freundlich (LF) model, presenting quite homogeneous binding sites for a bulk material and high capacity in the imprinting pH range. In addition, the affinity spectrum (AS) method has been represented showing the typical profiles of LF isotherm for both sub-saturation and saturation levels, being in general agreement with the encountered values for fitting coefficients. The covalent molecular imprinted polymer has been succesfully evaluated in a SPE process for further OTC determination in the certified mussel tissue (CRM 477)
Introduction
Molecularly imprinted polymers (MIP) are increasingly used for analytical applications, such as liquid chromatography, solid phase extraction, chemical sensors, catalysis, etc. MIP's are formed in the presence of a template molecule that can be linked covalently or not to functional monomers in a pre-polymerization complex.1 The template-monomer system is chosen such that in solution the imprint molecule complexes one or several functional monomers, which then become spatially fixed during the polymerization. Once the polymer is formed, the template is removed, leaving binding cavities, which are complementary in shape and functionality with the template molecule and therefore, able to selectively rebind this molecule. A benefit of imprinted polymers is the possibility to prepare adsorbents with selectivity pre-determined for a particular substance or a group of structural analogues.2One of the most practical applications of molecular imprinting in analytical separation science is solid phase extraction (SPE), especially for the application to biological and environmental samples. This technique has been denominated molecularly imprinted solid phase extraction (MISPE).2,3 In this context, MIPs should be suitable for the selective determination of organotin compounds in environmental samples. Organotin compounds (OTC) have been extensively used for many years as stabilizers in PVC, in anti-fouling paints and coatings for the protection of ship hulls and as precursors for SnO2-coatings on glass.4,5 Tributyltin (TBT) based compounds are one of the most widely used antifouling systems, since they have an acute toxicity to the target organisms.6,7 Due to the worldwide use of TBT-derivatives, high concentrations of organotin compounds still can be found in aquatic environments, either in sea-water or in marine organisms (mussels, oysters, etc.). The International Maritime Organization (IMO) has different pending proposals to further regulate the use of TBT-based antifouling paints. A ban on application of TBT-based paints from January 2003 and a complete ban in January 2008 have been suggested.6
The above facts prompted us to design molecularly imprinted polymers as a new tool for the determination of TBT-derivatives. The heterogeneity of MIPs leads to a high number of binding sites with a wide variation in structure, affinity and selectivity. This variation typically yields a much higher percentage of non-selective, low affinity sites, opposed to the desired high-affinity sites. This is one of the main drawbacks of the most common imprinting approach, the non-covalent method. The majority of strategies for reducing heterogeneity choose the modification and optimization of the imprinting process.8,9 Another strategy is to employ covalent imprinting, that it is generally perceived to yield better defined and more homogeneous binding sites. It employs reversible covalent bonds, usually involving a prior step of chemical synthesis to link the monomer to the template.10
The existence of MIPs showing weak non-covalent interactions between the functional monomer (Na-metachrylate) and TBT used as template has been previously reported.11 However, while selective imprinted sites were observed, these MIPs suffered from low homogeneity and capacity factor. Here we report the synthesis, characterization and chemical behavior of MIPs based on covalent complexes obtained from (Bu3Sn)2O and Bu2SnO, respectively, and m-vinylbenzoin. This compound was selected on the base of its likeliness to tropolone (Fig. 1), which has been employed before for OTC extraction from environmental samples, due to its high complexation capacity towards tin-derivatives.12,13 The resulting polymer was expected to have a high percentage of high-affinity sites, improving recognition capacity in terms of homogeneity and average binding affinity.
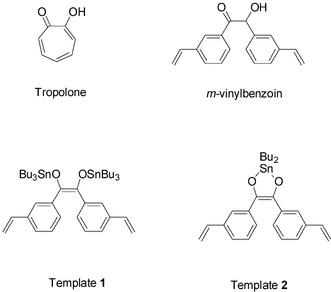 | ||
| Fig. 1 Chemical structures of tropolone, m-vinylbenzoin, Template 1, and Template 2. | ||
The selective recognition of the new polymer towards OTC are evaluated and discussed.
