Nanoshells and nanotubes from block copolymers
I. W.
Hamley
Centre for Self-Organising Molecular Systems, University of Leeds, Leeds, LS2 9JT, UK. E-mail: I.W.Hamley@leeds.ac.uk
First published on 10th March 2005
Abstract
Recent work exploring the use of block copolymer vesicles and tubules is reviewed. The stability and toughness of block copolymer vesicles are enhanced compared to those formed by low molar mass amphiphiles. Functionality can also readily be introduced through the polymer chemistry or by incorporating additional components (for example pore-forming membrane proteins). This design flexibility leads to numerous potential applications in encapsulation, in targeted drug delivery, templating of inorganic materials and many others.
 I. W. Hamley | Ian Hamley, born in 1965, is a Professor of Polymer Materials and Director of the Centre for Self-Organising Molecular Systems at the University of Leeds. He received his BSc degree from the University of Reading in 1987 and PhD in 1991 from the University of Southampton. He was then Royal Society Leverhulme William and Mary postdoctoral fellow at the FOM-Institute for Atomic and Molecular Physics, Amsterdam. In 1992, he took up a postdoc position in the Dept of Chemical Engineering and Materials Science at the University of Minnesota. He returned to a lectureship in the Dept of Physics at the University of Durham in 1993, leaving to take up an appointment as lecturer in Physical Chemistry at the University of Leeds in 1996, where he was promoted to Senior Lecturer in 2000, to Reader in 2001, and to Professor in 2004. |
1. Introduction
Vesicles are spherical shell structures comprising a bilayer of amphiphiles, which may be low molar mass compounds (surfactants, lipids) or may be block copolymers. If there is a slight mismatch in the effective interfacial area per hydrophile compared to hydrophobe, the bilayer membrane can spontaneously close into a vesicle rather than forming a planar lamellar structure. This can be translated into a condition on the so-called surfactant packing parameter which is defined as p = V/al where V is the volume per molecule, a is the effective cross-sectional area per molecule and l is the chain length normal to the interface. Vesicles are observed for ½ < p < 1.1 This can also be expressed in terms of finite mean curvature.21.1 Notation
Block copolymers are indicated by an italic b, for example PS-b-PAA indicates a diblock of polystyrene and poly(acrylic acid). Other abbreviations used are listed in Table 1. Where a specific diblock is considered so that chain lengths are specified, the prefix “P” is omitted. For example S300-b-AA44 indicates a polystyrene-b-poly(acrylic acid) with degrees of polymerization 300 and 44 respectively for the two blocks. Note that we have used the terms PEG and PEO according to the notation used in the original research—we have not attempted to distinguish carefully between them (PEG differs from PEO by hydroxyl termination as opposed to methyl termination).| Abbreviation | Polymer/Systematic name (where used alternatively) |
|---|---|
| PAA | Poly(acrylic acid) |
| PB | Poly(butadiene) |
| PBMA | Poly(butyl methacrylate) |
| PCEMA | Poly(2-cinnamoyloxyethyl methacrylate) |
| PCL | Poly(ε-caprolactone) |
| PDMA | Poly((2-dimethamino)ethyl methacrylate) |
| PDMS | Poly(dimethylsiloxane) |
| PEE | Poly(ethylethylene) |
| PEO; PEG | Poly(ethylene oxide)/poly(oxyethylene); Poly(ethylene glycol) |
| PGMA | Poly(glyceryl monomethacrylate) |
| PHEMA | Poly(2-hydroxyethyl methacrylate) |
| PI | Polyisoprene |
| PIAT | Poly(L-isocyanoalanine-(2-thiophen-3-ylethyl)amide) |
| PLLA | Poly(L-lactic acid) |
| PMMA | Poly(methyl methacrylate) |
| PMOXA | Poly(2-methyloxazoline) |
| PPO; PPG | Poly(propylene oxide)/poly(oxypropylene); poly(propylene glycol) |
| PPQ | Poly(phenylquinoline) |
| PPS | Poly(propylene sulfide) |
| PS | Polystyrene |
| PSMA | Poly(solketal methacrylate) |
| PtBA | Poly(tert-butyl acrylate) |
| PtBS | Poly(tert-butyl styrene) |
2. Polymer vesicles—general aspects
Polymeric vesicles have advantages compared to conventional surfactant vesicles or liposomes in enhanced chemical, thermal and mechanical stability. Vesicle formation by block copolymers, leading to so-called “polymersomes”,3 has been the subject of several reviews.2,4,5 A wide variety of chemical approaches have been developed to produce polymeric vesicles using block copolymers, and conditions to prepare them have been reviewed.5 Vesicles have been prepared from diblocks containing a glassy component, such as PS-b-PEO and PS-b-PAA diblocks, intensively studied by Eisenberg and coworkers.5 In addition there are now numerous reports on PEO-based copolymers with an amorphous non-vitreous hydrophobic block such as commercial Pluronic-type PEO-b-PPO-b-PEO triblocks,6 PB-b-PEO diblocks7 and PEE-b-PEO diblocks.3Vesicles formed by low molar mass surfactants and amphiphiles are generally kinetically trapped, non-equilibrium structures.1 In contrast, it has been proposed that thermodynamically stable vesicles can be formed by diblock copolymers due to their intrinsic polydispersity.8,9 The polydispersity leads to selective segregation of short hydrophilic blocks to the inside of vesicles, whereas longer hydrophilic blocks segregate to the outside. The preferred curvature of the bilayer is stabilized in this way. The effect is enhanced for smaller vesicles, as expected since the tendency to segregate will be greater as interfacial curvature is increased.9 Fluorescence probe experiments on PS-b-PAA diblocks with varying hydrophilic block length, labelled with pyrene at the junction point, confirmed this hypothesis.8,9 Earlier data from this group had shown that vesicle size could be changed reversibly by varying solvent composition, pointing towards thermodynamic stability.10,11Fig. 1 shows TEM data illustrating reversibility of vesicle formation and growth process for a diblock in aqueous solution.9 Bilayer fluidity is also required to reach equilibrium, and this can be achieved using suitable fluidizing solvents which enable the flip-flop exchange of copolymer chains.4
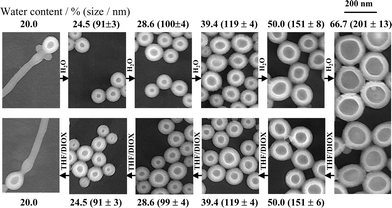 | ||
| Fig. 1 TEM images showing reversibility of vesicle formation and growth process upon changing water content for diblock PS300-b-PAA44 in a mixture of water + THF/dioxane.9 | ||
The vesicle shell can be cross-linked to create a hollow sphere, as discussed in section 6. Hollow tube structures have been prepared from core–shell cylindrical micelles. Hollow shells and tubes have numerous potential applications, especially in targeted delivery of solubilized compounds and these are reviewed here. The use of block copolymer assemblies as synthetic cell elements has been discussed.12 Vesicle bilayers are structurally analogous to cell membranes, and the analogy may be further developed by the incorporation of membrane proteins, as discussed in section 7. It should be noted that the present review is focussed mainly on block copolymer vesicles—information on micellar structures can be found elsewhere.13–16
Nanoshell structures have been observed for several rod–coil systems. PPQ-b-PS diblocks have been investigated, with a view to exploiting the photoactive and electroactive properties of the π-conjugated PPQ block.17 The self-assembly of PPQ-b-PS rod–coil diblocks in selective solvents for the PPQ rod block17 or the PS18 coil block results in very large aggregates (typically 3–5 µm across). The large size results from the formation of hollow structures (the fully extended length of the polymer chains is much smaller than the observed aggregate dimensions). For example, hollow micelles with a monolayer shell (Fig. 2), instead of a bilayer as in a vesicle, were formed as a result of the molecular shape. In a PPQ-selective solvent, a range of aggregate structures was observed, their size decreasing with decreasing fraction of rod block. Spherical aggregates were able to solubilize large molecules such as fullerenes. In a selective solvent for the PS block, hollow spherical aggregates were observed which formed ordered arrays in two and three dimensions when dried.18 Due to the size scale of the particles, iridescence was observed as a result of the spatial variation of refractive index. Potential applications as photonic band gap materials were highlighted. Giant vesicles have been prepared by electroformation using a diblock copolymer containing a thiophene-containing rod block.19,20 The thiophene units are attached laterally to the backbone which is helical, and stack on top of each other due to hydrogen bonding between amide units. Multilamellar vesicles were formed by these PS-b-PIAT diblocks with sizes up to 100 µm. The thiophene groups are polymerizable, enabling the rigidity of the vesicles to be controlled.
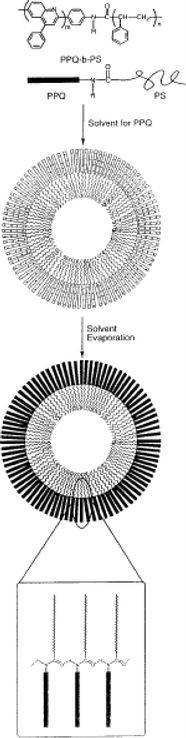 | ||
| Fig. 2 Schematic showing self-assembly of a PPQ-b-PS diblock into a hollow microsphere.17 | ||
Nanotubes (25 nm diameter, ∼µm length) have been prepared from giant diblock amphiphiles that comprise an end functionalized polystyrene chain reacted in situ with the disulfide moiety within a lipase protein.21 This is preliminary work, and it is likely that this type of approach will yield other interesting protein-coated nanostructures.
Since they are generally non-equilibrium structures, the preferred route to break up lamellar structures into vesicles is to apply shear. A number of studies have focussed on this process for block copolymers, as discussed in the following section.
3. Shear-induced vesicle formation
Unilamellar vesicles have been observed in dilute solutions of the PEO-b-PPO-b-PEO triblock with the commercial name Pluronic L121 in the two phase region between lamellar and micellar liquid phases.22 The high hydrophobe content in this copolymer is believed to favour the formation of vesicles, which were imaged by cryo-TEM. Later, Richtering et al. observed the shear-induced formation of multilamellar vesicles (“onions”) in the dilute lamellar phase formed in binary solvent (butanol–water) solutions of Pluronic P123 and F127.6,23 The presence of the cosolvent increased the interfacial area per block copolymer molecule and facilitated the mobility of molecules between different layers, both contributing towards an increased tendency for curvature of dilute lamellae into vesicles. Comparison was made to the shear-induced reorientation of a lamellar phase in the same system at higher polymer concentration.6 In this case the viscosity decreased steadily with increasing shear rate. In a more dilute system, increasing shear rate led to an increase in viscosity up to![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ∼ 1, followed by shear thinning at higher shear rates. The increase in viscosity signalled the formation of vesicles, as confirmed by SANS and SALS.
∼ 1, followed by shear thinning at higher shear rates. The increase in viscosity signalled the formation of vesicles, as confirmed by SANS and SALS.
4. Vesicles with internal structure
Compartmentalized vesicles or micelles are of interest due to the analogies with the partitioning of the cell into compartments surrounded by lipid membranes. The formation of aggregate structures containing internal arrays of tubes was observed for a PS-b-PAA diblock (S410-b-AA13), initially dissolved in DMF and then dialysed against water.24 Since different projections of the structure were imaged by TEM, a hexagonally-packed hollow hoop morphology could be proposed. Giant compound vesicle structures have also been reported for PB-b-PEO diblocks in aqueous solution.7 Polymersomes comprising a bilayer containing a lattice of passages or a network of tubules were observed. The formation of such high genus objects was analysed in terms of the elastic properties of copolymer membranes. Examples of optical micrographs and calculated membrane surfaces are shown in Fig. 3. | ||
| Fig. 3 Optical micrographs for compound vesicles formed by PB-b-PEO diblocks in aqueous solution (a, c, e) and calculated membrane surfaces (b, d, f, g).7 The scale bar represents 10 µm. The polymersomes contain (a) small passages, (b) large passages, (e) budded vertices. | ||
Porous polymer microspheres have been prepared by using PI-b-PAA diblocks to solubilise chloromethane containing PI-b-PtBA and homopolymer PtBA as oil droplets in water.25 The PtBA was then hydrolysed and extracted to leave porous spheres (diameter 0.3–1 µm). The shape and connectivity of pores could be controlled through the relative content of PtBA in the homopolymer vs. diblock.
5. Mechanical properties and micropipette aspiration
Due to their enhanced toughness and rigidity, block copolymer polymersomes are much more robust than conventional liposomes. The ability to cross-link the chains of course enables significant additional enhancement in mechanical properties, as discussed below.Polymer vesicles or polymersomes based on PEO-b-PEE diblocks in aqueous solution (Fig. 4) show greater toughness than conventional phospholipid vesicles, although the bending and area expansion moduli are comparable.3 The enhanced toughness (defined as the integral of the tension with respect to areal strain) and reduced permeability of the polymersomes could lead to applications in encapsulation. The elastic behaviour was examined via micropipette aspiration of vesicles generated by electroformation (Fig. 5). In this process, giant vesicles (20–50 µm in radius) are prepared from a thin film of polymer on adjacent electrodes subjected to an alternating current. Giant vesicles were also prepared by micropipette aspiration of PEO-b-PB diblocks.26 The interfacial viscosity and elasticity have been measured.26 The surface shear viscosity is about 500 times larger than that found for common phospholipid vesicles. On the other hand, the bending and stretching elastic constants are similar to those for lipid membranes. By pulling out a tether from the vesicle and monitoring its relaxation, it was possible to study the viscous coupling between the two monolayers comprising the polymeric membrane. The corresponding friction coefficient was about an order of magnitude larger than that found for typical fluid phospholipid membranes. The bending rigidity constant, kc, was measured via single and dual pipette aspiration techniques for PEO-b-PB diblocks.27 For a diblock with a lengthy hydrophobic block (B125), kc = 466 ± 157 kBT was more than an order of magnitude larger than values for typical lipids or a PEO-b-PB diblock with a short B46 block, where values range from 13–25 kBT. A quadratic scaling of kc with hydrophobic layer thickness, d, was reported, in agreement with the theoretical expression28,29kc = βKAd2. Here KA is the area elastic modulus and β is a constant, for which values have been calculated depending on whether the bilayer contains coupled or interdigitated monolayers.27
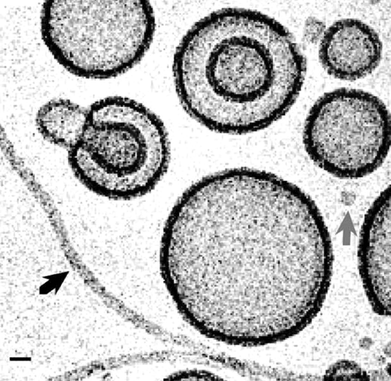 | ||
| Fig. 4 Cryo-TEM image of PEO-b-PEE diblock copolymer vesicles in water.3 The scale bar indicates 20 nm. | ||
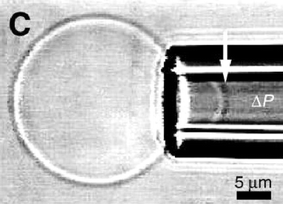 | ||
| Fig. 5 Aspiration of a polymer vesicle into a micropipette. The arrow marks the tip of a projection of a vesicle being sucked in by a negative pressure ΔP.3 | ||
An adaptation of the micropipette aspiration technique involves simultaneous application of voltage pulses to tensioned membranes, with the aim to rupture the membranes in a process termed electroporation.30 Polymersome membranes are able to withstand voltage pulses much higher than those at which lipid membranes rupture. Increasing the mechanical tension reduces the rupture voltage in a parabolic fashion. This can be understood using existing models for interfacial thermodynamics.30 The post-poration dynamics of high molecular weight diblock membranes is however significantly retarded.12 The formation of pores induced by puncturing giant vesicles with a sharp tip,31 or the extrusion of tubes from giant vesicles has been studied for giant unilamellar phospholipid vesicles,32 and extension of these techniques to examine the deformation behaviour and hydrodynamics of block copolymer vesicles should follow.
The diffusion of probe molecules within PEO-b-PEE and PEO-b-PB polymersome membranes has been studied by FRAP (fluorescence recovery after photobleaching).33 Diffusivity decreases with polymer molar mass, as the membrane rigidity increases. The decrease is particularly marked when the chains become entangled. A transition from Rouse dynamics to reptation dynamics is observed as polymer molar mass increases sufficiently for chains to become entangled. In the Rouse regime the total hydrodynamic friction on a chain is just the accumulated friction ζ on each of the N subsegments, DRouse = KBT/Nζ. The friction factors obtained are consistent with those expected based on composition, and extrapolate to those obtained for the polymer melt.
6. Cross-linked vesicles
Ding and Liu have investigated a number of routes to the preparation of hollow polymer nanospheres for controlled release applications using cross-linked block copolymer vesicles.34 They prepared PI-b-PCEMA vesicles in THF–hexane solutions. The PCEMA shell was photo-crosslinked.34 The PI chains were then hydroxylated to give water soluble vesicles with a hydrophilic coating on both sides of the shell. These were used to solubilize a dye compound, the release of which was studied via measurements of fluorescence intensity. The potential of nanocapsules with a cross-linked diblock shell for drug delivery applications was highlighted.34 The enhanced stability of cross-linked nanoparticles (for example in the bloodstream) and the ability to tune the capsule size and to incorporate responsive and/or functional moieties are additional advantages offered by the use of polymers. In an alternative approach, micelles of PSMA-b-PDMA were formed in aqueous solution with PDMA coronas and PSMA cores.35 The PDMA coronas were then chemically cross-linked. Finally the cores of the nanospheres were made hydrophilic by hydrolysis of the nitroxide groups in PSMA. It should be noted that this approach does not produce hollow spheres, and it is not clear how the uptake and release of hydrophilic compounds across the cross-linked shell can be decoupled.Methods to prepare hollow nanospheres have also been developed for ABC triblocks. PI-b-PCEMA-b-PtBS micelles were formed in THF–methanol mixtures.36 The PCEMA shell was cross-linked, and the PI core removed by ozonolysis to leave PCEMA shells with a PtBS coating. Extending this chemistry to create tetrablock copolymers, it was possible to fabricate hollow nanotubes which were used as templates to create polymer–Pd hybrid nanofibres.37 Tetrablock PI-b-PtBA-b-poly(CEMA-ran-HEMA)-b-PGMA self-assembled into cylindrical aggregates in aqueous solution. The core comprised core–shell cylinders of PI surrounded by PtBA then P(CEMA-ran-HEMA). The PGMA formed the water soluble corona. The process used to prepare nanotubes is illustrated in Fig. 6. The P(CEMA-ran-HEMA) layer was cross-linked followed by ozone etching to remove the PI core to form nanotubes. An alternative approach has been used by this group in which bulk nanostructures formed by an ABC triblock are fixed by shell cross-linking.38 The nanocylinders formed are then dispersed in solvent. PBMA-b-PCEMA-b-PtBA triblocks formed cylinders with PtBA cores and PCEMA shells in a PBMA matrix. The PCEMA was photochemically cross-linked, prior to dissolving the polymer film in THF to produce nanofibers. The PtBA units were finally hydrolysed to PAA to produce PAA-lined nanotubes. It was shown that the nanotubes could be loaded with Fe2O3 due to ion exchange of Fe2+ with the protons of the acrylic groups. The concept of fixing bulk structures formed by ABC triblocks and then dissolving in a selective solvent was used earlier to prepare Janus micelles.39 These are micelles with two different faces. Spherical Janus micelles were prepared from an ABC triblock microphase separated in the melt into a so-called “ball at the wall” morphology, comprising spheres of the minority PB block at the interface between PS and PMMA lamellae. The PB domain was cross-linked, allowing the structure of PS and PMMA hemispheres to be retained after dissolution.39
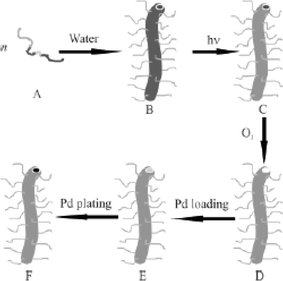 | ||
| Fig. 6 Schematic showing route to preparation of Pd nanotubes from a PI-b-PtBA-b-poly(CEMA-ran-HEMA)-b-PGMA tetrablock.37 The tetrablock (A) is dissolved in water to form cylindrical micelles (B). The PCEMA was photochemically cross-linked (C), prior to dissolving the polymer film in the THF to produce nanofibers. The PI was removed by ozonolysis (D) and the resulting nanotubes loaded with PdCl2 (E). The salt was then reduced to produce Pd nanoclusters (F). | ||
In contrast to the two-stage procedure used by Ding and Liu (cross-linking of the shell plus hydrolysis or ozone etching of the core), Meier and coworkers have pioneered a one-step route to prepare nanoshells from block copolymer vesicles.40,41 Vesicles were formed upon dispersion of an ethanolic solution of PMOXA-b-PDMS-b-PMOXA triblocks into aqueous solution. The PMOXA shell bears polymerizable end groups which can then be photo cross-linked,40 as illustrated in Fig. 7. The resulting nanocapsules are mechanically stable, and also retain their shape upon dispersion in organic solvents. Free-standing planar membranes with areas of approx. 1 mm2 were also prepared using this chemistry, and the enhanced mechanical properties (stability against electric field-induced rupture) of these films were compared to those of lipid bilayers.42
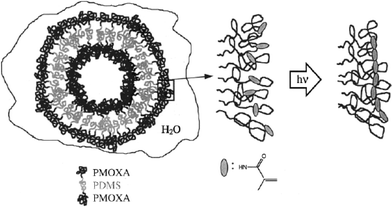 | ||
| Fig. 7 Schematic showing polymerization of PMOXA-b-PDMS-b-PMOXA block copolymer vesicles.40 | ||
Wang et al.43 have prepared vesicles from “diblocks” in which P4VP is hydrogen bonded to hydroxyl-containing polystyrene random copolymer, denoted PS(OH). The P4VP shell was cross-linked and the PS(OH) polymer in the core was dissolved.
7. Functionalised and biocompatible vesicles
In a beautiful concept demonstration, pore-forming transmembrane proteins have been incorporated within triblock copolymer planar membranes44 and vesicles (Fig. 8).45 The functionality of the bacterial porin OmpF is retained after their incorporation into the block copolymer membrane. When incorporated into the membrane, this protein forms channels that exclude molecules with molecular weights above 400 Da. Responsive nanocapsules can be fabricated containing these channels, and these have great potential for targeted drug delivery and diagnostics.41 It has further been shown that such porous nanocapsules can be used to prepare calcium phosphate nanoparticles, where the Ca2+ is transported across the shell through the ion channels.46 Another application uses the cross-linked vesicles as nanoreactors. The enzyme β-lactamase was encapsulated and used to hydrolyse ampicillin, which is a β-lactam antibiotic.45,47 The concept is illustrated in Fig. 8. Application of a transmembrane voltage above a certain threshold causes a reversible gating action of the OmpF.45 Polyelectrolyte added to the solution was shown to trigger the gating transition. The Donnan equilibrium established leads to a concentration difference of polyelectrolyte chains either side of the membrane. If this exceeds the gating potential, the channels are closed and the nanoreactor is deactivated. Incorporation of the bacterial channel forming protein LamB into block copolymer vesicle membranes enables DNA to be transported into the nanocontainer, since LamB serves as a receptor for λ phage to trigger the ejection of λ phage DNA.48 If the compositional asymmetry is large enough, PMOXA-b-PDMS-b-PMOXA triblocks self-assemble into hollow nanotubes in aqueous solution (Fig. 9).49 The tube diameter is relatively uniform, being 40 nm, with a length extending to several tens of microns.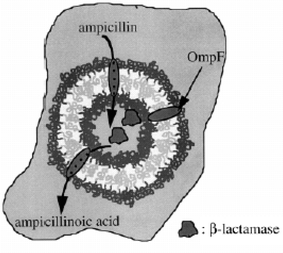 | ||
| Fig. 8 Nanoreactor comprising a cross-linked block copolymer vesicle encapsulating the enzyme β-lactamase which catalyses the hydrolysis of ampicillin.47 | ||
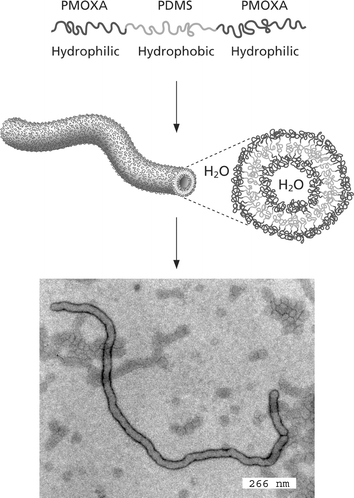 | ||
| Fig. 9 Nanotube formation using an asymmetric PMOXA-b-PDMS-b-PMOXA triblock in aqueous solution.49 The bottom picture is a TEM image of a nanotube, schematically shown above. | ||
The use of PEG-b-PEE polymersomes in protein encapsulation has been comprehensively studied.50 The vesicles exhibit good long-term stability in blood plasma, i.e. phagocytosis is inhibited. Cell viability was also shown not to be adversely affected by addition of the polymersomes. In contrast to liposomes, the block copolymer vesicles can withstand harsh thermal treatments to some extent although the vesicles shrink and leak their contents. It was proposed that these properties result from steric stabilization conferred by PEG. Compared to PEG-lipids, the polymersomes offer much improved mechanical stability and stability against dilution.
Triblock copolymer vesicles which are unstable to oxidation have been prepared with the aim of developing a responsive system for drug delivery.51,52 PEG-b-PPS-b-PEG triblocks were used, PEG being hydrophilic and poly(propylene sulfide) being hydrophobic but readily oxidised to poly(propylene sulfoxide) and ultimately poly(propylene sulfone), both of which are hydrophilic. This conversion results in the breakup of the vesicles. The resulting individual copolymer chains were short enough to be capable of elimination from the body.
Biodegradable polymersomes have been reported based on PEG-b-PLLA diblocks.53 The poration of PEG-b-PLLA and PEG-b-PCL vesicles (and mixed vesicles) due to hydrolysis has been investigated.54 Encapsulation of the anti-cancer agent doxorubicin was also studied. Release kinetics from giant vesicles were studied for model hydrophilic fluorescent encapsulants via phase contrast and fluorescence microscopy. The release (over a time scale from hours to days) was found to occur in a quantized manner, as each vesicle porates and disintegrates individually. A mechanism for the poration process is illustrated in Fig. 10. Release kinetics can be controlled via the PEG content of the copolymer and the hydrophobicity of the other block (PCL is more hydrophobic than PLLA).
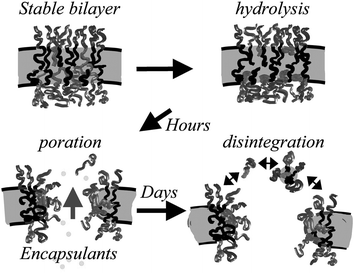 | ||
| Fig. 10 Mechanism proposed for membrane poration due to hydrolysis of polyester in PEG-b-polyester vesicles.54 Polyester chains are shown in light grey. | ||
8. Peptosomes
Polymer vesicles formed by block copolymers containing polypeptide blocks, i.e. so-called “peptosomes”, have been developed based on PB-b-poly(γ-L-glutamic acid) (Fig. 11).55–57 In the study by Kukula et al., the helix–coil transition of the peptide does not change the dimensions of the peptosome. In contrast, Checot et al. showed that the size of their peptosomes could be reversibly manipulated as a function of both pH and ion strength. The vesicle hydrodynamic radius increased by 50% when the peptide underwent a transition from an α-helix to a random coil conformation, driven by increasing pH.56,57 These authors also demonstrated UV-induced cross-linking of the PB to produce “shape persistent stimuli-responsive nanocapsules”. Block copolymers containing two distinct peptide blocks have also been shown to self-assemble into peptosomes.58 Poly(Nε-2-(2-(2-methoxyethoxy)ethoxy)acetyl-L-lysine)-b-poly(L-lysine) diblocks self-assembled into giant unilamellar vesicles. Vesicle size could be controlled by adjustment of hydrophobic content or overall copolymer chain length. No release of solubilized dye was observed, over a period of weeks. A copolymer was prepared in which 70% of the L-leucine residues of the hydrophobic domain of the block copolymer were replaced in a statistical manner with L-lysine. At high pH, poly(L-lysine) is not water soluble and it adopts an α-helical conformation. Vesicles similar to those formed by the unmodified copolymers were observed. However, a helix-to-coil transition driven by a reduction in pH caused destabilization of the vesicles and release of their contents. Thus pH-responsiveness was demonstrated. The co-operative self-assembly of poly(L-lysine)-b-poly(L-cysteine) block copolymers in the presence of silica and gold nanoparticles can be used to prepare hollow spheres coated with a shell of gold and a shell of silica (Fig. 12).59 The cysteine block facilitates disulfide links and these act as attachment points for gold nanoparticles. Introduction of silica nanoparticles then drove the formation of a silica coating. These hollow nanoshells were formed without the use of emulsions or a sacrificial core.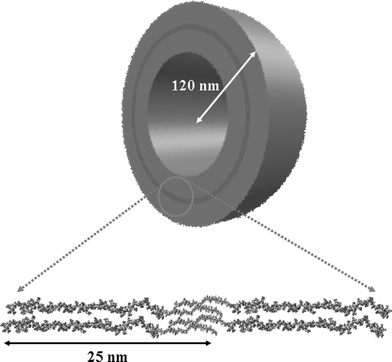 | ||
| Fig. 11 Proposed structure of a PB-b-poly(γ-L-glutamic acid) vesicle.56 | ||
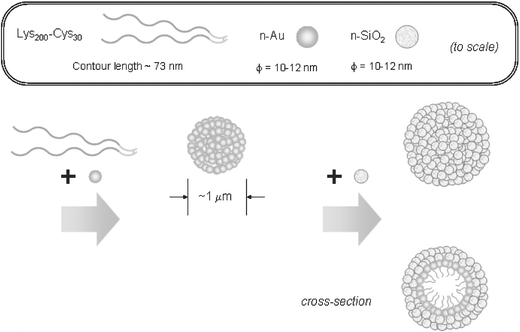 | ||
| Fig. 12 Schematic suggested for the hierarchical self-assembly of gold and silica nanoparticles into hollow spheres with a two-layer shell structure, using poly(L-lysine)-b-poly(L-cysteine) block copolymers.59 | ||
9. Summary
This review has highlighted the advantages of block copolymers in the preparation via self-assembly of hollow nanostructures. These offer great potential in fabricating relatively rigid and chemically inert structures with controlled dimensions up to the µm scale. Aspects of polymer chemistry such as the ability to cross-link the nanoshells or nanotubes, or the ability to functionalize the bilayer, either by incorporation of membrane proteins or using suitable biocompatible or biomimetic polymer components, offer exciting prospects in many applications. These systems will in the future be used in imaginative ways to fabricate tailored and responsive systems for drug delivery, gene therapy and other medical applications and to produce nanoreactors to template inorganic materials (metals, silica and other oxides etc.). We are particularly excited by the prospects to fabricate multi-layer structures using multiblock (ABC, ABCD…) copolymers (see contents entry) or by the use of mixed copolymers, possibly exploiting electrostatic, hydrogen-bonding or other noncovalent interactions.References
- I. W. Hamley, Introduction to Soft Matter, John Wiley, Chichester, 2000 Search PubMed.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143 CrossRef.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci. B: Polym. Phys., 2004, 42, 923 CrossRef CAS.
- J. Zipfel, P. Lindner, M. Tsianou, P. Alexandridis and W. Richtering, Langmuir, 1999, 15, 2599 CrossRef CAS.
- C. K. Haluska, W. T. Gozdz, H.-G. Döbereiner, S. Förster and G. Gompper, Phys. Rev. Lett., 2002, 89, 238302 CrossRef.
- L. Luo and A. Eisenberg, J. Am. Chem. Soc., 2001, 123, 1012 CrossRef CAS.
- L. Luo and A. Eisenberg, Langmuir, 2001, 17, 6804 CrossRef CAS ; erratum L. Luo and A. Eisenberg, Langmuir, 2002, 18, 1952 Search PubMed.
- H. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473 CrossRef CAS.
- H. Shen and A. Eisenberg, Macromolecules, 2000, 33, 2561 CrossRef CAS.
- P. Dalheimer, F. S. Bates, H. Aranda-Espinoza and D. E. Discher, C. R. Phys., 2003, 4, 251 Search PubMed.
- I. W. Hamley, The Physics of Block Copolymers, Oxford University Press, Oxford, 1998 Search PubMed.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef.
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688 CrossRef CAS.
- I. W. Hamley, Block Copolymers in Solution, Wiley, 2005, in press Search PubMed.
- S. A. Jenekhe and X. L. Chen, Science, 1998, 279, 1903 CrossRef CAS.
- S. A. Jenekhe and X. L. Chen, Science, 1999, 283, 372 CrossRef CAS.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772 CrossRef CAS.
- D. M. Vriezema, A. Kros, R. de Gelder, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Macromolecules, 2004, 37, 4736 CrossRef CAS.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224 CrossRef CAS.
- K. Schillén, K. Bryshke and Y. S. Mel'nikova, Macromolecules, 1999, 32, 6885 CrossRef CAS.
- J. Zipfel, J. Berghausen, G. Schmidt, P. Lindner, P. Alexandridis, M. Tsianou and W. Richtering, Phys. Chem. Chem. Phys., 1999, 1, 3905 RSC.
- L. Zhang, C. Bartels, Y. Yu, H. Shen and A. Eisenberg, Phys. Rev. Lett., 1997, 79, 5034 CrossRef CAS.
- Z. Lu, G. Liu and F. Liu, J. Appl. Polym. Sci., 2003, 90, 2785 CrossRef CAS.
- R. Dimova, U. Seifert, B. Pouligny, S. Förster and H.-G. Döbereiner, Eur. Phys. J. E, 2002, 7, 241 Search PubMed.
- H. Bermúdez, D. A. Hammer and D. E. Discher, Langmuir, 2004, 20, 540 CrossRef CAS.
- W. Helfrich, Z. Naturforsch., C: Biosci., 1975, 30, 841 CAS.
- E. Evans, Biophys. J., 1975, 14, 923.
- H. Aranda-Espinoza, H. Bermudez, F. S. Bates and D. E. Discher, Phys. Rev. Lett., 2001, 87, 208301 CrossRef CAS.
- O. Sandre, L. Moreaux and F. Brochard-Wyart, Proc. Natl. Acad. Sci. USA, 1999, 96, 10591 CrossRef CAS.
- N. Borghi, O. Rossier and F. Brochard-Wyart, Europhys. Lett., 2003, 64, 837 CrossRef CAS.
- J. C.-M. Lee, M. Santore, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 323 CrossRef CAS.
- J. Ding and G. Liu, J. Phys. Chem. B, 1998, 102, 6107 CrossRef CAS.
- Z. Zhang, G. Liu and S. Bell, Macromolecules, 2000, 33, 7877 CrossRef CAS.
- S. Stewart and G. Liu, Chem. Mater., 1999, 11, 1048 CrossRef CAS.
- Z. Li and G. Liu, Langmuir, 2003, 19, 10480 CrossRef CAS.
- X. Yan, F. Liu, Z. Li and G. Liu, Macromolecules, 2001, 34, 9112 CrossRef CAS.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-Hintzen, G. Krausch, V. Abetz and A. H. E. Müller, Macromolecules, 2001, 34, 1069 CrossRef CAS.
- C. Nardin, T. Hirt, J. Leukel and W. Meier, Langmuir, 2000, 16, 1035 CrossRef CAS.
- M. Sauer and W. Meier, in Colloids and Colloid Assemblies, ed. F. Caruso, Wiley-VCH, Weinheim, 2004 Search PubMed.
- C. Nardin, M. Winterhalter and W. Meier, Langmuir, 2000, 16, 7708 CrossRef CAS.
- M. Wang, M. Jiang, F. Ning, D. Chen, S. Liu and H. Duan, Macromolecules, 2002, 35, 5980 CrossRef CAS.
- W. Meier, C. Nardin and M. Winterhalter, Angew. Chem., Int. Ed., 2000, 39, 4599 CrossRef.
- C. Nardin, J. Widmer, M. Winterhalter and W. Meier, Eur. Phys. J. E, 2001, 4, 403 Search PubMed.
- M. Sauer, T. Haefele, A. Graff, C. Nardin and W. Meier, Chem. Commun., 2001, 2452 RSC.
- C. Nardin, S. Thoeni, G. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433 RSC.
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. USA, 2002, 99, 5064 CrossRef CAS.
- J. Grumelard, A. Taubert and W. Meier, Chem. Commun., 2004, 1462 RSC.
- J. C.-M. Lee, H. Bermudez, B. M. Discher, Y.-Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135 CrossRef CAS.
- A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nature Mater., 2004, 3, 183 CrossRef CAS.
- A. Napoli, M. J. Boerakker, N. Tirelli, R. J. M. Nolte, N. A. J. M. Sommerdijk and J. A. Hubbell, Langmuir, 2004, 20, 3487 CrossRef CAS.
- F. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37 CrossRef CAS.
- H. Kukula, H. Schlaad, M. Antonietti and S. Förster, J. Am. Chem. Soc., 2002, 124, 1658 CrossRef CAS.
- F. Chécot, S. Lecommandoux, Y. Gnanou and H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1340.
- F. Chécot, S. Lecommandoux, H.-A. Klok and Y. Gnanou, Eur. Phys. J. E, 2003, 10, 25 Search PubMed.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nature Mater., 2004, 3, 244 CrossRef CAS.
- M. S. Wong, J. N. Cha, K.-S. Choi, T. J. Deming and G. D. Stucky, Nano Lett., 2002, 2, 583 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |