Polypeptide hydrogels via a unique assembly mechanism
Timothy J.
Deming
*
Department of Bioengineering, University of California, Los Angeles, CA 90095, USA. E-mail: demingt@seas.ucla.edu; Fax: +1 310 794 5956; Tel: +1 310 267 4450
First published on 10th March 2005
Abstract
There is a long history of man's use of materials derived from peptides and proteins. These natural materials possess sophisticated mechanisms of nanoscale self assembly, which have inspired the design of many synthetic and biosynthetic amino-acid based materials. These materials are attractive since they can have exceptional properties, environmental responsive behavior, biological activity, and can be metabolized. With all of their complexity, peptides and proteins rely primarily on two fundamental modes of self assembly: association of β-strands and the coiling of helices. In this context, a class of recently synthesized and characterized polypeptide materials are reviewed here, which were found to self-assemble by a fundamentally different process. This new mode of assembly was found to give rise to polypeptide hydrogels with a unique combination of properties (e.g. heat stability and injectability) making them attractive for applications in foods, personal care products, and medicine.
 Timothy J. Deming | Timothy J. Deming received a B.S. in Chemistry fom the University of California, Irvine in 1989, and graduated with a Ph.D. in Chemistry from the University of California, Berkeley, under Bruce Novak in 1993. After a NIH postdoctoral fellowship at the University of Massachusetts, Amherst with David Tirrell, he joined the faculty in the Materials Department at the University of California, Santa Barbara in 1995. He held a joint appointment in the Materials and Chemistry Departments where he was promoted to Associate Professor in 1999 and Professor in 2003. In 2004 he moved to his current position as Professor in the Bioengineering Department at the University of California, Los Angeles. Current research interests include polypeptide synthesis, self-assembly of block copolypeptides, and biological activity of polypeptides, for which he has received Young Investigator Awards from the National Science Foundation, the Office of Naval Research, The Arnold and Mabel Beckman Foundation, the Alfred P. Sloan Foundation, the Camille and Henry Dreyfus Foundation, the Materials Research Society, and the IUPAC Macromolecular Division. |
Introduction
Natural proteins possess many mechanisms for the physical association of peptide segments. The two motifs most widespread are β-strand H-bonding for sheet and fibril assembly, and the coiling of helices to form suprahelical bundles.1 Both assemblies are capable of linking peptide or protein chains together to form gel networks in aqueous solution, classic examples being the gels from gelatin and acid-denatured insulin.2 The general features of β-sheet rich peptide gels include association primarily via H-bonding, high thermal stability (i.e. up to 100 °C), and chain directions perpendicular to fibril axes.3 In gels from helical bundles, association is mainly through hydrophobic interactions; they are typically disrupted at elevated temperature,4 and, in cases of fibrillar assembly, the chains usually lie along fibril axes.5 Hydrogels have also been prepared from synthetic hybrid materials utilizing peptide components that associate via similar mechanisms.4Here, we describe a new motif for peptide gel assembly not found in nature: the association of amphiphilic block copolypeptides via entirely hydrophobic α-helical domains. The association of completely hydrophobic helical polypeptides is certainly well known, as in the case of liquid crystal phases of poly(alkylglutamates),6 however they have rarely been employed for self-assembly in aqueous systems due to their poor water solubility. In fact, long uninterrupted sequences of α-helix favoring hydrophobic residues, e.g. leucine, are quite rare in nature, and examples of known function are utilized as transmembrane domains that span lipid bilayers, typically not for protein–protein interactions.7
We have found that conjugation of polyelectrolyte segments to hydrophobic helical domains can stabilize formation of hydrated membranes and fibrils that form a robust hydrogel network.8 The assembly mechanism, elucidated via analysis of structure–property relationships and the use of a range of characterization tools,8,9 was found to occur via an unprecedented association of α-helices perpendicular to fibril/membrane long dimensions. This motif contrasts greatly to helix orientation in coiled-coil fibrils as well as the structures of organogels formed from hydrophobic α-helical polypeptides.10 The assemblies described here more closely resemble β-sheet fibrils in structure and stability, but without the interstrand H-bonding. In these materials, we have identified a new, general means of peptide assembly, which should allow the development of a wide-range of nanoscale materials with unprecedented capabilities.