Experimental
General procedures
Synthesis of template 1: A solution of m-vinylbenzoin (0.4 g) and (Bu3Sn)2O (0.88 g) in anhydrous benzene (150 mL) was heated in a Dean–Stark apparatus for 5 h under argon.14b The solvent was removed under reduced pressure and the residue was taken up with pentane. The white solid precipitate was filtered off and the process was repeated several times. Finally, the residue was distilled in vacuum yielding 57% of pure compound as clear, orange oil (0.72 g). 1H NMR δ 8.04 (s, 2H), 7.89–7.84 (m, 2H), 7.48–7.25 (m, 4H), 6.7 (m, 2 H), 5.74 (d, J = 17.5 Hz, 2H), 5.21 (d, J = 11.0 Hz, 2H). 13C NMR δ13.6, 16.3, 27.1, 27.9, 114.5, 127.9, 128.2, 129.4, 129,6, 132.4, 136.3, 137.5. IR (film): 2957, 2924, 2872, 2854, 1578, 1522 cm−1. C42H68O2Sn2: Calculated C, 59.88; H, 8.14, Found C, 60.30; H, 7.95.
Synthesis of template 2: A solution of m-vinylbenzoin (1.34 g) and Bu2SnO (1.6 g) in anhydrous benzene (40 mL) was heated in a Dean–Stark apparatus for 5 h under argon. The solvent was removed under reduced pressure and the residue was taken up with chloroform. The white solid precipitate was filtered off and the process was repeated several times. 51% of pure compound was obtained as clear yellow oil (1.28 g). 1H NMR δ 7.96–7.30 (m, 8H), 6.64 (m, 2H), 5.74 (d, J = 16, 2H), 5.25 (d, J = 11, 2H), 1.63–0.78 (m, 18H). 13C NMR δ 13.6, 26.8, 27.5, 27.8, 116.0, 126.9, 129.1, 133.1, 134.9, 135.4, 138.5. IR (film): 2926, 2856, 1672, 1554, 1438, 1353, 1236, 1153 cm−1. C26H32O2Sn: Calculated C, 63.06; H, 6.51, Found C, 62.79; H, 6.44.
Preparation of bulk polymers: All the reactions were done under argon atmosphere at 60 °C. Different co-monomers: (1) m-vinylbenzoin, (2) 4-vinylpriridine; (3) styrene, (4) Na-metachrylate, or (5) methyl-methacrylate, cross-linker ethylene glycol dimethacrylate (EGDMA) and templates (template 1 or template 2) were dissolved in 5 mL of porogen ACN (acetonitrile). The molar ratio employed was 1 ∶ 10 ∶ 30 (template : monomer : cross-linker). Initiator (AIBN) was added to the mixture to start the radical polymerization and the solution was stirred at 60 °C for 12 h. Non-imprinted polymers (NIPs) were obtained following the same general procedure without addition of any template. The different synthetized polymers are included in Table 1. The monolith bulk polymer was isolated by crashing the tube reaction until a thin powder was formed. This powder was sieved, and those particles smaller than 100 µm were selected. The solid was washed with Et2O and CH2Cl2 to remove residues of non-reactive species. For polymers derived from sodium methacrylate (NaMA) water was also used. Polymers were then taken into a Soxhlet apparatus, refluxed with 0.1 M HCl in MeOH during 8 h and dried under vacuum. The amount of template removed in order to prepare the polymers for rebinding experiments has been estimated to be 4% for MIP1; 70% for MIP2, MIP3 and MIP4; and 90% for MIP5 (expressed as Sn-concentration), calculated from the initial amount employed during the polymerization.
| Polymer | Template | Comonomer |
|---|---|---|
| a VB: m-vinylbenzoin; 4-VP: 4-vinylpyridine; MMA: methyl-methacrylate; NaMA: sodium methacrylate. Ethylenglycol dimethacrylate (EGDMA) was used as cross-linker and acetonitrile as porogen in all cases. b Ratio VB: comonomer : cross-linker 1 : 10 : 30. c Ratio template : comonomer : cross-linker 1 : 10 : 30. | ||
| NIP-1 | Noneb | 4-VP |
| MIP-1 | Template 1c | 4-VP |
| NIP-2 | Noneb | VB |
| MIP-2 | Template 1c | VB |
| NIP-3 | Noneb | NaMA |
| MIP-3 | Template 2c | NaMA |
| NIP-4 | Noneb | Styrene |
| MIP-4 | Template 2c | Styrene |
| NIP-5 | Noneb | MMA |
| MIP-5 | Template 2c | MMA |
Rebinding experiments: about 50 mg of polymer was placed in an empty solid-phase extraction cartridge and conditioned with 10 mL of Milli-Q water before loading. Then, different volumes of TBT standard solution, containing from 400 ng to 10 mg, were loaded onto the cartridge; washing-off and elution steps were performed similarly to the experiments described before for the MISPE method.
Data analysis: the analyte concentration measured after elution represented the amount of analyte specifically bound to the polymer (B) and the analyte measured after washing-off corresponded to the amount non-specifically bound (F). Both were measured by GFAAS and fitted to the different chosen isotherm models by using the non-linear curve fit of Solver Excell software.
Chemical modification of MIP3 and NIP3: to a 10 mL round bottomed-flask containing 20 mg of MIP3 in 3 mL methylene chloride, was added 1 mg of sodium acetate anhydrous and 10 µL of phenyl isocyanate. The mixture was heated in a water bath under reflux during 4 h. The same procedure was carried out with NIP3. After reaction, the remaining reagents were eliminated washing MIP3* and NIP3* in SPE cartridge with methylene chloride and water.
Extraction and MISPE of biota sample: 0.1 g of the mussel CRM 477 were directly weighed into a 20 mL amber glass vial. Then 10 mL of a mixture of acetic acid and methanol (3 ∶ 1) were added and the vials were introduced in an ultrasonic bath for 2 h;15 then the MISPE was loaded with 2 mL of extract (previously diluted to 50 mL at pH = 3). Washing steps with 10 mL of water–HCl (pH = 3) and 4 mL toluene were necessary to remove the interferent matrix; finally, elution was carried out with 6 mL of 0.3 M HCl in methanol. The eluted solution was ethylated and analysed by GC-FPD, following the procedure of Carlier-Pinasseau et al.16
Safety considerations: All chemical used throughout this work have been manipulated following strictly safety protocols. Benzene was used for the synthesis of the tin templates following the literature procedure for similar derivatives (ref. 14). Chloroform was the optimum solvent for further purification of the products, other solvents might be used but with poorer results.
Results and discussion
Different polymers were prepared by radical polymerization using templates 1 or 2; m-vinylbenzoin, 4-vinylpyridine, styrene, Na-methachrylate, or methyl-methacrylate as comonomers; and cross-linker (EGDMA). Table 1 summarizes the reagents and reagent ratios used in the preparation of the polymers. In each case, the corresponding non-imprinted polymer (NIP) was prepared under identical conditions , substituting the template 1 or 2 by m-vinylbenzoin.Evaluation of the imprinting effect: first of all, the imprinting effect was evaluated by comparing the TBT amount rebind by the different synthesized polymers in the presence or not of the template. A first batch of MIPs (MIP1, MIP2), polymerized in the presence of template 1, was loaded with 400 ng of TBT, observing low affinity binding sites as the behavior of imprinted and non-imprinted polymers was quite similar. Further analyte binding depends on pH, which was checked by preparing the loading aqueous solution containing TBT at different pH's (1–5). The retention was always quantitative at a pH between 2–5, and the maximum difference obtained for NIP and MIP during the washing step was 30% at pH = 4. Elution of the analyte bound to MIP was found to be difficult even at high acidic conditions (1 M HCl in MeOH). In fact, only the use of HCl in acetone resulted in quantitative analyte recovery, attributed to the formation of a complex, similar to tropolone–acetone.17 However, acetone is not a good eluent because it damages the polymeric and the SPE material.
Based on these results, a second batch of polymers, MIP3-MIP5, prepared using template 2 (Table 1) was tested. A set of rebinding experiments, analogous to those discussed above, were carried out. Retention was around 100%, specific binding sites were observed in the pH range of 2.5–3.5. The pH dependence for all the different co-monomers employed was similar. Comparison of this second batch with MIP1 and MIP2 resulted in higher differences between the imprinted and non-imprinted polymers, once the retained non-specific analyte was washed-out (around 70%). Elution of the template was now in the range of 85–90%, instead of 60–70% obtained before. This indicates reversible and weaker covalent interactions with template 2 derived polymers. As no significant differences in TBT rebinding were observed among this second batch, polymers made with NaMA as co-monomer (MIP3 and NIP3) were selected for further studies, based on their low shrink–swell effect compared with the other polymers. Fig. 2 shows the schematic representation of the imprinted cavity formed in MIP3.
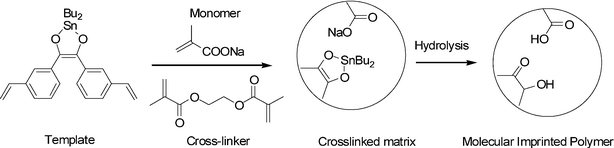 | ||
| Fig. 2 Schematic of the cavity formation in MIP3. | ||
Evidence of the imprinted cavities pH dependence: The observed pH dependence of the imprinting effect prompted us to pursue a more detailed study on this issue. The binding properties of MIP3 and NIP3 with respect to different pH values have been plotted in Fig. 3.
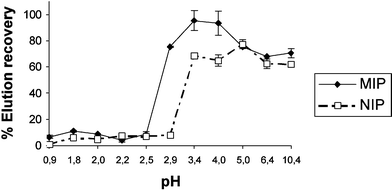 | ||
| Fig. 3 Binding properties modification of MIP3 induced by pH. | ||
The low elution recovery obtained at pH ≤ 2.5 (90% of analyte is washed-out) responds to the high percentage of non-specific interactions occurring both in NIP and MIP, due to the high H3O+ concentration present in the medium. The inverse situation is observed at pH ≥ 3,5 where strong acidic hydrolysis is needed in order to quantitatively recover TBT. Subsequently, in the pH range of 2.5–3.5, the imprinted polymer efficiently retains TBT and satisfactory differences between NIP and MIP are observed. The optimum situation occurs at pH = 3, with 80% of the analyte specifically bound. Here the MIP selectively links to the organotin compound whereas the corresponding NIP does not. Thus, the binding cavities present in MIP3 are pH dependent cavities. This effect can be considered a selective chemical modification to reduce binding site diversity discussed in more detail latter.
MIP3 isotherm characterization: this was performed by loading TBT in the range 400 ng–10 mg, as explained in the Experimental section. Calculation of the binding properties (affinity constant, binding site capacity and heterogeneity) was carried out using different models, ranging from simple ones (Freundlich, Langmuir, Langmuir–Freundlich) to low-high affinity models (Bi–Langmuir, Bi-Jovanovic–Freundlich).18,19 Three pH values (1.0; 3.0 and 5.0) have been selected for the characterization study of MIP3, based on the differences observed during the evaluation of the imprinting effect. The measurement of isotherms to characterize materials is usually carried out in equilibrium conditions to minimize the kinetic aspects involved in the guest–host interaction. Rebinding experiments were performed independently at different incubation times on-column (t = 0, 1, 15 and 24 h) after TBT loading as described in the Experimental section. Loading at pH = 5.0 provided, after 1 h of incubation, an increase of 30% washing-out amount related to the TBT washed at t = 0, and an additional 10% increase after 15 h. From 16 to 24 h it remained constant. However, the elution recovery remained constant during the total incubation time applied. This may be an indication that some kinetic effects occurred through the low affinity sites. By contrast, no significant differences were found at pH = 1.0 and 3.0 for the amount of template bound at different incubation times, meaning that equilibrium was immediately reached. These results indicate that diffusion mass transfer is really fast under these conditions, representing a clear advantage for the practical use of these materials, and also for isotherm fitting.
Fig. 4 shows the log plot [log B (bound analyte concentration) versus log F (free analyte concentration) of the experimental adsorption isotherms and the fitting to the five models mentioned above. The data demonstrates that both Langmuir–Freundlich (LF) and Bi-Jovanovic–Freundlich (bJF) isotherms fit at pH = 3 better than the other models, where the imprinting effect has been evidenced experimentally. Also, at pH = 5.0 LF and bJF fittings are satisfactory, whereas at pH = 1.0 the experimental data does not adjust (R2 < 0.9) to any of the models studied. In fact, the identical behaviour of MIP and NIP shown at high acid condition, shows evidence for the exclusive existence of low affinity interactions between template and monomer. The RSS and the modified Fisher test19—F calc—values of bJF at pH = 3.0 and 5.0 may suggest the existence of two different cavities, corresponding to a low–high affinity model. However, it seems more likely to assume a chemical modification of the cavities, induced by the enol equilibrium (see later for the explanation of this hypothesis), which strongly depends on pH; LF fitting also gave good RSS and F values. Considering this model has been suggested by many authors to have the advantage of modelling MIP behavior20 for homogeneous and heterogeneous systems as well as to simultaneously model both sub-saturation and saturation, further studies have been evaluated using LF-isotherms. Binding coefficients including the same parameters for a non-covalent approach11 are summarized in Table 2. Data from this table show as expected higher homogeneity, affinity and capacity factor (50 times higher) for covalent polymers than non-covalent polymers.
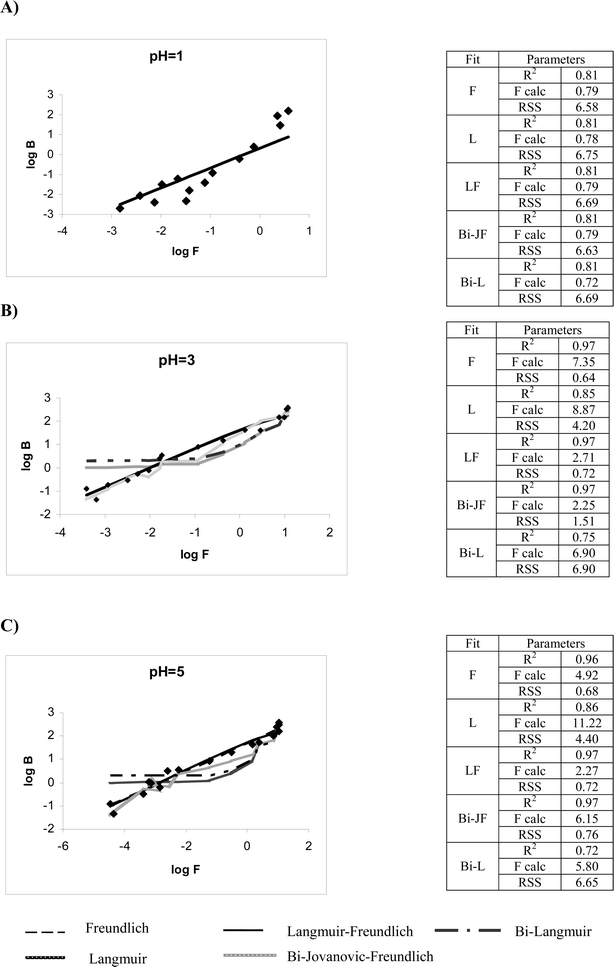 | ||
| Fig. 4 Experimental adsorption isotherms for MIP3 (data points), and fitting models at different pH's. (A) pH = 1.0; (B) pH = 3.0; (C) pH = 5.0. | ||
| pH | N t/µmol g−1 | m | a/mM−1 × 10−3 | K 0/mM−1 × 10−3 (Kmin–Kmax) | |
|---|---|---|---|---|---|
| a N t: binding site density; m: heterogeneity parameter; a: affinity; K0: affinity constant. b Unpublished results. | |||||
| A | 1.0 | 150 | 1 | 14.04 | 14.04 (0.264–677) |
| 3.0 | 370 | 0.83 | 126.59 | 83.90 (0.084–1545) | |
| 5.0 | 300 | 0.64 | 220.49 | 93.34 (0.094–22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 164) 164) |
|
| Bb | 6.15 | 0.73 | 7.19 | 14.92 (2.04–29673) |
|
The affinity spectrum (AS): this has been plotted in Fig. 5. The affinity spectrum is defined as the number of sites (N) versus log association constant (K) and yields the distribution of sites over a continuous range of binding constants and also characterizes the heterogeneity present in a MIP. The N(K) general equation21 was then solved considering LF our best fitting model by following Umplebý’s considerations:22
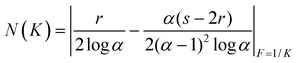 | (1) |
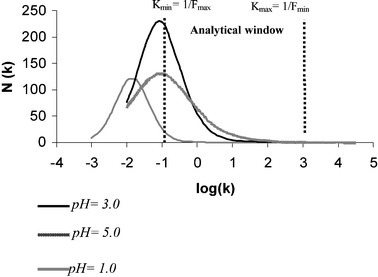 | ||
| Fig. 5 Affinity spectrum distribution for MIP3. N: number of sites; K: association constant. | ||
The use of the affinity distribution to characterize MIPs has two main advantages: the distribution can be generated directly from binding isotherms and these can be re-examined to yield corresponding AD; this method models accurately the heterogeneous distribution of MIPs and then allows a quantitative comparison of their binding properties. Solutions to the N(K) equation revealed an asymptotic relationship between the number of binding sites and the association constant within the concentration range studied (analytical window) at three pHs values. This profile is typical for non-covalently imprinted polymers or covalent polymers within the sub-saturation region.20 The latter is the situation of MIP3 that can be corroborated by the experimental adsorption isotherm (Fig. 4), where the characteristic curvature of a LF isotherm is not reached within the F and B measured. Experimentally, it is not possible to reach saturation of the polymers by loading in water because of the limited TBT solubility in this medium. Thus, the situation has been modeled beyond the analytical window, observing the unimodal distribution expected for a covalent polymer fitted by LF.21 Comparing distribution plots of different pHs with the fitting parameters given in Table 2, the highest capacity (Nt) is obtained at pH = 3, fitting with the highest value (N = 370). This is closely followed by Nt = 300 at pH = 5, and finally the lowest value at pH = 1 (Nt = 150), where the polymer has lost any recognition capacity but still binds the analyte by the non-specific sites. Looking at the plot shapes, the narrower the distribution the better the homogeneity and m approaches 1. This value has been obtained for pH = 1 (m = 1), in clear agreement with the literature, strengthening the fact that non-imprinted polymers are more homogeneous than the imprinted ones.23 At pH = 3 the distribution is narrower than at pH = 5, verified once again by m values (0.83 and 0.64, respectively). Finally, the distribution maximum at pH = 3 enters the analytical window (log K0 ≈ −1.1), fitting well with calculations given in Table 2, corresponding also to the highest affinity constant value. Therefore, a relatively high population of sites has been formed with medium association constants at that pH. However, the distribution maximum at pH = 1.0 (log K0 ≈ −2) means that TBT recognition by the imprinted cavities can be significantly thwarted, depending on the pH conditions employed. The hypothesis of cavities modification by pH is again reinforced.
Site selective chemical modification (SSCM): examples of MIPs experiencing changes of the binding properties by chemical modification after the polymerization process are scarce.24,25 This has also been achieved by thermal treatment26 or by variation of the pH.27 The pH dependence of the MIP3 presented derived from benzoins and might be explained by considering that protonation in the acid media is acting as a site selective chemical modification of the cavities. As it is well known, α-hydroxycarbonyl compounds can be enolized, and under acidic conditions they form protonated ene-diols.28 Thus, we can hypothezise that enol forms are responsible for reversible tin interactions. As decreasing the pH of the medium the non-specific cavities are easily protonated, selective cavities are then ready for tin recognition. At pH = 5 the protonation did not occur due to the low proton concentration of the medium; the optimum situation occurs at pH 3, with most of the non-specific interactions prevented. At lower pHs all the specific and non-specific sites are completely protonated, avoiding the association between the benzoin groups and the tin species. These equilibrium situations have been represented in Fig. 6.
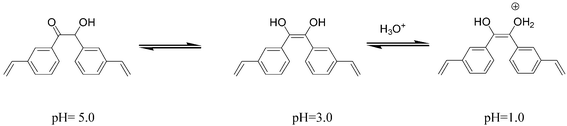 | ||
| Fig. 6 Enolization and protonation of m-vinylbenzoin. | ||
In favor of the role of the benzoin groups as the sole responsibility of this pH dependence plays on the fact that MIPs, synthesized under the same conditions but using other monomers such as styrene, and methyl methacrylate, showed analogous behavior within this pH range. Moreover, chemical modification of NIP3 and MIP3 by using phenylisocyanate, in order to transform OH groups in the benzoin moieties into carbamates derivatives (NIP3* and MIP3*), evidenced two facts shown in Table 3. First, the effect of protonation is overcome since no free hydroxyl groups remain, demonstrated by observed similar differences between MIP3* and NIP3* (40%) within the pH range 3–5. Second, association of TBT species with modified MIP3* is considerably weaker at the optimum pH for recognition (pH = 3), as a consequence of the OH modification (see washing and elution data in Table 3). Therefore, the chemical modification carried out in polymers MIP3 and NIP3 demonstrate the active role of benzoin on the pH dependence of the cavities that can not complex organotin in the same way. The free hydroxyl group in vinylbenzoin is exerting an active role in the recognition properties and in the pH dependence of the polymers. This can only be explained in terms of protonation and enolization of the vinylbenzoin units present in the polymer matrix.
| pH | Polymer | R% | W% | E% | Differencesb |
|---|---|---|---|---|---|
| a These data are based on three independent measures. R%: retention percentage; W%: washing percentage; E%: elution percentage. b Differences measured between NIP and MIP in each case. | |||||
| 5 | NIP3* | 88 ± 2 | 30 ± 1 | 50 ± 1 | 40 ± 3 |
| MIP3* | 100 ± 3 | 3 ± 1 | 90 ± 2 | ||
| NIP3 | 100 ± 3 | nd | 70 ± 1 | nd | |
| MIP3 | 100 ± 2 | nd | 70 ± 1 | ||
| 3 | NIP3* | 88 ± 1 | 60 ± 3 | 20 ± 2 | 40 ± 3 |
| MIP3* | 100 ± 4 | 30 ± 1 | 60 ± 3 | ||
| NIP3 | 100 ± 3 | 80 ± 2 | 20 ± 2 | 70 ± 3 | |
| MIP3 | 100 ± 2 | 10 ± 1 | 90 ± 3 | ||
Cross-reactivity among organotin compounds: experiments were conducted to evaluate the cross-reactivity among TBT, DBT, MBT and TPhT. The compounds were loaded separately at a concentration of 200 µg L−1 in water acidified with HCl at pH 1, 3 and 5. Subsequently, washing was performed with 4 mL of toluene and elution with 6 mL of 0.3 M HCl in methanol. These results showed again the same pH dependence of the polymers for all the OTC tested; the imprinted cavities are able to selectively re-bind all the organotin compounds at the optimum pH = 3 (Table 4). In addition, retention efficiency is affected depending on the OTC form: the tri-substituted species are quantitatively retained in both imprinted and non-imprinted polymers with subsequent washing-off from the non-imprinted whereas di and mono-substituted species present a loading recovery of only 50% in NIP. This behavior seems to be in response to the relatively high amount of medium–high selectivity cavities formed in the imprinted material which establishes strong reversible covalent interactions between benzoin functional groups and the organotin compounds. By contrast, in the non-imprinted polymer, electrostatic binding plays an important role similarly to the non-covalent polymers reported in a previous paper.11 This fact, together with the higher homogeneity affinity constants and capacity factor represents as expected the main improvement in comparison with the non-covalent approach.
Interferences evaluation: the covalent imprinted polymer has been tested for interference evaluation using the mussel tissue CRM 477 considered as a very interesting environmental sample for OTC determination (Table 5). The procedure for OTC extraction and MISPE has already been explained in the Experimental section. The presence of matrix interferences (coming from fat and other components) which may affect the derivatization and detection steps, was demonstrated by the discrepancy of recovery between the results obtained applying the external calibration approach (A in Table 5) and the certified value. However, when MISPE, by employing MIP3, was applied to the mussel extract the level of interferences drastically decreased and a remarkable improvement of recoveries was achieved (C in Table 5), even better than the addition standard method (B in Table 5). As a result, the values obtained are in good agreement with the certified values of the CRM. These results are very valuable since the covalently imprinted polymer can be employed as an easy and efficient way for clean-up and specific retention of organotin from biota samples. This may open new pathways for easy sample treatment, providing interference removal from complex matrices.
| MBT/mg Sn kg−1 | DBT/mg Sn kg−1 | TBT/mg Sn kg−1 | |
|---|---|---|---|
| a These data are based on three independent measures | |||
| A | 0.60 ± 0.09 | 0.29 ± 0.06 | 0.66 ± 0.05 |
| B | 0.98 ± 0.20 | 0.66 ± 0.12 | 0.80 ± 0.10 |
| C | 1.11 ± 0.06 | 0.80 ± 0.08 | 0.94 ± 0.10 |
| D | 1.01 ± 0.28 | 0.78 ± 0.12 | 0.90 ± 0.19 |
Conclusions
This work shows the successful synthesis of a covalent imprinted polymer able to recognise organotin compounds. The recognition is strongly influenced by pH and this dependence has been explained in terms of site selective chemical modification by an enolization/protonation model. The thermodynamic parameters measured give quite homogeneous binding sites and high capacity in the imprinting range (pH = 2.5–3.5) as well as an improvement in selectivity compared with a previous polymers synthesised following the non-covalent method. Also, the role of electrostatic interactions in the binding behaviour has been reduced in the present material, the main interactions being reversibly covalent. Finally, matrix interference evaluation by testing a complex environmental matrix like mussel tissue (CRM 477) showed good selectivity of these materials for environmental analysis.Acknowledgements
This work has been carried out with the financial support of the CICYT (projects BQU-2002-01348 and CTQ2004-06250-CO2-01-BQU). The authors gratefully thank the Spanish Ministry of Science and Technology for a Ramón y Cajal contract to Riansares Muñoz-Olivas and for the grant from the Spanish Ministry of Education to the PhD student Mercedes Gallego-Gallegos. Luis González McDowell is gratefully acknowledged for his important contributions in the isotherm and AS fitting. Also, C. Dietz is acknowledged for help with the English.References
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812 CrossRef CAS.
- L. I. Andersson, J. Chromatogr., B: Biomed. Appl., 2000, 739, 163 CrossRef CAS.
- J. Olsen, P. Martin and I. D. Wilson, Anal. Commun., 1998, 35, 13H RSC.
- R. Heinz, Ecotoxicol. Environ. Saf., 2003, 56, 180 CrossRef.
- M. Hoch, Appl. Geochem., 2001, 16, 719 CrossRef CAS.
- Ambient Aquatic Life Water Quality Criteria for TBT–Final, United States Environmental Protection Agency, Office of Water, 4304T EPA-822-B-02-001, 2003.
- A. Michael and P. Champ, Sci. Total Environ., 2000, 258, 21 CrossRef CAS.
- F. G. Tamayo, J. L. Casillas and A. Martín-Esteban, Anal. Chim. Acta, 2003, 482, 165 CrossRef CAS.
- D. Spivak, M. A. Gilmore and K. J. Shea, J. Am. Chem. Soc., 1997, 119, 4388 CrossRef CAS.
- G. Wulff and A. Biffis, in Molecularly Imprinted Polymers, ed. B. Sellergren, Elsevier, 2001, p. 71 Search PubMed.
- M. Gallego-Gallegos, R. Muñoz-Olivas, A. Martín-Esteban and C. Cámara, Anal. Chim. Acta, 2005, 531, 33 CrossRef CAS.
- J. M. Fernández-Escobar and J. M. Bayona, Anal. Chim. Acta, 1997, 355, 269 CrossRef CAS.
- M. Ceulemans, S. Slaets and F. Adams, Talanta, 1998, 46, 395 CrossRef.
- (a) W. Tagaki and W. H. Hara, J. Chem. Soc., Chem. Commun., 1973, 891 RSC; (b) A.G. Davies and J. Hawari, J. Chem. Soc., Perkin Trans., 1983, 875 Search PubMed.
- P. Rodríguez-González, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2004, 19, 767 RSC.
- C. Carlier-Pinasseau, G. Lespes and M. Astruc, Talanta, 1997, 44, 163.
- H. Hamabe, H. Sekiya, N. Nakano, K. Nishi and Y. Mishimura, Chem. Phys. Lett., 1997, 280, 390 CrossRef CAS.
- H. Andersson and A. Nochols, in Molecularly Imprinted Polymers, ed. B. Sellergren, Elsevier, 2001, p. 1 Search PubMed.
- C. Baggiani, G. Giraudi, C. Giovannoli, C. Tozzi and L. Anfossi, Anal. Chim. Acta, 2004, 504, 43 CrossRef CAS.
- (a) G. Vlatakis, I. Andersson, R. Müller and K. Mosbach, Nature, 1993, 361, 645 CrossRef CAS; (b) R. J. Umpleby, S. C. Baxter, Y. Chen, R. N. Shah and K. D. Shimizu, Anal. Chem., 2001, 73, 4584 CrossRef.
- D. L. Hunston, Anal. Biochem., 1975, 63, 99 CrossRef CAS.
- R. J. Umpleby, M. Bode and K. D. Shimizu, Analyst, 2000, 125, 1261 RSC.
- M. J. Whitcombe, M. E. Rodriguez, P. Villar and E. N. Vulfson, J. Am. Chem. Soc., 1995, 117, 7105 CrossRef CAS.
- N. Kirsch, C. Alexander, M. Lübke, M. J. Withcombe and E. N. Vulfson, Polymer, 2000, 41, 5583 CrossRef CAS.
- R. J. Umpleby, G. T. Rushton, R. N. Shah, A. M. Rampey, J. C. Bradshaw, J. K. Berch and K. D. Shimizu, Macromolecules, 2001, 34, 8446 CrossRef.
- Y. B. Chen, M. Kele, P. Sajonz, B. Sellergren and G. Giochon, Anal. Chem., 1999, 71, 928 CrossRef CAS.
- Y. B. Chen, M. Kele, I. Quiñones, B. Sellergren and G. Guiochon, J. Chromatogr., A, 2001, 927, 1 Search PubMed.
- (a) J. R. Keffe and A. J. Kresge, in The Chemistry of Enols, ed. Z. Rapport, John Wiley & Sons, New York, 1990, p. 399 Search PubMed; (b) J. Toullec, in The Chemistry of Enols, ed. Z. Rapport, John Wiley & Sons, New York, 1990, p. 323 Search PubMed.
Footnote |
| † This paper is dedicated to the memory of Dr Juan C. del Amo, a victim of the terrorist attack in Madrid on 11 March 2004. |
| This journal is © The Royal Society of Chemistry 2006 |