Hydrogel molecular parameters
Hydrogel formation was first discovered in a series of diblock copolypeptides containing a charged, water solubilizing domain (poly(L-lysine·HBr), K; or poly(L-glutamate Na salt), E) and an α-helical hydrophobic domain (poly(L-leucine), L), i.e. KmLn or EmLn, where m and n represent the number of amino acid residues in each segment (Table 1).8 Through a comprehensive study of a diverse range of samples where both overall chain length and block composition were varied, it was found that chain length modification of both polyelectrolyte and hydrophobic segments had significant effects on solution properties.9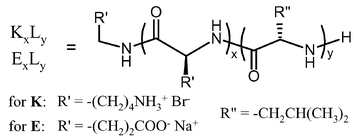
| Sample | Gelation concentration [wt%] | Gel strength [G′ at 3 wt%, 1 rad/s] | Gel strength 100 mM NaCl [G′ at 3 wt%,1 rad/s] |
|---|---|---|---|
| K80L20 | No gel at 6% | NA | NA |
| K190L10 | No gel at 5% | NA | NA |
| K180L20 | 2% | 12 Pa | 26 Pa |
| K170L30 | 0.75% | 590 Pa | 519 Pa |
| K160L40 | 0.25% | 4273 Pa | 299 Pa |
| K160(rac-L)40 | 2.5% | 36 Pa | NA |
| K380L20 | 0.25% | 146 Pa | 158 Pa |
| K370L30 | 0.031% | 940 Pa | 380 Pa |
| K360L40 | 0.125% | 480 Pa | 242 Pa |
| E180L20 | 0.5% | 124 Pa | 469 Pa |
| E160L40 | 0.25% | 265 Pa | 47 Pa |
To study the role of the hydrophobic domain in gel formation, a series of four block copolypeptides was prepared with overall average degrees of polymerization (DP) of 200, but with hydrophobic segments ranging from 10 to 40 leucine residues in length. It was observed that the K190L10 sample did not form a hydrogel, even at high concentrations (5 wt%). The K180L20, K170L30, and K160L40 samples all formed hydrogels, with minimum gelation concentration decreasing, and gel modulus increasing, as the oligoleucine domain increased (Table 1).8,11 The most telling distinction between these samples was that all oligoleucine domains of 20 residues and greater were found to be completely α-helical, while the 10 residue segment was found to be highly disordered. This finding showed that hydrogel formation was tied strongly to the helical conformation of the hydrophobic domain, and that the strength of the gel scaled proportionately to the length of the helical segment. As further verification of this point, a copolymer was prepared of identical composition to a strong gel former, but with disorder in the oligoleucine segment obtained by polymerization of racemic leucine monomers. This copolymer, K160(rac-L)40, although compositionally identical to K160L40, was found to only form very weak hydrogels at low concentration, confirming the importance of the regular α-helical conformation.8
The role played by the polyelectrolyte segments in hydrogel formation was examined by analysis of a series of polypeptides where the oligoleucine domain was held constant at 20 residues, the minimum amount to give a stable helical domain, and the polylysine domain was varied from 80 to 380 residues.11 From above, it was known that the K180L20 sample was a weak hydrogel former with a minimum gelation concentration of 2 wt% (Table 1). The sample K80L20 was found to not form a hydrogel, even at much higher concentrations (6 wt%). Thus, even though this sample possessed a helical hydrophobic domain, that alone was not sufficient to drive formation of a gel network. This result was similar to that found in our lab for uncharged amphiphilic diblock copolypeptides containing helical oligoleucine segments.12 These copolymers were found to assemble into flat membranes, which form vesicular structures, but not hydrogels. It appears that the short polylelectrolyte segments in K80L20 do not provide enough repulsive forces to distort a flat membrane structure into the gel network.9
Examination of the larger copolymer, K380L20, seemed to confirm this hypothesis. This sample was able to form stronger hydrogels at concentrations much lower than K180L20 (0.25 wt% vs. 2 wt%). The increase in gel strength for K380L20vs. K180L20 was insignificant by comparison to changes made in the hydrophobic domains. In the example above, addition of 200 lysine residues increased G′ from 12 to 146 Pa, while an increase of only 10 leucine residues (K180L20 to K170L30) increased G′ from 12 to 590 Pa. However, the increase in gel forming ability with increased polylysine length was substantial. It is thought that longer polyelectrolyte segments increase interchain repulsions such that the packing of the hydrophobic helices, which appear to prefer formation of a flat 2D sheets,12 must distort to minimize the overall energy of the system. The best way to do this, while maintaining favorable helix packing, is to twist the sheets into fibrillar tapes, where tape width is determined by the degree of twist.13 In this model, the helices are still able to pack perpendicular to the fibril axis, but with a slight twist between planes of parallel packed helices (Fig. 1).
 | ||
| Fig. 1 Drawings showing (A) representation of a block copolypeptide chain and (B) proposed packing of block copolypeptide amphiphiles into twisted fibrillar tapes, with helices packed perpendicular to the fibril axes. Polylysine chains were omitted from the fibril drawing for clarity (Reprinted with permission from ref. 9. Copyright 2004 American Chemical Society). | ||
Hydrogel structure
To test the hypotheses discussed above, which were based on molecular features, many characterization tools were applied to the polypeptide hydrogels. Surprisingly, the hydrogels were found to display structure over a wide range of length scales. Self-assembly of the polypeptide chains was expected to occur at the tens of nanometers scale, based on the size of individual polymer chains. Cryogenic transmission electron microscopy (CTEM) was used to visualize the assemblies in this size regime.14 By imaging vitrified, nonstained samples, we hoped to obtain a fairly accurate representation of the nanoscale gel structure. Using this technique, 3D gel networks were observed (Fig. 2a), with a network density that increased with sample concentration. Even at very low concentrations, the exact nature of the assembled structures could not be discerned, although the apparent highly anisotropic struts and curved surfaces were not inconsistent with fibrillar and twisted membrane morphologies. The irregularity of the structures, combined with high beam sensitivity, has, to date, limited further high resolution CTEM studies. | ||
| Fig. 2 (A) Cryogenic transmission electron microscopy image of K170L30 at 3.0 wt% in H2O showing an interconnected nanostructure of polypeptide matrix (dark) surrounded/filled by vitreous water (light). (B) Laser scanning confocal micrograph of K160L40 at 1.0 wt% in H2O with a heterogeneous microstructure visualized using DiOC18 hydrophobic dye (Reprinted with permission from ref. 8. Copyright 2002 Nature Publishing Group). | ||
X-Ray scattering experiments on the hydrogels have yielded very little data. The poor contrast of the samples against water, and the very low sample concentrations gives rise to poor signal to noise. To circumvent these problems, small angle neutron scattering (SANS) experiments on samples prepared in D2O were undertaken, which revealed regularity in the hydrogel mesh porosity.14 In a K180(LV)20 sample, where LV represents an equimolar statistical sequence of L-leucine and L-valine, there appeared to be a nanoscale porosity of ca. 140 to 210 nm that increased in size with decreasing sample concentration. In addition to this nanostructural organization, the hydrogels were also found to possess structure on the micron length scale. Examination of hydrogels that had been stained with hydrophobic, fluorescent dye using laser scanning confocal microscopy showed the presence of irregular microscopic voids in the network (Fig. 2b).8,14 Micro-rheology experiments, where the diffusion of colloidial particles in the hydrogel were monitored over time, revealed that the voids were essentially domains of polymer-free water.8 SANS studies also showed the presence of a sharp interface between polymer and water, indicating a strong segregation of the polypeptides from solvent.14 These data verified that the hydrogels phase separate into polymer-rich, and polymer-free domains of ca. 1 to 50 µm in diameter. This bicontinuous state was found to be very stable against centrifugation, shear, aging, and heat. Overall, the polypeptide hydrogels were found to exhibit complex self-assembly, with structural features ranging from nanometers to microns.
Rheological properties
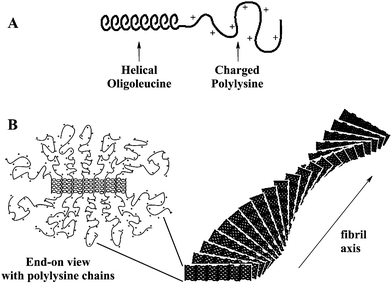
Initially, rheology was used to confirm the elastic gel nature of the polypeptide solutions, as well as determine minimum gel concentrations and storage moduli of different samples.8 The most notable result from these studies was the low sample concentrations at which gelation was observed, in many cases below 1.0 wt%. This feature contrasts greatly to other charged, amphiphilic block copolymers, which typically only form hydrogels at orders of magnitude higher concentrations.15 The key difference in the polypeptide samples is the helix packing motif that prevents these materials from forming spherical micelles and vesicles and drives assembly into a fibrillar network. More detailed rheological analysis of an array of samples gave further insights into hydrogel properties.9 The gels were found to break down (as measured by G′) at high strain amplitudes (i.e. shear thinning) in a concentration dependent manner. That is, the hydrogels tended to become more brittle (gel thins at smaller strain amplitudes) as polypeptide concentration increased.Since the gels break down under high strain, the kinetics of their recovery could be measured. Hydrogel samples were subjected to large oscillatory strains causing G′ to drop two orders of magnitude. Gel recovery was then monitored by measuring G′ at small, non-destructive strains. Very fast recovery (<10 s) of elastic properties (i.e. >80% of original G′) was observed when compared to hours typically required for similar recovery in conventional biopolymer hydrogels (e.g. gelatin) (Fig. 3).8 The reason behind the fast recovery found in these polypeptide hydrogels is not obvious. Initially, it was believed that rapid amphiphile reorganization was due to the short chain lengths of the block copolymers as compared to larger biopolymers, which allows greater chain mobility. Also, as these are physically associating gels, the associations would be expected to be somewhat reversible. However, the observed microscale phase separation in the polypeptide gels argues against any appreciable diffusion of free polypeptide chains. Furthermore, critical micelle concentrations of these amphiphilic block copolymers have been found to lie in the region of 10−11 M, consistent with little network disassembly.16
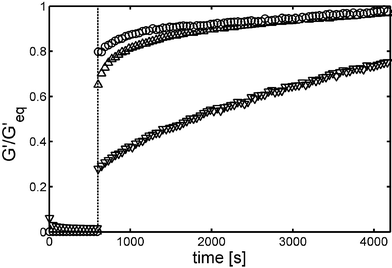 | ||
| Fig. 3 Recovery of gel strength G′ for (△) 1.0 wt% K160L40, (○) 0.75 wt% K160V40 and (∇) 2.0 wt% gelatin. Large amplitude oscillatory breakdown (1000% at 6 rad s−1 for 600 s) was followed by linear recovery measurements (0.3–1.0% at 6 rad s−1). G′ was normalized to the equilibrium value G′eq to facilitate sample comparisons (Reprinted with permission from ref. 8. Copyright 2002 Nature Publishing Group). | ||
Optical microscopy studies gave some insight into recovery of these hydrogels. Simple pressure on a coverslip over a thin gel sample allowed observation of micron-sized gel domains sliding past each other as whole units, with the aqueous phase acting as a lubricating and compliant layer.16 These observations suggest that the local gel network was not disrupted under large strain, but that perhaps only the interconnects between gel domains were broken. Since the bulk of the polymer gel does not need to reorganize after strain, as only the interconnects need to reform, recovery is rapid. This hypothesis requires interconnecting regions that can reversibly and rapidly dissociate then reform, without much loss in integrity. Such cross-links are not widely known in conventional hydrogels. A note of distinction in the block copolypeptide gels is that their recovery is much faster than even those composed of physically associated small molecules, such as β-sheet forming peptides.3 The main difference between these two systems is the association mechanism: β-sheet H-bonding versus hydrophobic α-helix packing. It is reasonable to assume that alignment and registry are much more critical in β-sheet formation than in helix packing, as H-bonds must be precisely located, as in crystal formation. The peptide chains can also unfold upon β-sheet rupture, or form disordered H-bonds, which may also slow reassembly compared to hydrophobic helices, which can pack together effectively with less precision.
Related to hydrogel recovery after strain is stability to thermal treatment. The thermal breakdown of many physically associating hydrogels (e.g. gelatin) is well known.2 In these systems, helical bundles typically dissociate at elevated temperature resulting in loss of crosslinks and conversion from a gel to a fluid. Hydrogels based on β-sheet fibrils are more robust at elevated temperature, with only a few examples being known to break down.3 Rheological measurements on block copolypeptide hydrogels have been performed at temperatures up to 90 °C in H2O, where no changes in mechanical properties compared to ambient conditions were observed.9 Thus, with regard to thermal stability, the helix-based polypeptide hydrogels are more like β-sheet gels than those formed from coiled helices. This similarity likely arises since the polypeptide gels are composed of fully hydrophobic helices, which have no tendency to become solvated by polar water molecules at any temperature. It is worth noting that β-sheet fibril-like properties apparently have been realized in these block copolypeptide hydrogels using an entirely different structural motif. We expect this new assembly mechanism will provide an additional means of functionality to self-assembling peptides, since the α-helical structure is more tolerant of amino acid substitution, and is more readily prepared at different lengths compared to β-sheets.
Block architecture
To probe the molecular interactions in block copolypeptide hydrogels further, the role of block architecture on assembly has been investigated.9 A key issue concerning hydrogel nanostructure is the orientation of helical oligoleucine segments in the self-assembled networks. Based on studies of flat membranes formed using similar helical copolypeptides,12 it was hypothesized that the hydrophobic helical segments would align perpendicular to either 2D membranes or 1D fibrils, not unlike the orientation of the chain axes in β-sheet fibrils.13 However, helical polypeptides are typically found to align parallel to long fibril axes, as was found in organogels made from hydrophobic α-helical polypeptides.10 In the hydrogel system described here, the amphiphilic nature of the block copolymers provides a soluble charged domain that limits the packing possibilities of the hydrophobic helices, and should favor chain alignment perpendicular to an interface (Fig. 4).9 Although such assembly would allow greatest access of the charged segments to the polar solvent, an arrangement of helices parallel to the interface, with charged segments bending out away from the aligned rods, must also be considered.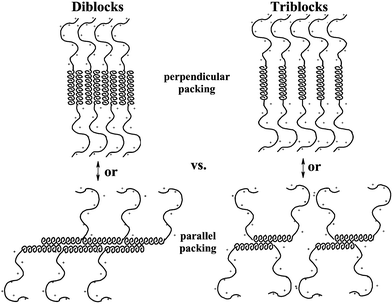 | ||
| Fig. 4 Drawings showing different possible packing motifs of helical segments in block copolypeptide hydrogels. Diblock (KnLm) and triblock (KnLmKn) architectures are shown for comparision in both perpendicular and parallel packing arrangements. Note that the parallel arrangement should greatly disrupt helix packing for the triblock copolymers. | ||
To help resolve this issue, symmetric triblock copolymers were prepared, where a hydrophobic helical segment was capped by polyelectrolyte on both ends. It was envisioned that, based on packing arguments, this architecture would favor perpendicular orientation of the helices and disfavor packing of helices parallel to a fibril axis (Fig. 4). While it is difficult to make direct, quantitative comparisons between diblock and triblock copolypeptides, it was found that, for equivalent hydrophobic domains, the triblock copolymers formed stronger hydrogels than corresponding diblock copolymers.9 This result confirmed, at least, that the triblock architecture did not destabilize hydrogel formation, and so the model of helix alignment perpendicular to self-assembled fibril axes seems plausible. The increased gel strengths of the triblock samples was most likely due to higher polyelectrolyte content, due both to larger molecular weights of the triblock samples, as well as increased polyelectrolyte density per helix segment. Note that the polylysine “brush” is twice as dense in an assembled triblock copolymer fibril compared to one formed from diblock copolymers, since there are twice as many polyelectrolyte chains per helix in the triblocks (Fig. 4). The resulting increased polyelectrolyte interchain repulsions likely enhance network formation for the reasons discussed above.
Chain length polydispersity
Another means to probe helix packing in this system is to examine the effects of helix length polydispersity. As the hydrophobic helical domains contain all the same amino acids, it would be expected that there is no inherent sequence information guiding helix–helix alignment and registry, as long as the leucine side-chains can interdigitate. The main driving force for registry of helices arises from maximization of helix–helix overlap, which avoids exposure of the hydrophobic domains to the charged polylysine segments and the polar aqueous environment. Since these samples are synthetic, and thus possess finite chain length distributions, it would be expected that some of the longer helical segments would extend into the polar environment, leading to less stable assemblies. It is also possible that, since defect structures may form to stabilize such exposed helices, polydispersity gives rise to disorder in the assemblies, driving formation of the observed gel networks rather than well-ordered membrane or fibril structures.The block copolypeptides used to prepare hydrogels possessed chain length distributions (Mw/Mn) of 1.1 to 1.3,8 which are generally considered narrow for synthetic polymers.17 To test if chain length heterogeneity was influencing structure, polydispersity was artificially increased by mixing samples of different hydrophobic segment length. In one experiment, L40 and L20 hydrophobic segments were combined by mixing copolymers of the same overall length: K160L40 and K180L20. A solution of K160L40 at 0.25 wt% in water is just able to form a hydrogel. However, a mixture containing K160L40 at 0.25 wt% and K180L20 at 0.50 wt% in water was found to be liquidlike.9 Thus, even with more overall polymer present, the mixture, which contained a bimodal distribution of helix lengths, was unable to form the hydrogel structure. In another experiment, a mixture containing K160L40 at 0.25 wt% and K160(rac-L)40 at 0.25 wt% in water, where the racemic leucine domain is known to be disordered,8 was also found to prevent hydrogel formation, confirming the absolute requirement for helicity in all of the hydrophobic segments.8 These results show that both large chain length distributions and loss of helicity in the hydrophobic segments prevent formation of the hydrogel structure. Furthermore, they also show that although the samples described here are not monodisperse, their narrow chain length distributions are critical for hydrogel formation. Since truly monodisperse samples are presently not available, it cannot be said whether such samples would also form hydrogel networks, of different structures altogether.
The experiments above were focused on modifications to the hydrophobic domain, since this is the one that drives chain assembly. However, the polyelectrolyte segments have also been shown to have a strong influence on hydrogel formation, and so polydispersity in this domain was investigated as well.9 For these studies, a hydrogel of K180L20 at 2.5 wt% in water was compared to 4 : 1 (molar composition) mixtures of K180L20 with either K380L20 or K80L20, which were prepared at the same overall molar concentrations as the pure K180L20 sample. K380L20 was known to form a stronger gel than K180L20, while K80L20 was found to be unable to form hydrogels at concentrations below 6 wt%.9 Both sample mixtures formed hydrogels (Fig. 5), with the K380L20 mixture being stronger than pure K180L20, and the K80L20 mixture being weaker, as might be expected.9 The unexpected result was that polydispersity in the polyelectrolyte segments was found to have no adverse effects on hydrogel formation. In fact, it appeared that mixtures of different polyelectrolyte chain lengths gave additive effects, which provides a useful means to finely adjust gel properties via mixing of different polymer compositions. The results of all the mixing experiments point to highly specific, conformation and length dependent association in the hydrophobic domains, but very little interaction in the polyelectrolyte domains. The major parameter relating to the polyelectrolyte segments is the presence of a critical amount of polyelectrolyte at the block interface, which can be realized by a combination of chain length and brush density. Gel formation in these materials is critically dependent on the presence of hydrophobic helical segments of low polydispersity. Hence, even though these materials are synthetic polymers that may appear heterogeneous when compared to natural biopolymers, they do possess the ability to interact in both a conformation and size-specific manner.
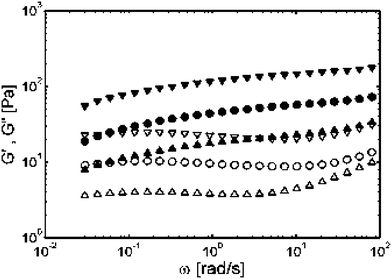 | ||
| Fig. 5 Storage modulus G′ (solid symbols) and loss modulus G" (open symbols) for pure samples and mixtures with different lysine segment lengths. Frequency sweeps of (●) 2.5 wt% K180L20, (▼) mixture with 2 wt% K180L20 and 1 wt% K380L20 (▲) mixture with 2 wt% K180L20 and 0.25 wt% K80L20 (Reprinted with permission from ref. 9. Copyright 2004 American Chemical Society). | ||
Applications in ionic media
A potential important issue in the use of block copolypeptide hydrogels for applications, including cosmetics, food additives and as biomedical materials, is their compatability with buffers, salts and serum. All of the studies of the hydrogels described above were performed in deionized (DI) water.8 Based on the properties of other polyelectrolyte hydrogels,18 it was possible that the gels would collapse in the presence of added ions (e.g. 100 mM NaCl). Added salt generally acts to screen the many like charges along the chains, weakening the forces responsible for chain stretching and interchain repulsion that typically support these gel networks. Such behavior is seen in chemically cross-linked poly(acrylic acid) gels, for example.18 Since our hydrogels are supported by an entirely different mechanism based on helix assembly, we hoped that they might be able to tolerate high ionic strength solutions.In order to evaluate the stability of polypeptide hydrogels to added salts, we first examined the rheological properties of the 200 residue copolymers (K160L40, K170L30, and K180L20) in the presence of NaCl (Fig. 6).11 The strongest gel former in DI water, K160L40, was surprisingly the least stable to added salt. At a concentration of 1.0 wt%, this copolymer was seen to precipitate upon the addition of even small amounts of NaCl (ca. 50 mM). In many ways the behavior of this sample was similar to the properties of the more hydrophobic material, K140L60, in salt-free water. K140L60 is partially insoluble in DI water and only forms viscous solutions, despite the large hydrophobic domain.11 The polyelectrolyte segments in this sample are not large enough to effectively solubilize and stabilize the hydrophobic gel network. Qualitatively, it appeared that added salt had a similar effect on the K160L40 sample by “salting out” or reducing the solubility of the polylysine chains, resulting in collapse of the network.
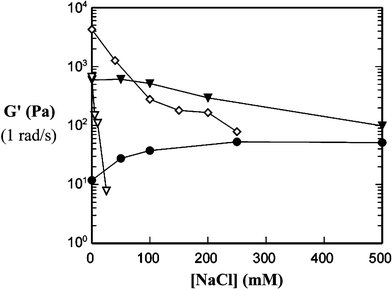 | ||
| Fig. 6 Hydrogel strength (G′) as a function of salt concentration for different KmLn diblock copolypeptides. Gel strengths were measured at 1 rad s−1: (●) K180L20 3.0 wt%, (▼) K170L30 3.0 wt%, (⋄) K160L40 3.0 wt%, and (∇) K160L40 1.0 wt% (Reprinted with permission from ref. 11. Copyright 2003 American Chemical Society). | ||
The inherently much weaker gel formed from K180L20 showed the most surprising behavior with added salt. At 3.0 wt%, this weak gel actually became stronger when salt was added, and was able to remain homogeneous at NaCl concentrations above 0.50 M. Thus it appeared that salt stability and inherent gel strength were somewhat mutally exclusive. Advantageously, the K170L30 copolymer showed intermediate behavior at 3.0 wt%, forming a relatively strong gel that was able to maintain most of its strength and show very little turbidity upon addition of salt. In the K180L20 sample, interchain repulsions of the large polyelectrolyte segments in DI water were believed to hinder efficient packing of the small hydrophobic domains, resulting in very weak gels.11 Observance of strain hardening in K180L20, but not in K170L30 and K160L40, also hinted that the hydrophobic helices were not inherently well packed, but could be packed into a stronger network under strain.9 Accordingly, K180L20 hydrogels displayed increasing brittleness as salt concentration was increased, indicating that the hydrophobes became better packed into stronger networks in ionic solutions.9 It seems plausible that the addition of salt relaxes the polyelectrolyte repulsions in this sample to allow better assembly of the hydrophobic network, and thus form a stronger gel.
The K170L30 sample, with intermediate composition, was mainly unaffected by ionic strength, as salt only seemed to affect samples with lysine : leucine ratios at the extremes of the gel forming region.11 A hypothesis for this lack of salt sensitivity can be based on the trade off between electrostatic and steric repulsions as salt is added. At low ionic strengths, the polyelectrolyte chains should be highly stretched due to intrachain electrostatic repulsions, and should repel each other for interchain electrostatic reasons. This interchain repulsion is the basis for our model on gel formation, as described above. As salt is added, the electrostatic interactions are screened. The loss of interchain repulsions would be expected to disrupt the hydrogel structure, however, loss of intrachain repulsions should result in chain coiling, which effectively increases chain size at the block interface (Fig. 7). The resulting increased interchain steric repulsions may act to replace the interchain electrostatic repulsions, resulting in only a small change in hydrogel structure and properties.
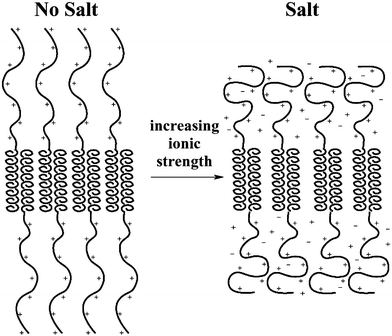 | ||
| Fig. 7 Drawing depicting changes in polyelectrolyte conformation as salt is added to block copolypeptide hydrogels. Electrostatic repulsions (left) are counterbalanced by steric repulsions (right) as polyelectrolyte chains coil in ionic media. | ||
Since K170L30 was found to be the most salt tolerant, 3.0 wt% hydrogels of this copolymer were subjected to a variety of buffers and ionic media.11 The most destabilizing media were those containing multivalent anions such as phosphate or sulfate. These ions are more efficient at condensing and possibly crosslinking the polyelectrolyte segments, effectively decreasing their solubility in water.19 Once the solubility limit of the polyelectrolyte is reached, the gels will collapse, as seen with K160L40. Even so, there is enough compositional flexibility in polypeptide synthesis to allow preparation of hydrogels stable to almost any ionic media. For example, K180L20 hydrogels were found to strengthen dramatically in the presence of divalent sulfate, and E180L20 was found to strengthen in divalent calcium.11 The K170L30 hydrogels were also stable over a broad pH range (4 to 9), essentially remaining intact as long as the polyelectrolyte segments were not neutralized. Gel strengths in these media were found to vary moderately, but seemed to depend more on the composition of the buffer than the pH of the solution.11 This result is not too surprising since each buffer contains ions of different valency, and since the charge density of the polyelectrolyte segments was unaffected in this pH range (pKa(avg.) of polylysine ≈ 10.5).
Hydrogel samples were also prepared in DMEM (Dulbecco's Modified Eagles Medium) and DMEM with 5% fetal calf serum and penicillin as representative cell culturing media.11 The K170L30 hydrogels were stable and remained transparent, which was somewhat surprising, since these media contain numerous multivalent ions and anionically charged proteins. It is likely that the proteins coat the polylysine segments in the gel since it is known that polylysine homopolymer will coagulate many serum proteins in solution.20 Apparently, the resulting polyelectrolyte complexes retain enough charge or hydrophilicity to solubilize the hydrophobic gel scaffold and prevent precipitation and collapse of the network. Overall, these copolypeptide hydrogels display remarkable stability in both the presence and absence of ionic species. Hydrogels formed from helical or β-sheet-forming proteins and peptides typically show some sensitivity to ions, either requiring them to form gels, or disrupting in their presence.2,3 Likewise, hydrogels prepared from synthetic polyelectrolytes (e.g. crosslinked polyacrylic acid) are very sensitive to salts, shrinking dramatically as ionic strength is increased.18 The gelation mechanism for our polypeptides, the association of hydrophobic helices, provides a robust structure that is unperturbed under a variety of conditions, including variation of pH, ionic strength, and temperature. These properties illustrate the novelty of the gelation mechanism in these amphiphiles, and how it can give rise to properties that have not been realized in other materials.
Conclusions
This review described a new class of amino acid derived hydrogel materials that assemble via a mechanism substantially different from those of other protein and peptide based hydrogels. This hydrophobic helix relies on the same amino acid components as the well-known “leucine zipper” motif found in α-helical coiled-coils.1,5 However, in addition to assembling via a different packing geometry in fibrils, the amphipathic nature of coiled-coils gives rise to instability in water at elevated temperatures, while the packing of entirely hydrophobic helices is unaffected even under the forcing conditions of an autoclave.The stability observed in copolypeptide hydrogels is similar to that found in H-bonded β-sheet fibrils. In many ways, the twisted fibrillar model proposed for the assembly of the helical polypeptide domains is based on the core structure of β-sheet fibrils. A key distinction is that the dimensions of a β-sheet fibril are dictated primarily by the twist associated with the interstrand H-bonding pattern as well as the nature of peptide side-chains, which may present hydrophobic residues that promote fibril bundling, yielding thicker fibers.13 Due to the regularity of the β-sheet twist, these fibrils are usually quite thin, typically 5 to 10 nm in diameter.3 Use of short β-strands (∼10 residues), necessitated since longer sequences will usually intramolecularly chain-fold or form disordered aggregates, also contributes to small fibril dimensions. Consequently, gel strength in these systems can typically only be adjusted by variation of peptide concentration.3
With the block copolypeptides, the associating domain is intramolecularly folded into a stable α-helical conformation, and thus can be readily varied in length to tune the strength of the interhelix associations, and, in turn, gel strength.8 As shown above, hydrogel strength can be adjusted not only by variation of the oligoleucine domain size, but also via modification of block architecture (i.e. triblocks) as well as through mixing of samples with different polyelectrolyte segment lengths. Thus, the change in assembly motif from highly specific H-bonding of β-sheets to less precise packing of helices allows tuning of macroscopic hydrogel properties simply and intuitively via a number of molecular parameters.
The helix assembly motif gives rise to an additional practical feature in that the samples can rapidly recover from shear thinning. Most H-bonded β-sheet networks recover only slowly after shear thinning, if they recover at all.3 This may be a consequence of chain unfolding, chain misfolding, or the time required for strands to align properly to form the required H-bonds. The reassembly of hydrophobic helices is a much faster process, possibly since the individual chains do not unfold, and exact chain alignment is likely not required. As such, the the copolypeptide assemblies may be intrinsically less ordered than some of the structures found in biopolymer hydrogels, yet consequently may possess many practical advantages over these materials. Overall, we believe the new assembly motif found in these hydrogels has tremendous potential as an alternative method for nanoscale assembly of biomolecular materials.
Acknowledgements
This work was supported by grants from the National Science Foundation (Chemical and Transport Systems CTS-9986347, and MRSEC Program DMR-0080034). The author thanks Dr Andrew P. Nowak (University of California, Los Angeles) and Professor Darrin Pochan (University of Delaware) and Professor Victor Breedveld (Georgia Institute of Technology) for invaluable collaborations and insightful discussions on this work.References
- D. Voet and J. G. Voet, Biochemistry, 2nd edn, Wiley, NY, 1995, ch. 7 Search PubMed.
- A. C. Clark and S. B. Ross-Murphy, Adv. Polym. Sci., 1987, 83, 57–192 CAS; , The Science and Technology of Gelatin, eds. A. G. Ward and T. A. Courts, Academic Press, London, 1977 Search PubMed; Biorelated Polymers and Gels, ed. T. Okano, Academic Press, San Diego 1998 Search PubMed.
- D. A. Kirschner, H. Inouye, L. K. Duffy, A. Sinclair, M. Lind and D. Selkoe, Proc. Natl. Acad. Sci. USA, 1987, 84, 6953–6957 CAS; S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. USA, 1993, 90, 3334–3338 CAS; A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS; J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. M. Pakstis and J. Gill, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS; K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 7146–7147 CrossRef CAS; J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS; C. Wang, R. J. Stewart and J. Kopeček, Nature, 1999, 397, 417–420 CrossRef CAS.
- Y. Zimenkov, V. P. Conticello, L. Guo and P. Thiyagarajan, Tetrahedron, 2004, 60, 7237–7246 CrossRef CAS.
- C. Robinson and J. C. Ward, Nature, 1957, 180, 1183–1184 CAS; C. Robinson, Tetrahedron, 1961, 13, 219–234 CrossRef CAS.
- N. J. Gay, L. C. Packman, M. A. Weldon and J. C. J. Barna, FEBS Lett., 1991, 291, 87–91 CrossRef CAS.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS.
- V. Breedveld, A. P. Nowak, J. Sato, T. J. Deming and D. J. Pine, Macromolecules, 2004, 37, 3943–3953 CrossRef CAS.
- E. R. Blout and R. H. Karlson, J. Am. Chem. Soc., 1956, 78, 941–946 CrossRef CAS; K. Tohyama and W. G. Miller, Nature, 1981, 289, 813–814 CrossRef CAS.
- A. P. Nowak, V. Breedveld, D. J. Pine and T. J. Deming, J. Am. Chem. Soc., 2003, 125, 15666–15670 CrossRef CAS.
- E. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. USA, 2001, 98, 11857–11862 CrossRef CAS.
- D. J. Pochan, L. Pakstis, B. Ozbas, A. P. Nowak and T. J. Deming, Macromolecules, 2002, 35, 5358–5360 CrossRef CAS.
- C. Tsitsilianis, I. Iliopoulos and G. Ducouret, Macromolecules, 2000, 33, 2936–2943 CrossRef CAS.
- E. Holowka, A. P. Nowak and T. J. Deming, unpublished results.
- L. J. Fetters, Encyclopedia of Polymer Science and Engineering, 2nd edn, Wiley-Interscience, New York, 1987, vol. 10, pp. 19–25 Search PubMed.
- T. Tanaka, Sci. Am., 1981, 244, 110–123; R. Dagani, Chem. Eng. News, 1997, 75, 26–37.
- E. Katchalski and M. Sela, Adv. Protein Chem., 1958, 13, 243–492 CAS; M. L. Tiffany and S. Krimm, Biopolymers, 1969, 8, 347–359 CAS.
- L. Richert, Ph. Lavalle, D. Vautier, B. Senger, J.-F. Stoltz, P. Schaaf, J.-C. Voegel and C. Picart, Biomacromolecules, 2002, 3, 1170–1178 CrossRef CAS; M. E. Carr, Jr., R. Cromartie and D. A. Gabriel, Biochemistry, 1989, 28, 1384–1388 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |