
Bulk COFs and COF nanosheets for electrochemical energy storage and conversion
Jie
Li†
,
Xuechun
Jing†
,
Qingqing
Li
,
Siwu
Li
,
Xing
Gao
,
Xiao
Feng
 * and
Bo
Wang
*
* and
Bo
Wang
*
Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science, Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: bowang@bit.edu.cn; fengxiao86@bit.edu.cn
First published on 5th May 2020
Abstract
Covalent organic frameworks (COFs) as an emerging class of crystalline porous materials have received much attention due to their tunable porosity, modifiable skeletons, and atomically precise structures. Besides, COFs can provide multiple high-rate charge carrier transport (electron, hole, and ion) pathways, including conjugated skeletons, overlapped π electron clouds among the stacked layers, and open channels with a variable chemical environment. Therefore, they have shown great potential in electrochemical energy storage (EES) and conversion (EEC). However, in bulk COFs, the defects always impede charge carrier conduction, and the difficulties in reaching deep-buried active sites by either electrons or ions lead to limited performance. To overcome these obstacles, numerous research studies have been carried out to obtain COF nanosheets (NSs). This review first describes the preparation strategies of COF NSs via bottom-up and top-down approaches. Then, the applications of bulk COFs and COF NSs in EES and EEC are summarized, such as in batteries, supercapacitors, and fuel cells. Finally, key challenges and future directions in these areas are discussed.
 Jie Li | Jie Li received her Bachelor's degree from Wuhan University of Technology in 2016. She then joined Prof. Bo Wang's group as a doctoral student at the School of Chemistry and Chemical Engineering, Beijing Institute of Technology. Her research interests include design, synthesis, and functional application of novel metal–organic frameworks and covalent organic frameworks. |
 Xuechun Jing | Xuechun Jing received her Bachelor's degree from Qingdao University of Science and Technology in 2018 and then joined Prof. Bo Wang's group as a MS student at the School of Chemistry and Chemical Engineering, Beijing Institute of Technology. Her current research focuses on the synthesis and application of covalent organic framework films. |
 Xiao Feng | Xiao Feng received his BS degree in materials chemistry in 2008 and PhD degree in materials science in 2013 from Beijing Institute of Technology. He pursued his study in Japan as a joint PhD student at the Institute for Molecular Science (2009–2012). He is now a professor at Beijing Institute of Technology and is mainly engaged in the research of crystalline porous materials (e.g., covalent organic frameworks and metal–organic frameworks) and their applications in membrane separation. |
 Bo Wang | Bo Wang obtained his BS and MS degrees from Peking University in 2004 and the University of Michigan in 2006, respectively, and his PhD from the University of California Los Angeles in 2008. He has been a professor in the School of Chemistry and Chemical Engineering, Beijing Institute of Technology, since 2011. His research interests focus on novel functional porous materials design and synthesis, energy storage, advanced battery materials, gas purification, toxicant capture, and sensing. |
1. Introduction
Demand for continued energy supply is one of the tremendous challenges that we are facing today due to the disparity between increasing energy requirements and worldwide energy shortage. Electrochemical energy conversion (EEC) and storage (EES) are represented as the most effective technologies for the utilization of energy. The EEC techniques can convert sustainable energy sources (e.g., wind energy and solar energy) into transportable and stable chemical energy.1–4 To obtain higher energy densities and energy conversion efficiency, developing novel materials with high performance is of vital importance.5–13Covalent organic frameworks (COFs), assembled by covalent bonds through reversible reactions, have the advantage of high porosity,14,15 adjustable and periodic pores,16–19 well-defined structures,20,21 and functional skeletons.22,23 The reversibility of the polymerization reaction allows “error corrections” to ensure the obtaining of thermodynamically stable networks with long-range order.20,24 COFs have been widely applied in storage and sorption,25–28 separation,29–32 catalysis,33–39 sensing,40–42 optoelectronics,43–47 and so on. The first COFs were synthesized by Yaghi and co-workers in 2005.48 The successful preparation of COFs has overcome the “crystallization problem”, i.e. the covalent bonding of building blocks often produces amorphous or poorly crystalline polymeric materials, and makes it possible to design and prepare polymers with highly ordered and predictable primary, secondary, and tertiary structures based on the reticular chemistry principle. Furthermore, the recent achievement of producing high-quality single crystals enables the deciphering of their structures with atomic precision.49,50
COFs can be categorized into two dimensional (2D) and three dimensional (3D) COFs depending on the geometric symmetry of the building blocks (Fig. 1).51–55 In 2D COFs, the organic units are covalently bonded and restricted in 2D sheets that further stack to form a layered structure via π–π interaction. Most of them are stacked in eclipsed mode with the formation of periodically aligned channels, while very few of them are stacked in a staggered fashion.15,51 Both stacking modes show well-defined arrangements. For 3D COFs, their building blocks including an sp3 carbon, silane, or boron atom help to expand the network to three-dimensional space. Furthermore, the diversity of building blocks creates numerous combinations, which endows the COFs with immense possibilities in structural design (Fig. 2).56
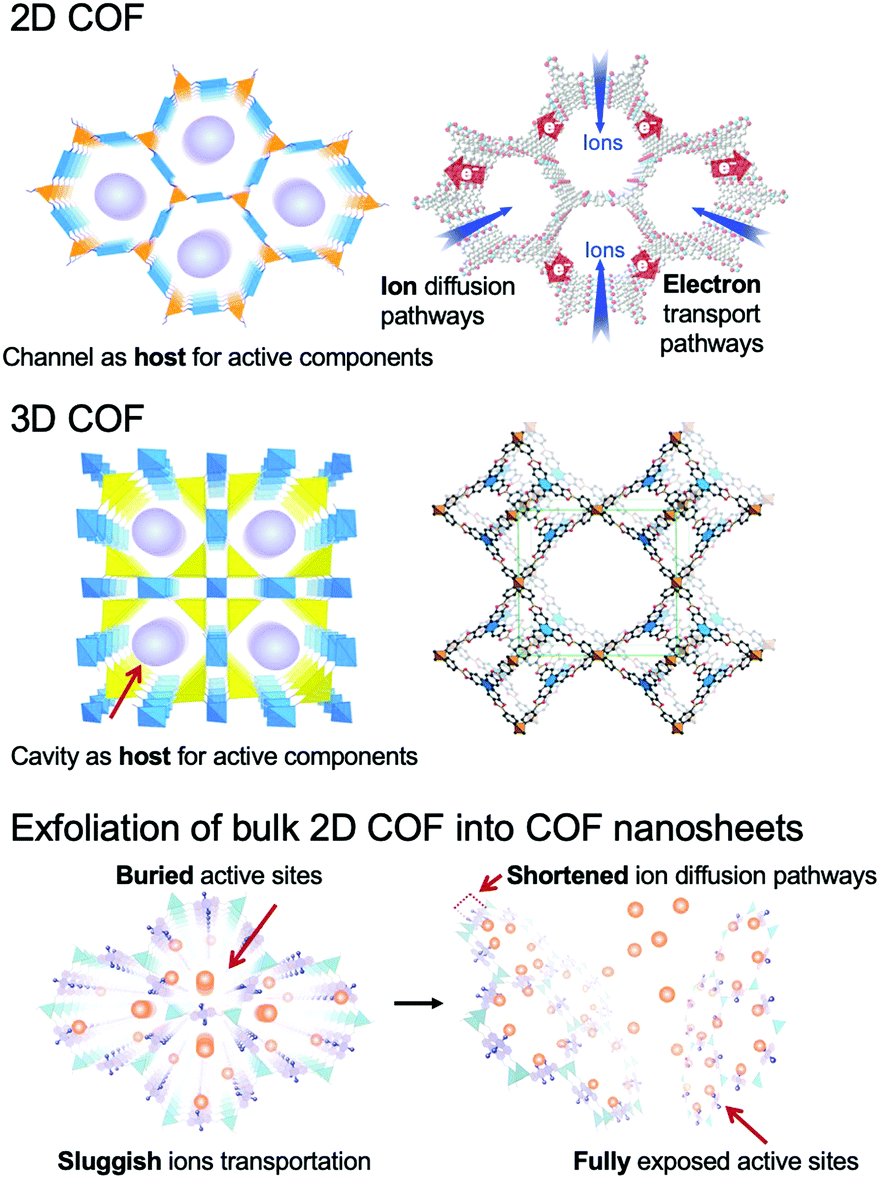 | ||
| Fig. 1 Schematic representation of 2D COFs, 3D COFs, and the exfoliation of the bulk 2D COF into COF nanosheets. The atomic connectivity and structure of 3D COFs (right) is reproduced from ref. 53 with permission from American Association for the Advancement of Science, copyright 2007. | ||
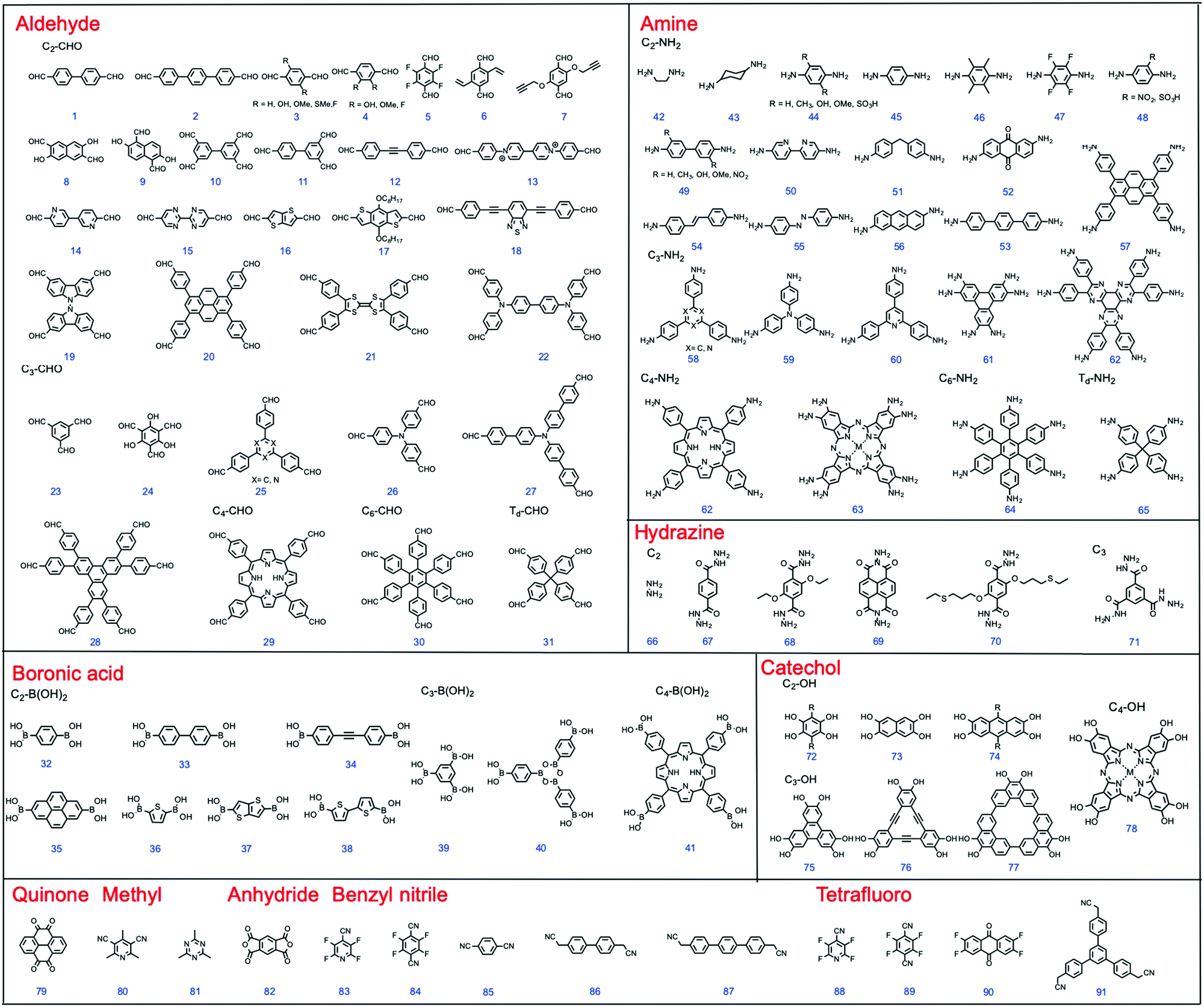 | ||
| Fig. 2 Chemical structures of building blocks with different geometries. | ||
The unique structural features of COFs endow them with great advantages and potential for application in EES and EEC. (a) Periodic arrangement. The redox/catalytic active sites can be accurately anchored to specific sites on the skeleton with the predesigned environment, and their electronic structures can be easily tuned, offering great opportunities for tailoring the performance of COFs and serving as platforms for the mechanism and structure–function relationship study. Moreover, in 2D COFs, vertical columnar arrays formed by the stacking of aromatic building units as well as conjugated polymeric layers can provide continuous channels for the hole and/or electron movement.57–60 In 3D COFs, the 1D conjugated segments interlace with each other and construct long-range conjugated systems that may also provide transport pathways. (b) Porous framework with well-aligned channels. Normally, both 2D and 3D COFs have the advantages of modified skeletons, multiple open sites, and a highly porous structure with large pore volume. Because of these advantages, COFs provide the possibility of incorporating active fragments into their skeletons or impregnating active molecules within their channels. The ability for selective adsorption or enrichment of particular reactants, like gases or ions,26 inside their channels is beneficial for electrochemical reactions. In addition, the open channels of COFs are capable of permitting ion migration;61 especially, the ionic COFs or COFs encapsulated with carriers can efficiently accelerate ion transport.
In the past few years, COFs have been broadly investigated in the EES and EEC fields (Fig. 3).62–65 Although 2D COFs are good electro-functional materials, long channels formed by stacked layers pose a great obstacle for ion diffusion and for reaching the active sites, and the defects of COFs and boundaries between particles restrict the transportation of electrons and ions. These issues lead to an increased mass transport resistance and inherent low conductivity of bulk COFs, which limit their applications. Remarkably, the sheet-like COFs with a thickness of single- or few-atom layers possess unique chemical, physical, electronic, and optical properties.66–68 They can minimize the transport length for the ions to reach the active sites, and provide sufficient electron conduction pathways from the electrodes or conductive additives to the active sites in COFs. Specifically, COF NSs with adjustable electronic/optoelectronic properties and fully exposed active sites are of particular interest for EES and EEC applications.
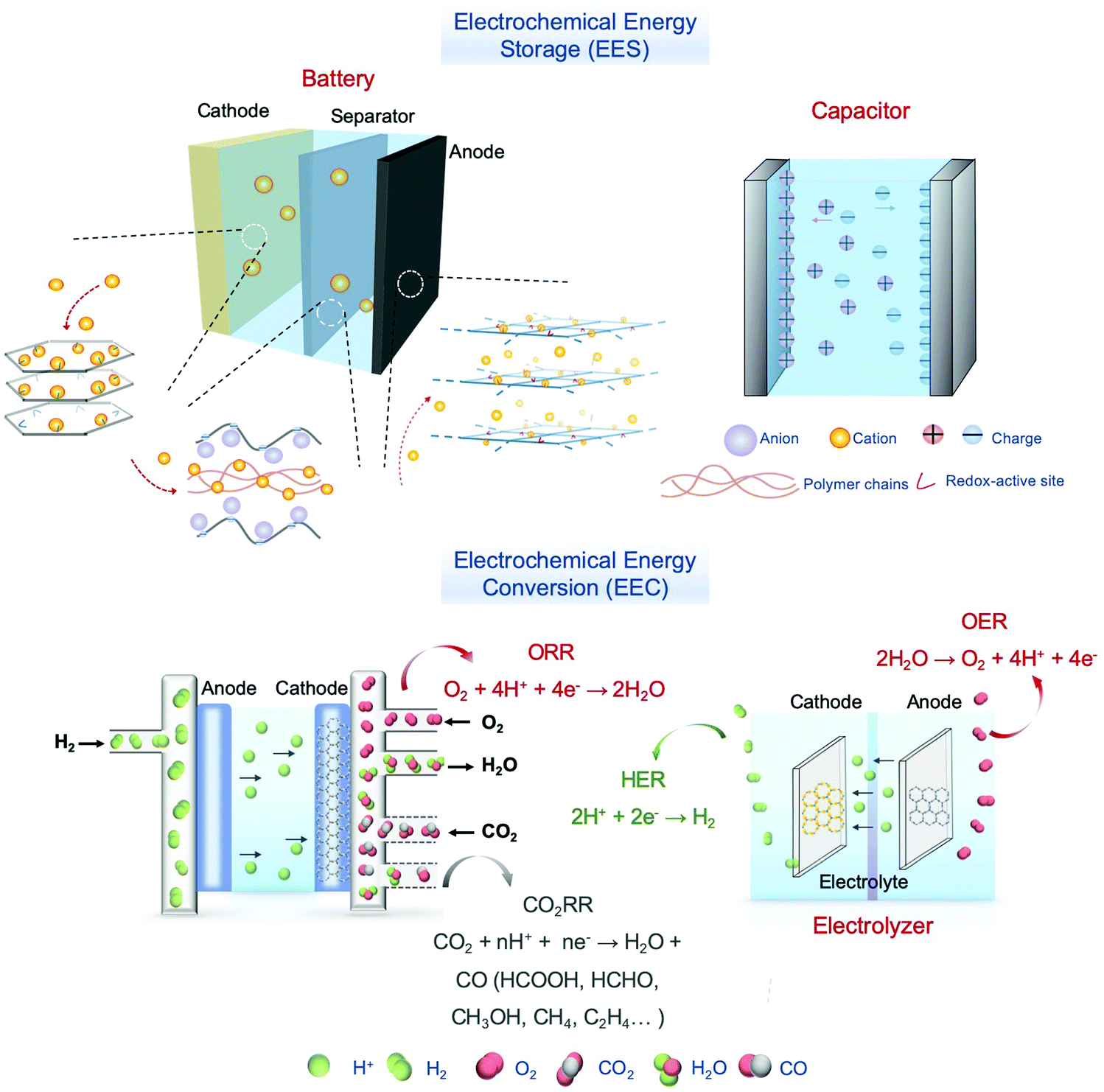 | ||
| Fig. 3 Conceptual presentation of an integrated system of EES and EEC. | ||
Specifically, for battery electrodes and supercapacitors, the shortened ion diffusion pathways and more accessible active sites in the COF NSs give rise to an elevated rate performance; for anode materials, the sheet-like structure provides the capability of accommodating ions between the layers based on the manner in which ions are inserted, even the single-layered COF NSs can follow the surface ion storage mechanism where ions are exchanged on the surface of electrodes during the charging–discharging processes; for supercapacitors, COF NSs by virtue of their porous nature and higher specific surface area are helpful for wetting the electrode surface by electrolytes and absorbing ions, resulting in improving their specific capacitances. As for electrocatalysts: (a) compared with the bulk COFs, COF NSs possess more accessible active sites on the surface that are able to directly contact substrate molecules as well as electrons with a lower diffusion/transportation barrier; (b) in terms of metal-containing COF-based electrocatalysts, the ultrathin nanosheet structures are conducive to the incorporation of metal atoms/clusters in a similar chemical and physical environment, benefiting the study of the structure–function relationship and the modulation of their performance; (c) generally, COF NSs exhibit a better dispersibility in solvents, making it easier to afford electrodes with uniform coverage of active materials.
Many reviews focusing on COFs in EES and EEC fields have been summed up,69–72 but few have discussed the fundamental understanding of the relationship between COFs’ structure and their electrochemical performance. In this review, we have presented the most recent progress, emphasized the importance of preparing 2D COF nanosheets for their electrochemical applications, highlighted the transport pathways of electrons and ions in COFs, established the association between the functions of COFs and their applications, and evaluated their performance from structural design. Firstly, we have summarized the synthetic methods for COF NSs and thin films which have been classified into bottom-up and top-down approaches; then we discussed the development of COFs in EES and EEC applications, including in rechargeable batteries, supercapacitors, as well as electrocatalysis; in the final part, we have discussed the challenges that still exist in these areas and have provided some perspectives.
2. Synthesis of COF nanosheets
Bulk COF materials can be prepared under solvothermal, microwave, ionothermal, or mechanical conditions, and many previous reviews have summarized and discussed these methods in detail.15,46,51 Thus, in the following sections, we have mainly introduced the strategies for the preparation of COF NSs and thin films, which are classified into bottom-up and top-down methodologies (Fig. 4).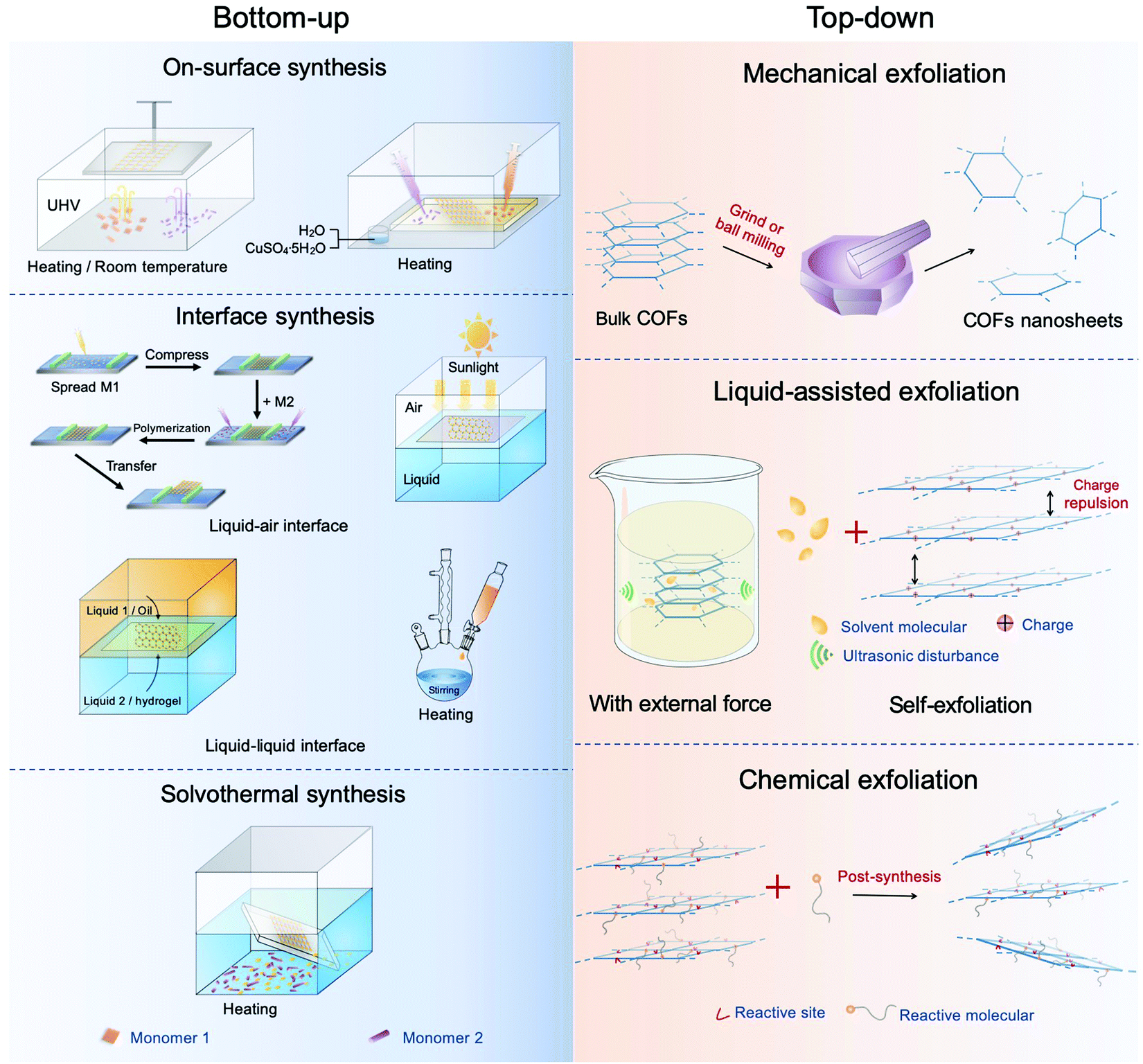 | ||
| Fig. 4 Diagram of the synthesis of CONs. The left represents the bottom-up method and the right top-down synthesis. | ||
2.1 Bottom-up synthesis
The bottom-up strategy is an approach of great importance to fabricate COF NSs and thin films, the key point of which is the preorganization of precursors and the restriction of condensation reactions between the monomers in well-confined spaces, such as the surface of smooth substrates or the interface between two phases.For the preparation of COF NSs with atomic thickness, generally, an on-surface strategy is employed. It is crucial to control the growth orientation of COF NSs or thin films and avoid random dispersion or accumulation of monomers on substrates. Therefore, the selected substrates, such as highly oriented pyrolytic graphite (HOPG) and metals, should possess a well-defined single-crystal surface to guide the polymerization reactions. Very recently, few-layered COF NSs were successfully prepared with the aid of surfactants or laminar assembly polymerization (LAP).73
In comparison, the conditions for the fabrication of COF thin films with multilayer nanosheet-stacked structures are not so strict, but it is required to carefully optimize the temperature, monomer concentration, solvent polarity, substrate, etc.
2.1.1.1 Chemical vapor deposition growth under ultrahigh vacuum (UHV). The pioneers of on-surface synthesis of 2D covalently bonded molecular nanostructures are Grill and Hecht et al.,74 who reported that molecular nanostructures containing porphyrin building blocks and linked by covalent bonds were successfully formed on Au(111) surface after thermal activation. Moreover, they also noted that the resulting nanoarchitectures with controlled shape and size could be precisely manipulated by changing the chemical structures of the construction units. To activate the molecular building blocks, they proposed two alternative methods but obtained similar products. The first one was depositing the intact molecules on the metal surface and then dissociating the Br substituents by heating. The second one was activating the molecules in the evaporator and then depositing them onto the metal surface at room temperature (Fig. 5a–d). By comparison, the former method was more efficient, attributed to the enhanced molecular diffusion and clustering of networks upon heating compared to that at room temperature. Similarly, other halogenated aromatic monomers have been utilized for the preparation of sCOFs under UHV conditions.75,76
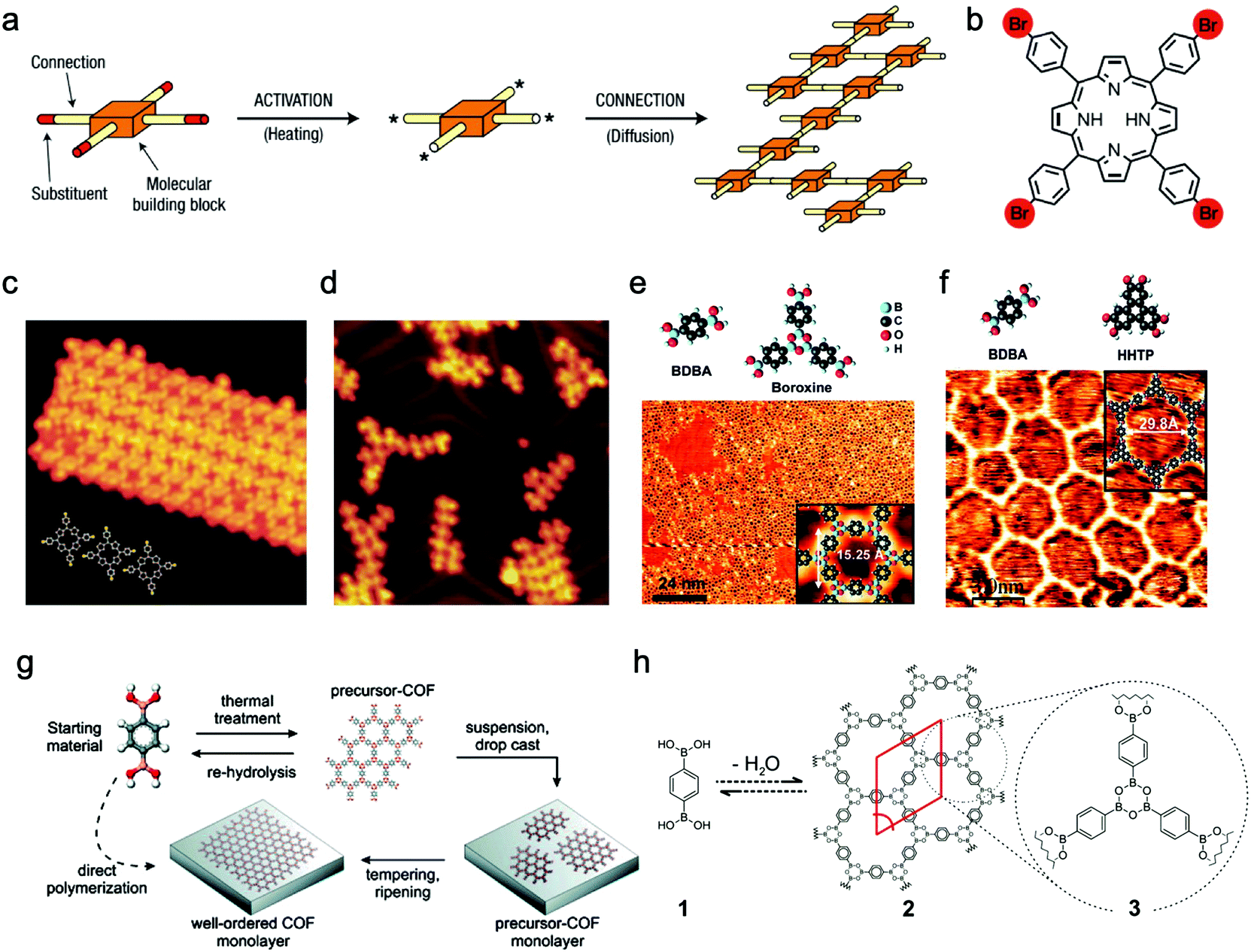 | ||
| Fig. 5 (a) Schematic illustration of activated building units interconnected to form the covalent networks. (b) Molecular structure of Br4TPP (Br atom is marked red). (c) STM image of unactivated Br4TPP deposited on Au(111) at a low evaporator temperature of 550 K. (d) In method (ii), the molecules are activated to form a network on the surface of Au(111) at an evaporator temperature of 610 K. Reproduced from ref. 74 with permission from Nature Publishing Group, copyright 2007. (e) Construction units (upper left: BDBA, upper right: three BDBA monomers form boroxine by dehydration condensation) and STM image of nearly single-layered sCOF-1 on Ag(111). (f) Top: Molecular structures of BDBA and HHTP; bottom: STM image of SCOF-2 on Ag(111). Reproduced from ref. 77 with permission from American Chemical Society, copyright 2008. (g) Schematic illustration of the two methods. (h) Reaction scheme for self-condensation of BDBA monomers. Reproduced from ref. 105 with permission from American Chemical Society, copyright 2011. | ||
Boronate linkage with planar molecular configuration and good reversibility has been applied for constructing sCOFs.77–81 In 2008, Abel et al.77 prepared sCOFs with tunable nanoporous structures on Ag(111) surface under UHV by using boronate-based building units. Both sCOF-1 (obtained by self-condensation of 1,4-benzenediboronic acid (BDBA) (32)) and sCOF-2 (obtained by co-condensation of BDBA and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) (40)) displayed energetically favorable structures and formed a hexagonal array with boroxine or triphenylene moieties at the nodes (Fig. 5e and f). Since the network of sCOF-2 showed fewer defects compared to that of sCOF-1, they thought that it could be attributed to the fact that the bimolecular reaction for dioxaborole formation was more kinetically favorable than the trimolecular boroxine reaction and the HHTP units may help to improve the rigidity of the networks. They found that the temperature of the substrate and evaporator, as well as the annealing process, greatly influenced the surface coverage by sCOFs. In 2012, Zamora, Gómez-Rodríguez et al. reported the formation of a sCOF on Au(111) based on polyester condensation under UHV.82 In 2015, Chi and Li et al. showed that the cyclotrimerization of acetyls to aromatics can also be used for the preparation of sCOFs on the surface under UHV.83 In addition, Schiff-base coupling between primary amines and aldehydes is of great interest in the on-surface synthesis of sCOFs.84–87
2.1.1.2 Thermal polymerization at ambient pressure. The process of implementing UHV conditions is complex, and the instrumentation and also single-crystal metallic surfaces have to meet high requirements. Alternatively, solution deposition may serve as a more efficient and simpler preparation route to obtain long-range ordered sCOFs. However, after solution deposition, the aryl–aryl coupling reaction proceeded on a heated gold surface only afforded dimers and trimers.88 The long-range ordered sCOFs with high surface coverage have been successfully prepared by reversible dehydration reactions with the manipulation of the amount of water.86,89–104
In 2011, Lackinger et al. first deposited BDBA monomers with boronic acid groups or nanocrystalline precursor COFs on the graphite surface by drop-casting (Fig. 5g and h), and then positioned the samples in a container placed within a preheated oven.105 Additional water was added in the reactor but did not directly contact the samples, and the container remained slightly open to maintain the ambient environment during the reaction. They found that the presence of water molecules was helpful for dominating the growth of COFs, which came to an end when all the water molecules were evaporated. STM characterization showed that the resulting monolayers of COF-1 formed on the graphite substrate with or without precondensation seemed to be indistinguishable regarding lattice parameters, domain size, and defect density. Then they obtained a series of high-quality sCOFs with pore sizes ranging from 1.0 to 3.2 nm by choosing building blocks with different lengths.91 Additionally, they investigated the impact of the activation temperature on molecule diffusion ability and reaction rate. They observed the structural characteristics of both sCOFs and noncovalently self-assembled structures depositing on the surface before the thermally activated polycondensation. Furthermore, Lei and Tian et al.106 reported that they successfully co-condensed triangular aromatic aldehydes and linear aromatic diamines based on Schiff-base reaction at the octanoic acid/HOPG interface either at room temperature or in a low vacuum with moderate heating.
In 2012, Wan and Wang et al. successfully fabricated well-ordered and large-scale sCOF-1 with a domain size of 200 × 200 nm2 on HOPG by modulating the chemical equilibrium via introducing CuSO4·5H2O into a closed reaction system. The controlled experiments without the addition of CuSO4·5H2O only resulted in products with disordered networks.89 Then they successfully produced another three ordered sCOFs with triangular, rhombille, or semi-regular Archimedean tilings, via Schiff-base reaction on the surface of HOPG with the aid of CuSO4·5H2O.107
Notably, most of the sCOFs usually possess single linkages. To further broaden the variety of sCOF structures, it is attractive to develop multi-component molecular covalent nanoarchitectures. Wang's group proved that Schiff-base reaction and boronic acid dehydration have orthogonality; in other words, they can take place simultaneously during on-surface synthesis without interference with each other.100 They adopted the gas–solid (HOPG) interface reaction methods to prepare highly ordered hybrid sCOFs linked by boroxine rings and imine bonds. Moreover, they successfully synthesized the first chiral sCOF with a windmill structure.
Kunitake et al. proved that extended 2D networks could be fabricated via a surface-mediated Schiff-base reaction on hydrophobic iodine-modified Au(111) surface in aqueous solution with precise pH and thermodynamic control.90 They emphasized the importance of choosing substrates and monomers. This method avoided complicated operations such as high temperature and ultrahigh vacuum. It paved the way for the synthesis of well-designed sCOFs via facile ways of “bottom-up” methodology.
2.1.1.3 Influencing factors. The on-surface synthesis of 2D sCOFs contains many kinetic processes such as adsorption, diffusion, nucleation, growth, etc. In order to obtain well-ordered and high-quality 2D sCOFs, the density of the nucleus, the diffusion rate of the precursors, and the growth rate should be taken into account. In most cases, the suitable growth conditions for the formation of 2D sCOFs include low nucleus density, high diffusion rate, and slow growth rate to guarantee a large domain size, high reaction efficiency, and thermodynamically stable structures, respectively. In this part, we have discussed the primary influencing factors for sCOF growth.
(a) Thermodynamic equilibrium control. The key to produce ordered and uniform molecular networks constructed with covalent bonds is the regulation of the reaction equilibrium and reversibility.
The substrate or system temperature will affect the reaction rate, and consequently, the quality of the sCOFs. Usually, the condensation reaction does not occur at low temperature; however excessively high temperature leads to a large number of defects caused by desorption or excessive polymerization of monomers.82,83 Therefore, a suitable temperature range that allows the reaction to proceed in a controllable and reversible manner is of great importance.
The reversible surface polycondensation is considered to be endothermic, and thus, elimination and release of water molecules will entropically promote the forward reaction. According to Le Chatelier's principle, the reversibility of the condensation reaction, which determines the domain size and defect degree, can be manipulated by water control. In the above-mentioned methodologies, the amount of water in the reaction system has been managed by additional water supplement, pressure control, or adding aquo-complex. For example, CuSO4·5H2O in the closed system acted as a water “reservoir”. During heating, the water molecules were released from CuSO4·5H2O to facilitate defect repair via decomposing and reconnecting mislinked units; on the other hand, during the cooling process, these water molecules would be soaked up again by anhydrous CuSO4 to prevent the sCOFs from decomposition.89,107
(b) Substrate. In addition to the ability to guide the polymerization process, substrates also have a great influence on the adsorption capacity and diffusion degree of the precursor molecules on them.75,76,79,108,109 Therefore, substrates play significant roles in increasing the coverage, regularity, and orientation of the resultant polymeric sCOFs. Substrates with single-crystal surfaces are required to produce high-quality sCOFs for the on-surface synthetic procedures. For example, Gutzler and Lackinger et al. polymerized halogenated aromatic monomers under UHV and found that the dissociation of covalent carbon–halogen bonds and subsequent association of radicals were different on graphite(001), Cu(111), and Ag(110), respectively.75 Bieri and Fasel et al. prepared polyphenylene networks by covalent bonding of the hexaiodo-substituted macrocycle on Cu(111), Au(111), and Ag(111).76 They found that the morphologies of the resultant products were significantly different on different substrates. Density functional theory (DFT) analysis revealed that the equilibrium of diffusion and coupling pathways of the precursors influenced by substrates were different. On Cu(111), the formation of covalent intermolecular bonds between radicals occurred spontaneously as soon as the primal diffusion barrier was overcome. In contrast, the diffusion progress proceeded prior to the intermolecular coupling on the Ag surface, thus promoting the motion of the radicals and resulting in well-ordered 2D networks.
(c) Monomer. Monomers, as well as their relative ratios, also have strong impacts on the structure, stability, uniformity, and application properties of sCOFs.86,92–98,110
Wan and Wang et al. investigated the effects of monomer diffusion and the molecule–substrate interaction by changing the length and rigidity of the monomer skeleton. They demonstrated that the increase of substrate–molecule interaction preferred to form macrocycle structures rather than sCOFs due to the poor mobility of the adsorbates.110 They also demonstrated that sCOFs synthesized on the surface by using tetrathiafulvalene equipped with four benzaldehyde groups (4ATTF) (21) and two linear diamine molecules with different lengths as precursors showed compelling differences.93 When reacted with p-phenylenediamine (PPDA) (45), 4ATTF was inclined to form long-range ordered rhombus structures. In contrast, 4ATTF when reacted with 1,1′-biphenyl-4,4′-diamine dihydrochloride (BPDA) (49) resulted in the formation of triangular and irregular rhombus structures.
Lei's group investigated the influence of precursor concentration on the production of sCOFs.95 They proved that, when reacting with the same aldehyde, the larger amine monomer demanded lower concentration to form a well-ordered crystalline 2D polymer. Wang's group used tetradentate monomer 1,3,6,8-tetrakis(p-formylphenyl)pyrene (20) with D2h symmetry and ditopic linear diamines to prepare sCOFs whose network shape varied from rhombus, parallelogram, and Kagome. They illustrated that high precursor concentration led to the formation of quadrate networks, while low concentration resulted in Kagome networks.97
2.1.2.1 Solvothermal method. The solvothermal synthesis method as a typical wet-chemical synthesis strategy is widely applied to produce ultrathin 2D nanomaterials. During the solvothermal synthesis, water or an organic solvent works as the reaction medium in a closed system and the heating temperature is set above the boiling point of the solvent. The heated solvents in high pressure can facilitate the reaction and enhance the crystallinity of nanocrystals.111 The solvothermal method has also been used to prepare COF multilayer nanosheets and thin films with or without the presence of substrate soaking in the solvents.112–119
In a typical example, Dichtel et al. reported that 2D COF-5 thin films can be formed on the surface of single-layer graphene (SLG) supported by copper, silicon carbide, or transparent fused silica (SiO2) substrates in a mixture of monomers and solvents at 90 °C (Fig. 6a–d).120 Compared with the corresponding COF powder sample, the COF film possessed a much higher orientation, in which the polymeric layers stacked normal to the SLG surface.
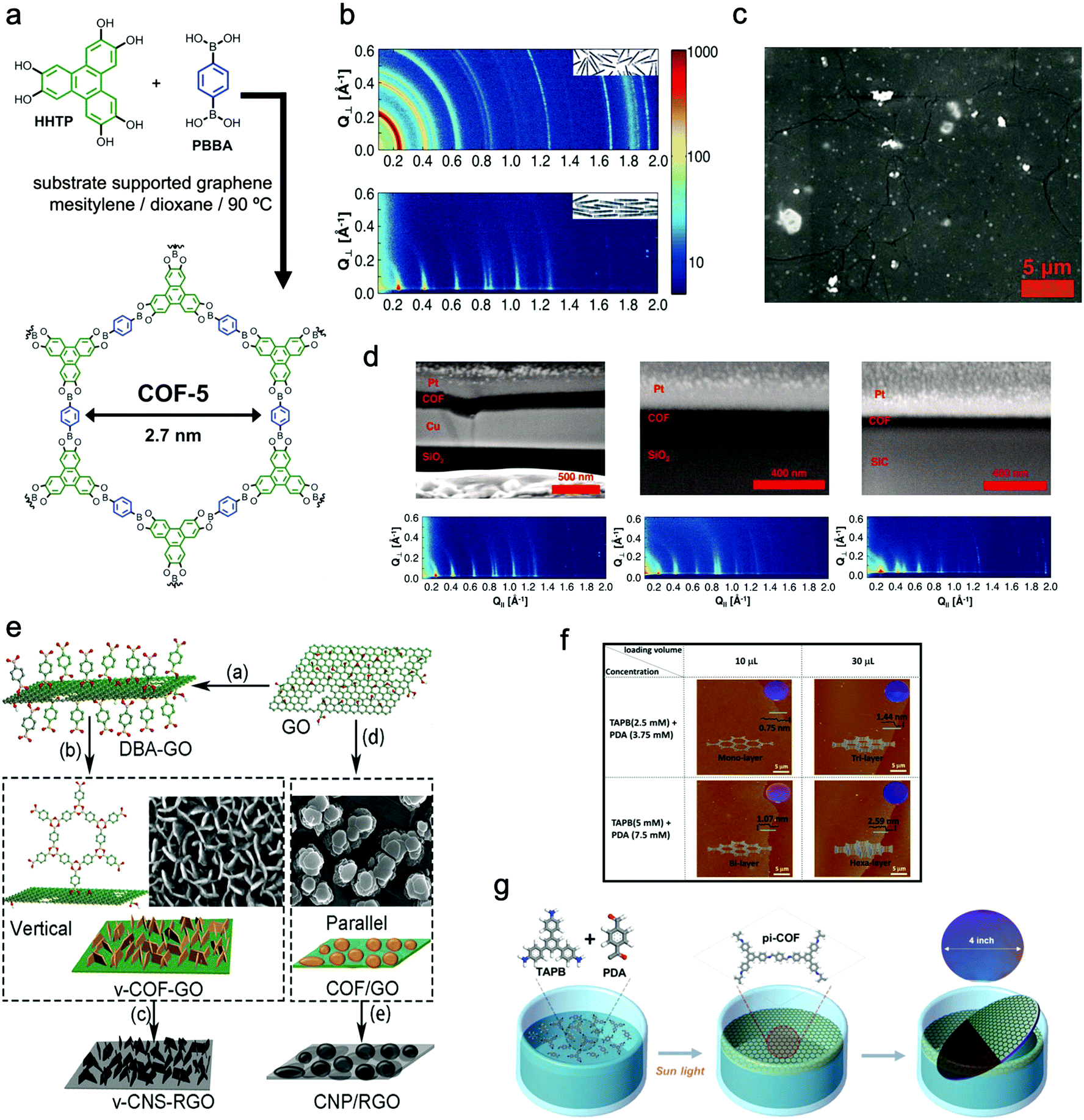 | ||
| Fig. 6 (a) Schematic illustration of the reaction of COF-5 films. (b) X-ray scattering data of the COF-5 powder (top) and GID data of the COF-5 film on SLG/Cu (down). (c) SEM image of the COF-5 thin film. (d) Cross-sectional SEM images and their corresponding GIDs of COF-5 films grown on SLG/Cu for 30 minutes, 2 hours, and 8 hours with film thicknesses of 195 ± 20 nm, 94 ± 5 nm, and 73 ± 3 nm, respectively. Reproduced from ref. 120 with permission from American Association for the Advancement of Science, copyright 2011. (e) Schematic illustration of the two methods: (a–b–c) functionalization of GO with DBA which worked as a vertically growing COF-1 nanosheet molecular nucleation site; (a–d–e) formation of COF-1 platelets parallel to GO in the absence of BDA for GO functionalization followed by carbonization to form carbon nanosheets oriented perpendicular or parallel to the RGO surface. Reproduced from ref. 112 with permission from Wiley-VCH Verlag GmbH & Co. KgaA, copyright 2018. (f) AFM images and thickness distribution of single, double, triple, and six 2D Lp-pi-COF films. (g) Schematic illustration of the reaction of Lp-pi-COF synthesized on the water interface with the assistance of photons. Reproduced from ref. 124 with permission from American Chemical Society, copyright 2018. | ||
Talyzin's group developed a hybrid 2D–2D material consisting of perpendicularly oriented COF-1 NSs and graphene (Fig. 6e).112 For directing the vertical growth of COF-1 NSs, they modified graphene oxide with diboronic acid molecules to function as nucleation sites. The thickness of COF-1 NSs was controlled from about 3 to 15 nm. Besides, Wang's group realized the growth of boronic ester-based COFs on carbon nanotubes (CNTs) to form COF-10@CNT with a few-layered structure. It can be seen from the high-resolution transmission electron microscope (HRTEM) images that the CNTs were covered by a COF-10 thin layered structure (about 6 nm) with a layer spacing of 0.35 nm.121
2.1.2.2 Interface synthesis. Liquid/air and liquid/liquid interfaces can provide confined spaces for the preparation of 2D COF nanosheets or films.
For instance, Zhang's group prepared Schiff-base 2D COF NSs with a thickness of about 0.7 nm at the air/water interface by the Langmuir–Blodgett method.122 Bao's group placed the reaction solution in a covered Petri dish for 2 days in an ambient environment and the COF films were formed at the solution/air interface. The surfaces of the resulting films were smooth and the thickness could be tailored from 1.8 nm to 29 nm by altering the monomer concentration and reaction time.123 Choi et al. found that the formation of the COF NSs at the air/water interface could be significantly shortened to less than 1 h by photon irradiation, and the thickness of the resultant COF NSs was 0.75–2.59 nm (Fig. 6f and g).117,124 They noted that photon energy played a vital role in both accelerating imine condensation and facilitating the transformation from generated amorphous imine to crystalline COFs.
Feng's group prepared crystalline quasi-two-dimensional (q2D) polyaniline (PANI) films at the air–water interface by using surfactant (sodium oleyl sulfate, SOS) monolayers as the template. The obtained crystalline q2D PANI films possessed lateral sizes of 50 cm2 and tunable thickness (2.6–30 nm).125 Furthermore, Feng, Zheng, and Kaiser et al. successfully proposed a surfactant-monolayer-assisted interfacial synthesis (SMAIS) strategy to synthesize crystalline few-layered 2D polyimide (2DPI) and 2D polyamide (2DPA) at the interface of water and air with the aid of surfactant monolayers (sodium oleyl sulfate, SOS). The obtained 2DPI possessed a thickness of about 2 nm and a crystal domain size of about 3.5 μm2, while these values for 2DPA were 10 nm and 0.4 nm μm2, respectively. They also identified that the amorphous areas connected with crystalline domains dominated about 40% area (Fig. 7a–e).126 Very recently, Feng, Dong, Zheng, and Kaiser et al. used a similar strategy to synthesize three fully crystalline PI-2DPs with tunable thickness (6 to 200 nm) and crystalline domain (100–150 nm in size), which were one order of magnitude larger than those of previously reported 2D polyimine films.127 Feng, Dong, and Cuniberti et al. further prepared 2D crystalline boronate ester covalent organic framework (BECOF) films with tunable thickness (6–16 nm) and large single-crystalline domain up to 60 μm2.128
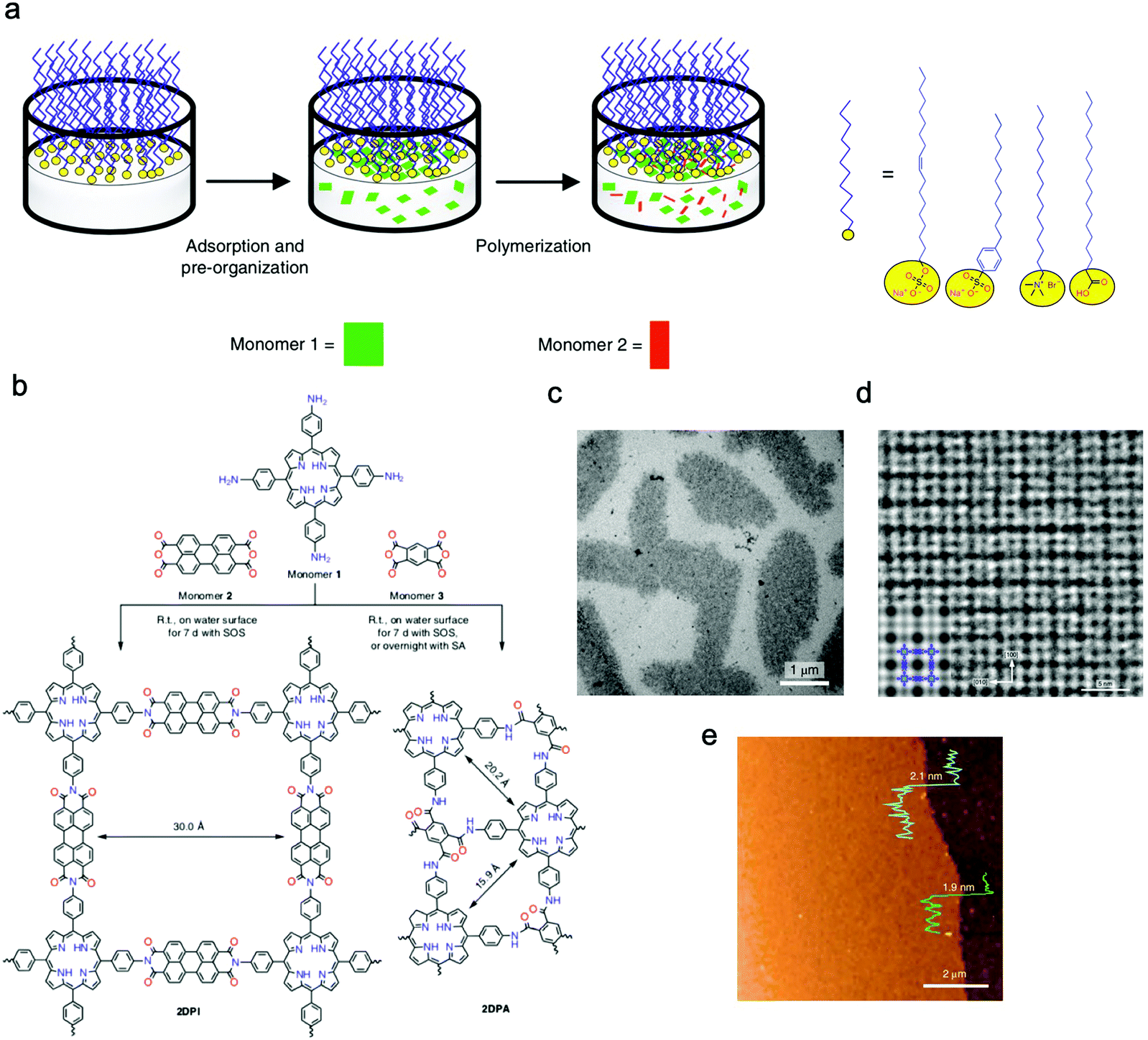 | ||
| Fig. 7 (a) Schematic illustration of the synthetic procedure for the 2D polymers at the interface of water and air with the aid of surfactant monolayers. (b) Schematic illustration of the reaction of 2DPI and 2DPA. (c) A TEM image of the 2DPI film. (d) An AC-HRTEM image of 2DPI. (e) An AFM image of the 2DPI film. Reproduced from ref. 126 with permission from Nature Publishing Group, copyright 2019. | ||
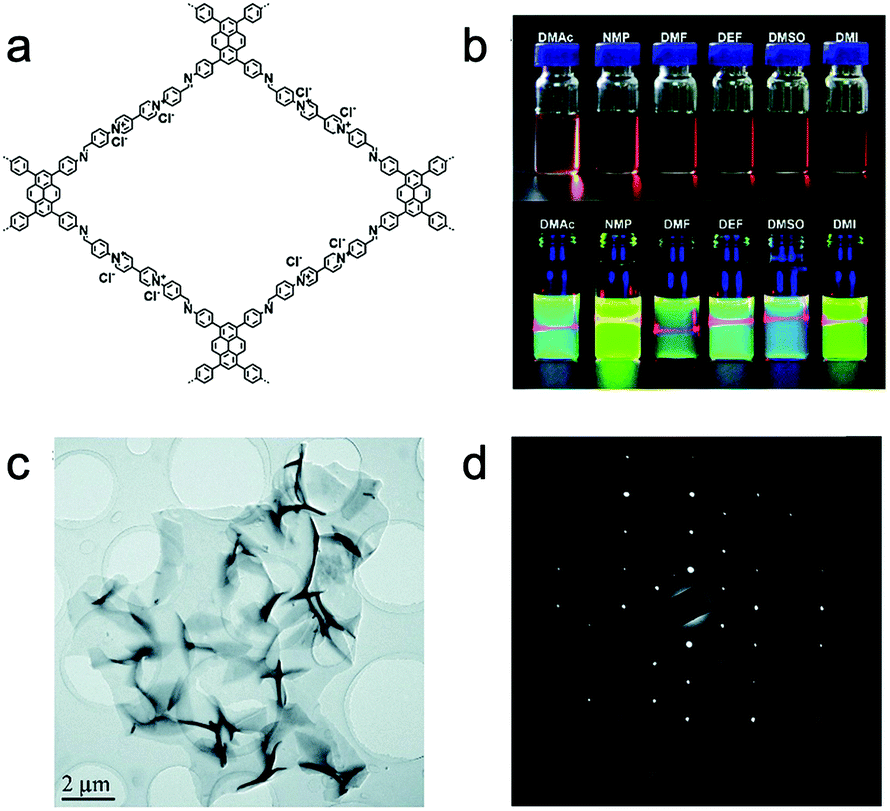
Li and Ma et al. reported a buffering interlayer interface (BII) method to synthesize high-quality 2D COF NSs at the interface of two miscible organic solvents.129 They introduced a low-density solvent interlayer between two miscible solvents to work as a buffer layer, in which the difference in density caused the directional movement of the interface.
Banerjee, Mukherjee, and Das et al. prepared a series of free-standing large-scale COF thin films with a thickness of sub-100 nm at the liquid–liquid interface by Schiff-base reaction.130 They introduced amine-p-toluene sulfonic acid (PTSA) salt to slow down the precursors’ diffusion rate for increasing the crystallinity of films (Fig. 8a–c). Zhang, Ma, and Chen et al. transformed polymeric covalent organic frameworks (polyCOFs) into defect-free and freestanding films. The linear polymers PEG400/600 were used as building blocks to prepare the polymer monomer (DTH-POLYMER) containing 2,5-diethoxy-terephthalohydrazide (DTH) (68) moieties, and then, DTH-POLYMER and DTH reacted with another monomer triformylbenzene (TB) (23) to form the final product polyxCOF-42 films at the interface between mesitylene and mixed solution of water and dioxane. What's more, they successfully synthesized films with a thickness of 4.5 ± 0.3 nm by adjusting the concentration of the reactants.131
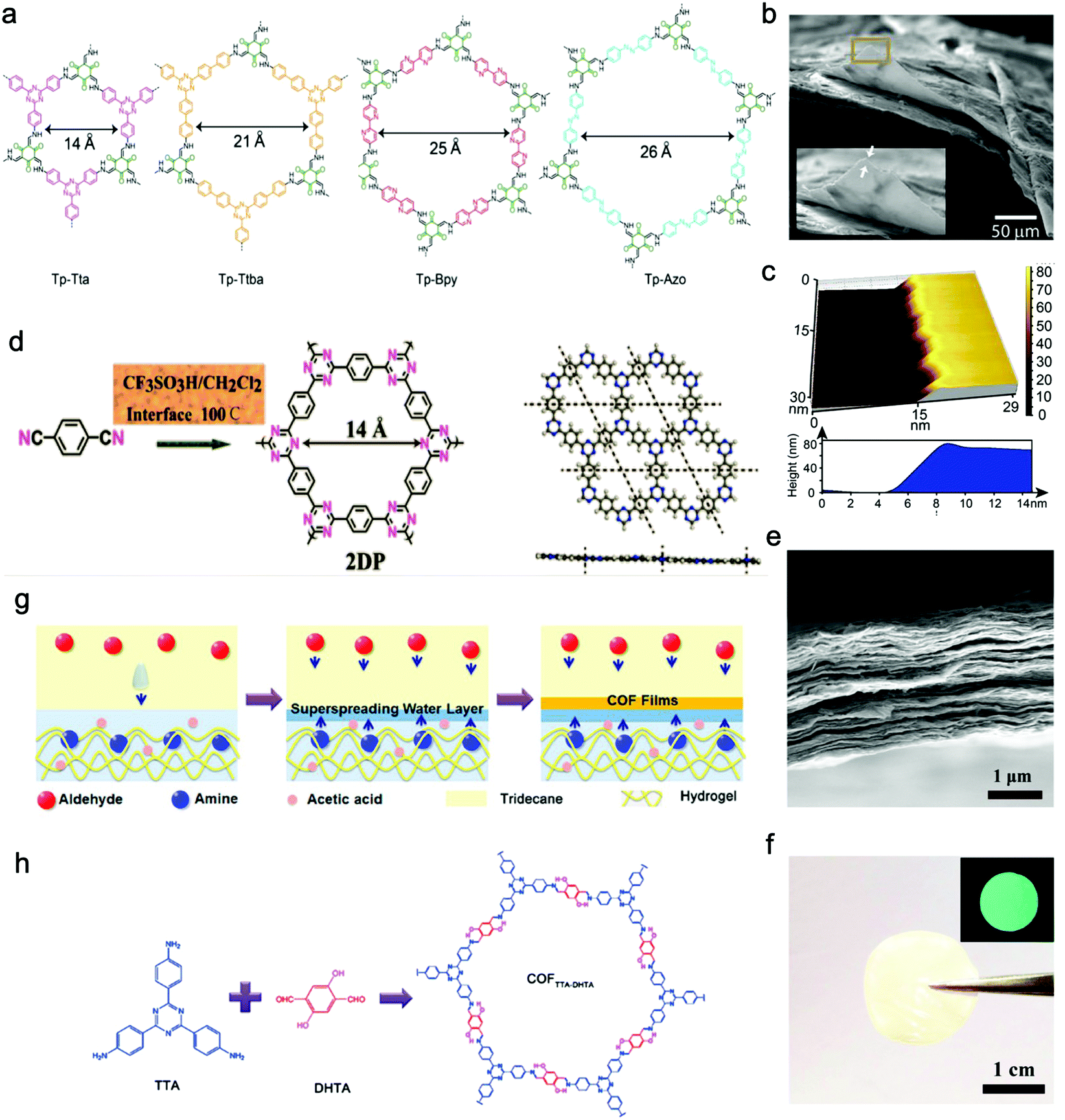 | ||
| Fig. 8 (a) Chemical structures of COF films (Tp-Tta, Tp-Ttba, Tp-Bpy, Tp-Azo). (b and c) SEM and AFM images of Tp-Bpy films. Reproduced from ref. 130 with permission from American Chemical Society, copyright 2017. (d) Schematic illustration of the reaction of triazine-based 2DP. (e) Cross-sectional SEM image of the 2DP film. (f) The photo of the self-supporting 2DP film. The inset is 2DP film's fluorescent picture under UV light. Reproduced from ref. 132 with permission from American Chemical Society, copyright 2017. (g) Schematic illustration of the formation of a self-supporting COFTpPA film at the oil/water/hydrogel interface. (h) Schematic illustration of the reaction of COFTTA-DHTA. Reproduced from ref. 133 with permission from American Chemical Society, copyright 2018. | ||
Besides, Xu's group successfully proposed a solution-synthesis strategy to prepare triazine-based COF NSs with single-/multi-layered structures.132 They first added CF3SO3H to a flask, and then the monomer solution (1,4-dicyanobenzene (85) in CH2Cl2) was introduced into the flask through a dropping funnel at a reaction temperature of 100 °C. Abundant liquid–liquid interfaces, where the cyclotrimerization reaction took place, were formed between the two insoluble solvent phases (Fig. 8d–f).
Wang and Liu et al. synthesized free-standing 2D COF films by using superspreading water layers between hydrogel and oil phase as a confined interface. In detail, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) (58) was dissolved in the hydrogel while 2,5-dihydroxyterethaldehyde (DHTA) (3) was dissolved in the oil phase; the two monomers encountered and reacted in the superspreading water layer to form COF films with tunable thickness ranging from 4 to 150 nm when changing the concentration of monomers (Fig. 8g and h).133
2.1.2.3 Influencing factors for few-layered COF NSs. In order to obtain highly qualified few-layered COF NSs for different applications, many factors should be taken into account, including monomer, solvent, reaction time, temperature, substrate, auxiliary reagent, and so on.
(a) Monomer. Types, concentration, ratio, and other related parameters of monomers greatly affect the preparation process, thickness, quality, number of layers, and other properties of COF NSs.112,115,117,124,127,130,132,133
Typically, the concentration of monomers plays an important role in controlling the thickness and layer number of COF NSs. In the process of preparing perpendicularly oriented COF-1 NSs on GO, Talyzin's group successfully controlled the thickness of films in the range of 3–15 nm by adjusting the loading of monomers.112 Wang's group synthesized COFTFPy-PPDA thin films on SLG by a solvothermal method, wherein they employed 1,3,6,8-tetrakis(p-formylphenyl)pyrene (TFPy) (20) and p-phenylenediamine (PPDA) (45) as building blocks. The thickness of the obtained thin films varied in the range of 47–55 nm, based on the concentration of TFPy from 2 to 5 mmol L−1.115 Choi et al. found that the number of layers for COF NSs at the air/water interface could be efficiently regulated by changing the concentration of the precursor solution (Fig. 6f).117,124
(b) Solvent. It is worth noting that the proper solvent combination is one of the key factors for preparing uniform and highly crystalline COF thin films. The solvent mixture with an appropriate ratio is beneficial for spreading the precursor solution homogeneously at the interface, which can be attributed to the polarity effect of the precursor solution.117,124,132 Choi et al. synthesized polyimine-based COF (pi-COF) NSs at the air/water interface. The solvent composition they used to dissolve the reactive monomers was 1,4-dioxane, mesitylene, chloroform, and acetic acid, by which they successfully controlled the polarity of the precursor solution, resulting in the formation of a uniform and stable layer on the water surface.124
(c) Reaction time. The duration of the reaction affects the thickness of the obtained COF thin films. For example, Bao's group prepared COF films at the solution/air interface in a covered Petri dish and they found that longer reaction time led to thicker films.123
(d) Substrate. Choosing a suitable substrate is regarded as one of the efficient ways to prepare COF thin films with a highly crystalline and ordered structure. The obtained COF thin films have been shown to form 2D layers oriented preferentially parallel to the substrate surface and vertically aligned via π–π stacking.120
Dichtel et al. demonstrated that 2D COF-5 thin films can be formed on the surface of SLG supported by copper, silicon carbide, or transparent fused silica (SiO2) substrates. They found that the COF-5 films exhibited a similar structure but a more uniform film/substrate interface and smaller thickness (94 ± 5 nm) on SLG/SiO2 compared with that on SLG/Cu, while the COF-5 film formed on SLG/SiC showed no visible grain boundaries, few bulk crystallites, and the smallest thickness (73 ± 3 nm) (Fig. 6d).120
(e) Auxiliary regent. In some cases, adding auxiliary reagents (e.g., surfactants125–128 and regulators119) to the reaction systems can effectively regulate the structures and improve the quality of COF NSs.
For example, Feng, Zheng, and Kaiser et al. successfully synthesized few-layered 2D polyimide (2DPI) and 2D polyamide (2DPA) at the water–air interface with the aid of a surfactant (sodium oleyl sulfate, SOS). They further tried other surfactants with different polar groups, including sodium 4-dodecylbenzenesulfonate (SDBS) and hexadecyl-trimethyl-ammonium bromide (CTAB). They found that in the pre-organization process, only when the surfactant (SDBS) and monomer 4,4′,4′′,4′′′-(porphyrin-5,10,15,20-tetrayl) tetraaniline (62) had electrostatic interaction it could lead to the formation of crystalline 2DPI. In contrast, if the surfactant (CTAB) and the monomers had electrostatic repulsion interaction, the obtained product was amorphous.126
In 2019, Jiang, Zeng, and Wang, et al. reported an imine-exchange synthesis strategy to prepare COF NSs by introducing excess 2,4,6-trimethylbenzaldehyde (TBA) into the reaction system under solvothermal conditions. During the formation of the COF, TBA molecules were attached to the edge of COF NSs, which hindered the accumulation of π–π stacking of COF NSs and promoted the growth of COF NSs along the 2D plane, realizing the control of NS layers, and the increase of yield as well as size. They successfully synthesized porphyrin-based COF-366, COF-367, COF-367-Co, TAPB-PDA COF, and TAPB-BPDA COF by using this method.119
2.2 Top-down method
Bulk 2D COFs are layered structures with periodically aligned channels. The individual polymeric layers are stacked along the vertical direction through relatively weak interactions (e.g. van der Waals force and/or hydrogen bonding). The top-down method can prepare multi-/mono-layered COF NSs by directly breaking the interactions between the neighboring layers of COFs, which is a facile and effective approach to obtain NSs. To date, the methodologies applied to exfoliate COFs can be divided into four classes, including mechanical exfoliation, liquid-assisted exfoliation, self-exfoliation, and chemical exfoliation.Compared with pestle grinding,134–136 ball milling137–139 is a scalable mechanical delamination method to obtain COF NSs. As an example, a series of aromatic β-ketoenamine linked COFs with intramolecular C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HN hydrogen bonding were exfoliated by mechanical milling without solvent, operating at 30 Hz for 30 min.139 The resultant powder was diluted with toluene (or water) and sonicated for 30 min. After removing big particulates by centrifugation, the visibly clear supernatant containing COF NSs was obtained. The COF NSs revealed approximately half-micron large sheets with the thickness in the range of 0.7–4.5 nm, corresponding to a stack of ∼2–15 COF sheets. Similarly, a Schiff-base COF was wet-ball milled in dimethyl formamide (DMF) (80 mL) at 225 rpm for 24 h. The resultant powder was dispersed by sonication for 30 min and then centrifuged, achieving few-layered nanosheets.138
O⋯HN hydrogen bonding were exfoliated by mechanical milling without solvent, operating at 30 Hz for 30 min.139 The resultant powder was diluted with toluene (or water) and sonicated for 30 min. After removing big particulates by centrifugation, the visibly clear supernatant containing COF NSs was obtained. The COF NSs revealed approximately half-micron large sheets with the thickness in the range of 0.7–4.5 nm, corresponding to a stack of ∼2–15 COF sheets. Similarly, a Schiff-base COF was wet-ball milled in dimethyl formamide (DMF) (80 mL) at 225 rpm for 24 h. The resultant powder was dispersed by sonication for 30 min and then centrifuged, achieving few-layered nanosheets.138
Firstly, the solvent system is an essential factor for achieving efficient exfoliation in liquid.140,144,149 The proper solvent system can both facilitate the exfoliation and inhibit the aggregation of the nanosheets. For instance, the relationship between different solvents and products for the exfoliation of 2D hydrazone-linked COF-43 with the expected stability of hydrazone linkages was investigated.140 When COF-43 was immersed in tetrahydrofuran (THF), trichloromethane (CHCl3), toluene, and methanol, it did not change its original diffraction patterns, whereas its exposure to dioxane, H2O, and DMF led to the loss of crystallinity as judged from PXRD patterns. The authors speculated that the loss of crystallinity might be attributed to the exfoliation of bulk COF-43 rather than breaking of its covalent linkages. The hydrolysis of COF-43 was excluded by solid and solution-state infrared spectroscopy (IR) measurements, demonstrating that COF-43 possessed good chemical stability in each solvent. The atomic force microscope (AFM) images of the suspensions sonicated in different solvent systems further confirmed the exfoliation behavior. When combined with dynamic light scattering (DLS), it was observed that there were significant size differences between the dispersed species in the different solvent systems. When COF-43 was sonicated in dioxane, high aspect ratio platelets with a width of 200 nm were achieved with a height of 1.32 ± 0.37 nm (related to 3–5 layers). Thin sheets with a height of 3.3 Å (bi-/single-layered) were realized after sonicating in H2O while much thicker sheets were obtained after deposition from non-exfoliating solvents, such as THF. These observations suggested that dispersing COF-43 in exfoliating solvents led to high aspect ratio platelets.
Secondly, the design of skeletons is another crucial factor that can influence the exfoliation products. The introduction of flexible building units into the skeleton of COFs can weaken the π–π interactions and make them easily exfoliated to nanosheets. Two C3v-symmetric flexible building units were used to form an [3+3] imine-linked COF, TPA-COF (Fig. 9a), which possessed a layered hexagonal structure with an interlayer distance of 4.1 Å and could be easily transformed to nanosheets by exfoliation.141 In a typical process, bulk TPA-COF was dispersed in 150 mL of ethanol and sonicated for 3 h. After sedimentation for 24 h, the upper colloidal suspension was collected and then centrifuged at 7000 rpm for 5 min, resulting in TPA-COF NSs with a thickness of 3.5 ± 0.3 nm (Fig. 9d). Notably, the fine crystallinity of the nanosheets was revealed by low-dose HRTEM (Fig. 9b). The arranged hexagonal structures were observed in the denoised HRTEM image, matching well the simulated one (Fig. 9c).
 | ||
| Fig. 9 (a) Schematic of TPA-COF. (b) High-resolution TEM image of a typical TPA-COF NS with the low-dose technique using a direct-detection electron-counting camera. Inset: FFT of the TPA-COF NS. (c) Denoised HRTEM image, and the inset image is the simulated HRTEM image. (d) AFM image of TPA-COF nanosheets. Reproduced from ref. 141 with permission from American Chemical Society, copyright 2017. (e) Schematic illustration of the exfoliation of NUS-30 by the temperature-swing gas method. Reproduced from ref. 142 with permission from the Royal Society of Chemistry, copyright 2019. | ||
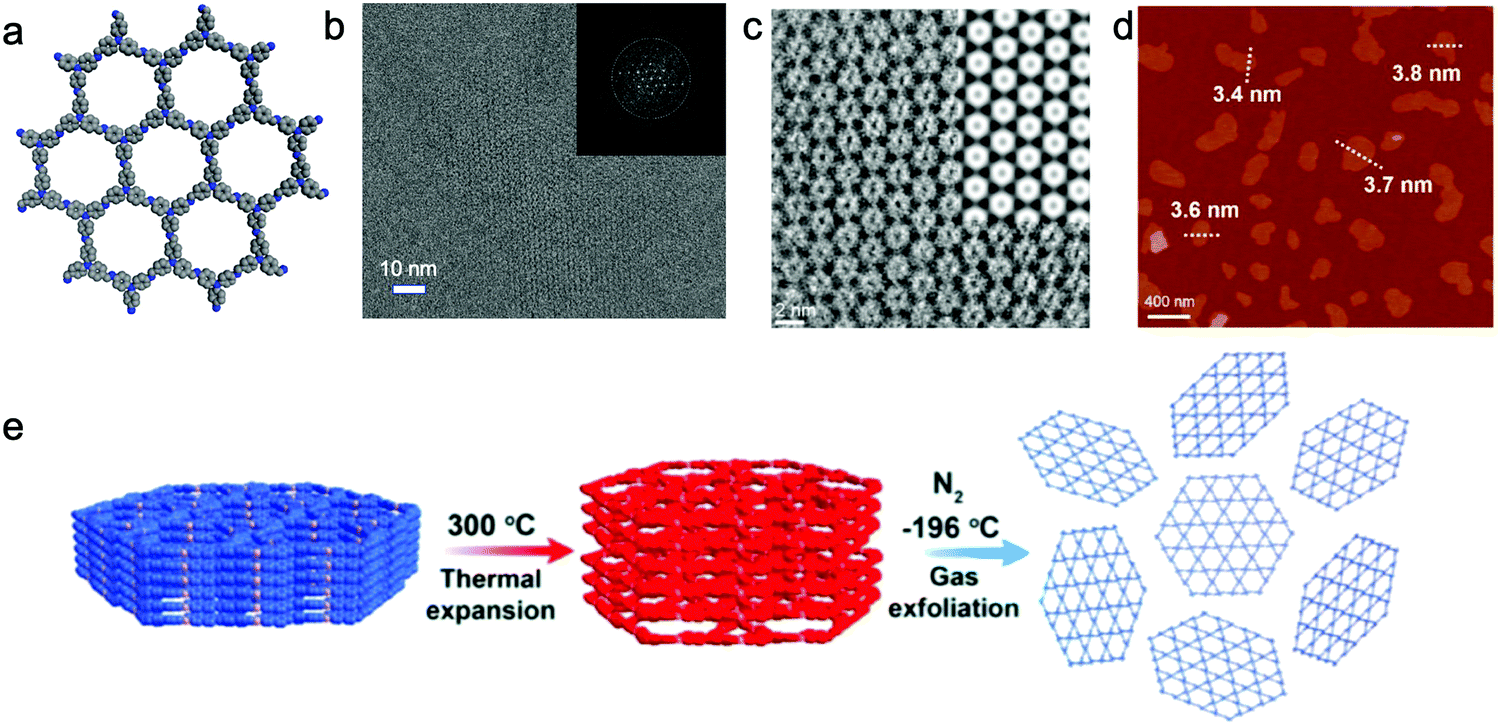
A temperature-swing gas exfoliation approach also transforms bulk COFs to NSs in liquid.142 Three azine-/imine-linked 2D COFs, NUS 30–32, were exfoliated by using this method. First, the bulk powder was heated to 300 °C for 10 min in air, and then immersed into liquid N2 quickly (Fig. 9e). These procedures were repeated 5 times, and large particles were removed by centrifuging the suspension at 3500 rpm for 10 min. The supernatant containing NSs was further collected and centrifuged at 8000 rpm for 10 min to remove bulk particles. The micrometer-sized CONs with a thickness around 2–4 nm were observed by AFM and transmission electron microscopy (TEM). The authors suggested that the interlayer distance was expanded during the heating processes, allowing the gasified liquid N2 to exfoliate the layered structure.
The behavior of self-exfoliation in different solvents was also investigated. PyVg-COF (Fig. 10a) was synthesized combining two building blocks with opposing properties.155 One of the monomers (4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline, Py (57)) tends to form crystalline stacks with strong π–π interactions; the other one (1,1-bis(4-formylphenyl)-4,4′-bipyridinium dichloride, Vg2+·2Cl− (13)) with inherently high charge density exhibits strong electrostatic repulsion upon stacking. The large-area monolayer or multilayer nanosheets were achieved after immersing PyVg-COF in various organic solvents, showing an obvious Tyndall effect (Fig. 10b). The HRTEM image and selected area electron diffraction (SAED) pattern revealed the micro-scaled size and high crystallinity of NSs, respectively (Fig. 10c and d). The self-exfoliation behavior appeared when attaining a higher skeleton–solvent interaction absolute value, that is, the successful self-exfoliation of PyVg-COF only happens when the skeleton–solvent interactions are higher than the interlayer interactions of the skeletons.
 | ||
| Fig. 10 (a) Schematic of PyVg-COF. (b) Photos represent PyVg-COF dispersed in various solvents. Top, under sunlight; bottom, under 365 nm UV light. (c) Low magnification TEM image of PyVg-COF nanosheets. (d) SAED of the PyVg-COF nanosheet. Reproduced from ref. 155 with permission from the Royal Society of Chemistry, copyright 2019. | ||
In addition, a 2D porphyrin-containing COF (DhaTph) was delaminated by introducing disruptions between the layers, which was implemented through coordinating the porphyrin centers with axial ligands.157 The NSs with a width of several hundred nanometers and a thickness of around 3 nm were obtained by refluxing DhaTph in pyridine solutions for 20 h.
Notably, Dichtel et al. presented a strategy to exfoliate 2D imine-linked COFs by acid. For example, the addition of excess trifluoroacetic acid (TFA) could temporarily protonate BND-TFB COF's imine linkages and lead to a disordered stacking, which minimized the electrostatic repulsion between adjacent sheets and further promoted exfoliation. The exfoliated NSs with the thickness ranging from 5 to 50 nm were capable of being dispersed in organic solvents. The retained imine-linked network, uniform six-fold symmetry, and well-matched diffraction peak (100 plane) were demonstrated by FT-IR, HRTEM, and FFT (fast Fourier transform), respectively.161
3 Electrochemical energy storage
3.1 Batteries
A battery is an energy storage device consisting of cathode, anode, electrolyte, and separator. The cathode and anode communicate with electrons under a potential difference, and thus the electrochemical energy is stored or released in the batteries. To prevent short circuits, a porous membrane as a separator is placed between cathode and anode to insulate electrons but allow ions to move across. At the same time, liquid or solid electrolytes with high ionic conductivity are employed to facilitate ion transport. Currently, the capacity, rate performance, and lifetime of electrode materials still cannot fulfill the demand of matching the supply and releasing energy anytime and anywhere; liquid electrolytes suffer from safety risk while solid electrolytes show low ion conductivity. As a highly porous crystalline polymer, COFs are capable of storing numerous ions and their inner surfaces can be metrically modified with redox-active sites.162,163 Thus, COFs are potential materials as battery electrodes. Besides, their electrical insulation nature and open channels make COFs an attractive option in solid electrolytes and separators.3.1.1.1 COFs as redox-active cathodes. Conventional electrode materials are dominated by inorganic materials such as transition metal oxides or phosphates (e.g., LiCoO2, LiMn2O4, and LiFePO4). Organic molecules as electrodes have been developed recently for their environmental benignity, adjustable theoretical capacity, and redox potential. However, their high capacity is generally accompanied with an inferior cyclic performance at high rates due to their partial dissolubility in the electrolyte and poor conductivity. Besides the merits from organic materials, highly crosslinked COFs exhibit high stability and are almost non-soluble in any solvent, and specifically, 2D COFs theoretically possess high charge carrier (electron and/or hole) conductivity in planar layers through π-conjugation as well as across the stacking sheets via overlapped π electrons. Moreover, few-layered 2D COFs are beneficial for ionic/electronic diffusion and are able to deliver a high rate capability.
For instance, 1,3,5-triformylphloroglucinol (Tp) (24) showed a capacity below 30 mA h g−1 and couldn’t be fully recharged during cycling. 2,7-Bis((E)-benzylideneamino)benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone (DANTB) (69) delivered an initial capacity of 125 mA h g−1, and then, the capacity decreased rapidly within 10 cycles. In sharp contrast, on incorporating these redox-active groups into a Schiff-base COF (Tp-DANT-COF), it displayed a capacity of 78.9 mA h g−1 at the current density of 1.5C (corresponding to 200 mA g−1) and retained the capacity for more than 200 cycles.164 It demonstrated the importance of robust networks. In addition, when the monomer Tp is replaced by 1,3,5-triformylbenzene (Tb) (23), the resultant Tb-DANT-COF exhibited lower voltage hysteresis and higher initial discharge capacity.
Despite the fact COFs exhibit high conductivity in and between the layers theoretically, the battery performance still suffers from their limited electron and ion conductivity. The reasons for this are (1) the defects and boundaries between particles greatly hinder the electron transport; (2) the 2D COF sheets tend to be packed closely in an eclipsed fashion due to strong π–π interactions, leading to difficulty in the diffusion of ions to the interior active sites through the long transportation paths even at a high current density.137,165
The electron conductivity can be enhanced by integrating COFs with conductive materials such as carbon nanotubes (CNTs) or graphene.166–169 Jiang et al.167 presented a strategy for improving the electron mobility via growing redox-active COFs on CNTs (DTP-ANDI-COF@CNTs). The capacity of DTP-ANDI-COF@CNTs was retained at 74 mA h g−1 after 700 cycles, corresponding to 90% utilization efficiency of the redox-active sites, and with a stable Coulombic efficiency reaching 100%.
Feng and Wang et al. proposed a strategy to reduce the ion/electron migration length and accelerate the ionic/electronic diffusion by exfoliating bulk COF materials into few-layered NSs with more accessible functional sites.137 An anthraquinone-based COF (DAAQ-TFP-COF) (Fig. 11a) was successfully exfoliated to ultrathin NSs with a thickness of 5 nm via ball milling (donated as DAAQ-ECOF) (Fig. 11c). The Li+ ion diffusion coefficient of DAAQ-ECOF is 6.94 × 10−11 cm2 s−1, which is three times higher than that of DAAQ-TFP-COF and higher than those of conventional inorganic materials. The electrochemical process of bulk DAAQ-TFP-COF was dominated by ion-diffusion, whereas that of DAAQ-ECOF got rid of the ion-diffusion restriction and was controlled by electron transport. Accelerated Li+ transport and shortened diffusion paths resulted in good battery performance. DAAQ-ECOF presented a capacity of 145 mA h g−1 at 20 mA g−1 over more than 70 cycles, delivering 96% of its theoretical capacity, while only 73% was retained for pristine DAAQ-TFP-COF (Fig. 11e). At a current density of 500 mA g−1, DAAQ-ECOF also exhibited a capacity retention of 98% (107 mA h g−1) after 1800 cycles. Attractively, by introducing benzoquinone building units or attaching 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) functional groups (Fig. 11b) via molecular design, the capacity could be increased to 210 mA h g−1 and the discharge voltage could be increased to 3.6 V (Fig. 11d).
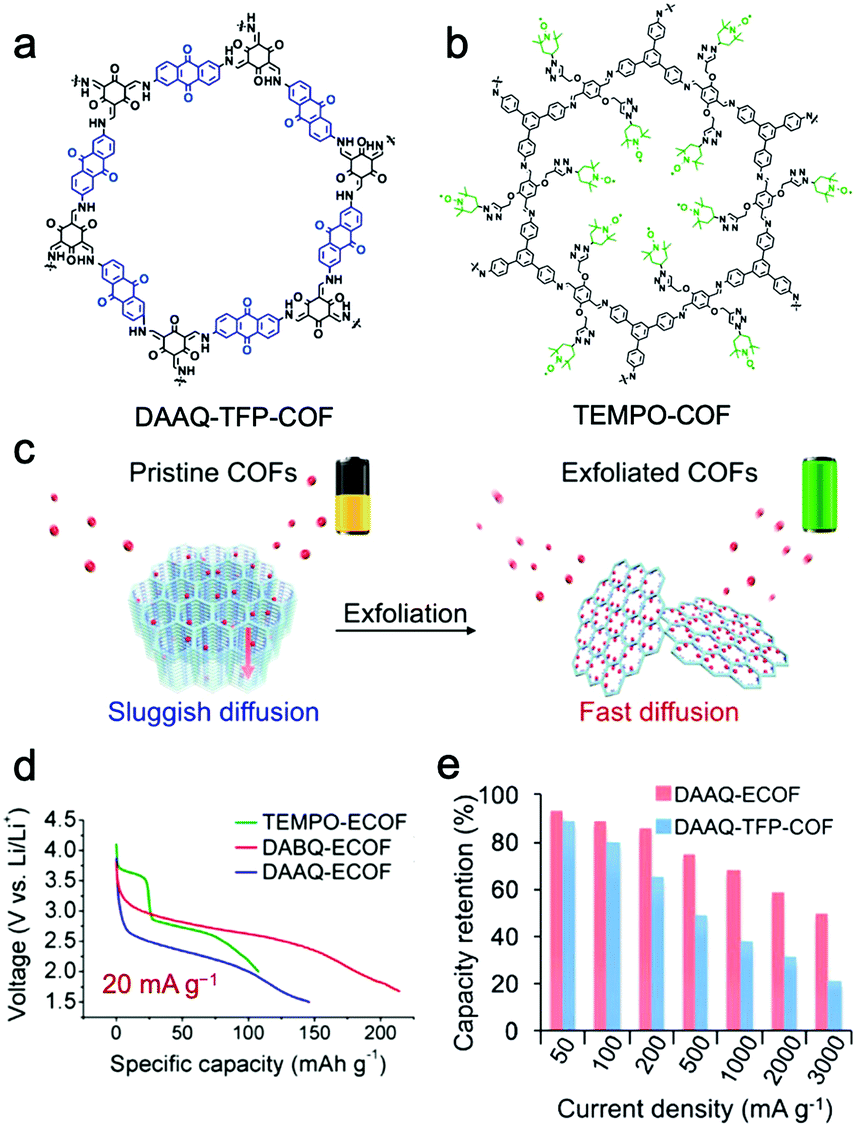 | ||
| Fig. 11 Chemical structures of DAAQ-TFP-COF (a) and TEMPO-COF (b). (c) Schematic representation of the exfoliated 2D redox-active COFs as cathodes for LIBs; (d) discharge curves; (e) capacity retention. Reproduced from ref. 137 with permission from American Chemical Society, copyright 2017. | ||
3.1.1.2 COFs as the host for phase-changing components. COFs can be considered as promising hosts for phase changing cathodes. Phase changing cathodes during charge and discharge processes involve chemical bond breaking and recombining as well as multiple phase transitions.3 The theoretical capacities of the phase changing cathodes are generally high because these reactions usually involve multi-electron transfer.
Taking lithium–sulfur batteries (LSBs) as an example, they are typical multi-electron transfer reaction systems with a high theoretical capacity of 1675 mA h g−1. Although LSBs are considered as one of the most promising candidates for next-generation batteries due to their exceptional theoretical capacity, low cost, and natural abundance, there are several drawbacks hindering their development: (1) element sulfur will produce large volume fluctuation (about 80%) during charge and discharge owing to the huge density mismatch between sulfur and final products, Li2S; (2) the insulating nature of sulfur and Li2S2/Li2S will influence electrochemical kinetics; (3) the intermediates (lithium polysulfides (PSs)) during operation are soluble and migrate freely between cathode and anode, leading to a “shuttle effect” and rapid capacity fading.170–172 COFs are suitable for LSBs for the following reasons: (1) the accessible cavities and adjustable apertures can be designed to accommodate volume changes and limit the migration of PSs; (2) the electronic interactions between the layers in 2D COFs can facilitate the transportation of electrons and ions; (3) the interior of pores can be introduced with specific functionalities to bind the soluble intermediates; (4) COFs consist of strong covalent linkages, which provide favorable thermal and chemical stability. Several efforts have been devoted to illustrating COFs as suitable hosts to encapsulate sulfur species.173–181
CTF-1 was the first COF used as the sulfur host for LSBs.173 It possessed a surface area of 789 m2 g−1 with a pore size of 1.23 nm. The composite CTF-1/S@155 °C was prepared by a melt-diffusion strategy with a sulfur loading of 34 wt%, which showed a discharge capacity of 1197 mA h g−1 at the 2nd cycle and maintained at 762 mA h g−1 after 50 cycles at 0.1C.
The sulfur loading is an important parameter, which can influence the electrochemical performance. A porphyrin-based COF (Por-COF) loaded with 55 wt% sulfur gave a capacity of 929 mA h g−1 at the 2nd cycle and held a capacity of 633 mA h g−1 after 200 cycles at 0.5C.174 Another porphyrin-based COF (Py-COF) with 70 wt% sulfur loading delivered a capacity of 963.4 mA h g−1 after 100 cycles at 1.0C.175
Compared with N-doped porous organic polymers, doping positively polarized elements or introducing active groups that can react with sulfur can trap PSs more efficiently, such as elemental B, fluorinated groups, or vinyl groups.176,177,179,180 Tang and Li et al. presented a boronate ester COF (COF-1) as the sulfur host for trapping PSs (Fig. 12a).176 With a sulfur loading of 40%, COF-1/S delivered an outstanding initial capacity up to 1628 mA h g−1, and a capacity of 929 mA h g−1 was achieved after 100 cycles at 0.2C. CTF-1 was selected for comparison because it had a similar pore size but a different pore surface environment. The calculated adsorption energy between Li+ ions with COF-1 and CTF-1 was similar, while that for linear S82− in COF-1 and CTF-1 delivered quite different values: 34 kcal mol−1 for COF-1 and 24 kcal mol−1 for CTF-1 (Fig. 12b). These differences suggested that COF-1 can trap Sx2− species more effectively, which may be attributed to the more positively polarized pore surface because of the electron-deficient B atoms in COF-1.
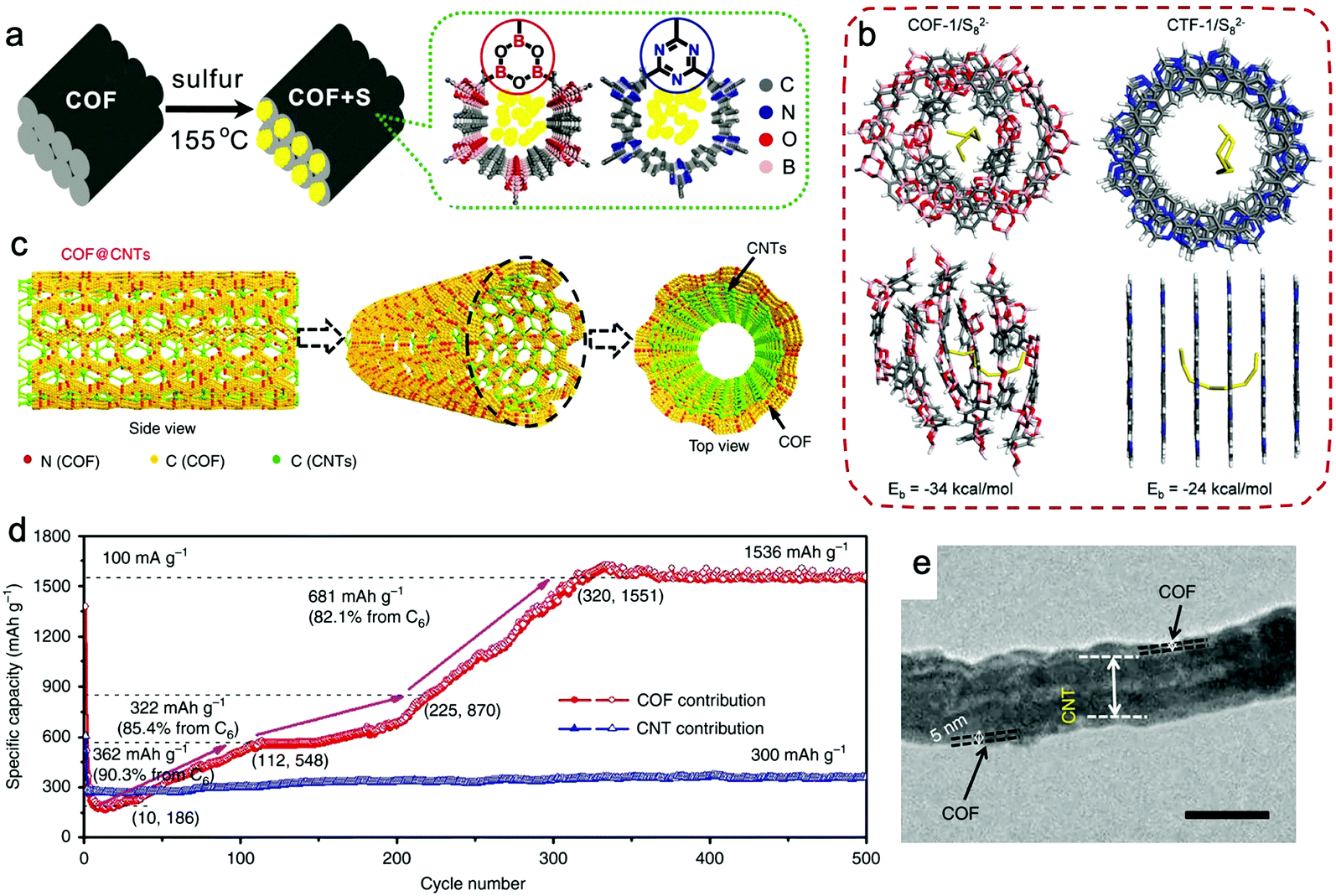 | ||
| Fig. 12 (a) Graphical representation of the synthesis of COF/S composites. (b) The adsorption energy for S82− anions in the pores of COF-1 and CTF-1, calculated by quantum mechanics/molecular mechanics. Reproduced from ref. 176 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, copyright 2016. (c) Schematic of COF@CNTs showing CNTs covered with a few COF layers. (d) COF's capacity contribution calculated based on its mass in COF@CNTs at 100 mA g−1 during the charge/discharge process. (e) TEM image of COF@CNTs with a scale bar of 100 nm. Reproduced from ref. 189 with permission from Nature Publishing Group, copyright 2018. | ||
The COF-derived material is another alternative to increase the conductivity of the composite cathode and hence improve its electrochemical performance.177,178 A boron/oxygen co-doped porous carbon (BOC) host material (BOC@CNT) was prepared by carbonizing the COF-10@CNT composite. BOC@CNT/S had a sulfur loading of 68.5% and exhibited an outstanding reversible capacity of 1077 mA h g−1 after 200 cycles at 0.2C, and 794 mA h g−1 after 500 cycles at 1C.177
Tables 1 and 2 summarize the electrochemical performances of COFs as redox-active materials or as hosts for phase-changing components used in cathodes, respectively. The COF-based cathodes possessed high capacity (up to ∼200 mA h g−1) and good cycling performance, which were comparable or even better than those of the commercialized cathodes (e.g., LiPFeO4, capacity of about 150 mA h g−1 in practice). However, due to the lack of investigations of COF-based cathodes in full cells these evaluations are not very appropriate and further efforts are still needed.
| Capacity (CC/DC/CD) | Cycling performance (RC/CD/CN) | Voltage range (vs. Li/Li+) | Ref. | |
|---|---|---|---|---|
| Note: CC: charge capacity (mA h g−1), DC: discharge capacity (mA h g−1), CD: current density (mA g−1), RC: reversible capacity (mA h g−1), CN: cycle number. | ||||
| PIBN-G | —/271.0/28 | 208.1/280/300 | 1.5–3.5 | 168 |
| NT-COF | —/124/20 | —/20/100 | 1.5–4.0 | 165 |
| 2D CCP-HATN | —/62.5/11.7 | —/—/— | 1.2–3.9 | 166 |
| 2D CCP-HATN@CNT | —/116/11.7 | —/58.5/1000 | ||
| PI-ECOF-1 | —/112/14.2 | 72/14.2/300 | 1.5–3.5 | 193 |
| PI-ECOF-1/rGO50 | —/167/14.2 | —/142/300 | ||
| 2D-PAI@CNT | —/104.4/100 | —/500/8000 | 1.5–3.5 | 169 |
| 2D-PAI | —/28.5/100 | —/—/— | ||
| DAAQ-ECOF | —/145/20 | 107/500/1800 | 1.5–4.0 | 137 |
| DABQ-ECOF | —/210/20 | —/—/— | ||
| TEMPO-ECOF | —/115/20 | —/—/— | ||
| DTP-ANDI-COF | —/42/200 | —/—/— | 1.5–3.5 | 167 |
| DTP-ANDI-COF@CNTs | —/74/200 | 67/200/700 | ||
| Tp-DANT-COF | 78.9/93.4/200 | 71.7/1000/600 | 1.5–4.0 | 164 |
| Tb-DANT-COF | 135.4/144.4/50 | 80.1/500/300 | ||
| PPTODB | —/198/20 | 135.2/20/150 | 1.5–3.5 | 194 |
| DAAQ-TFP | —/—/78.5 | 53.5/157/500 | 1.4–3.6 | 195 |
| DAPH-TFP | —/96.2/85.5 | 81.7/171/500 | ||
| PEDOT@DAAQ-TFP | —/—/78.5 | 59.8/157/500 | ||
| PEDOT@DAPH-TFP | —/99.2/85.5 | 93.2/171/500 | ||
| Loading (ML/PL) | Capacity (CC/DC/CD) | Cycling performance (RC/CD/CN) | Voltage range (vs. Li/Li+) | Ref. | |
|---|---|---|---|---|---|
| Note: ML: mass loading (mg cm−2), PL: percentage loading (%), DC: discharge capacity (mA h g−1), CD: current density (C), RC: reversible capacity (mA h g−1), CN: cycle number | |||||
| CTF-1/S@155 °C | —/34 | 1304/1497/168 | 762/168/50 | 1.1–3.0 | 166 |
| S/Azo-COF | —/39 | —/1536/167.2 | 741/167.2/100 | 1.5–2.8 | 196 |
| COF-F-S | 0.5/60 | —/1120/167.2 | 645/167.2/100 | 1.7–2.8 | 180 |
| S/TpPa-COF@MWCNTs | 1–2/— | —/1242.2/83.6 | 526.5/836/450 | 1.6–2.8 | 197 |
| S@TFPPy-ETTA-COF | —/— | —/723/167.2 | —/167.2/130 | 1.7–2.8 | 181 |
| poly-sulfide@TFPPy-ETTA-COF | —/— | —/1069/167.2 | —/167.2/130 | ||
| Por-COF/S | 1.8/55 | —/1166/840 | 633/840/200 | 1.8–2.7 | 174 |
| Py-COF/S | 0.8–1.2/70 | —/1145/836 | 877.2/3344/200 | 1.8–2.7 | 175 |
| S-COF-V | —/67 | —/1400/335 | 959/335/100 | 1.7–2.8 | 179 |
| COF-F-SeS2 | 1.2/40 | —/1703/167.2 | 970/167.2/100 | 1.7–2.8 | 198 |
| COF-1/S | —/40 | —/1628/334.4 | 770/836/200 | 1.5–3.0 | 176 |
| TAPB-PDA-COF/S | —/60 | —/1357/200 | 705/200/210 | 1.8–2.8 | 199 |
| HCPT@COF/S | —/69.3 | —/1224/334.4 | 1053/334.4/300 | 1.7–2.8 | 200 |
| Carbonized COFs | |||||
| BOC@CNT/S | 3.5/— | —/1434/334.4 | 797/1672/500 | 1.7–2.8 | 177 |
A COF denoted as IISERP-CON1 and incorporated with the triazole moiety was grown into self-exfoliated COF NSs and further used as the anode in lithium-ion batteries (LIBs).184 A reversible intercalation mechanism was proposed due to the following reasons: its electrochemical behavior did not show obvious redox peaks during cycling; based on computational studies, the distance between lithium and the ring nitrogen atoms of the triazole or the hydroxyl oxygens of the phloroglucinol was longer than the Li–N or Li–O distance; IR, PXRD, X-ray photoelectron spectroscopy (XPS) and 1H nuclear magnetic resonance (1H-NMR) characterization demonstrated the weak interaction between lithium ion and COF. A capacity of 720 mA h g−1 at 100 mA g−1 over 100 cycles was achieved and a capacity of over 500 mA h g−1 at 500 mA g−1 after 1000 cycles was retained. Such a good cycling performance was attributed to the mildly reversible interaction between lithium and IISERP-CON1. Wang et al.121 reported a few-layered structure of COF-10@CNT used in potassium ion batteries. The exposed active sites attributing to the few-layered structure facilitated the intercalation of K+, which was based on the π-cation interaction between K+ and conjugated π-electrons of benzene rings. Thus, an impressive reversible capacity of 288 mA h g−1 after 500 cycles at 0.1 A g−1 and 161 mA h g−1 after 4000 cycles at 1 A g−1 was obtained.
For achieving higher capacity, introducing more redox-active sites in the backbones of COFs is desired. A few-layered 2D imine-based COF fixed by carbon nanotubes was achieved and used as the anode in LIBs (Fig. 12c).189 The CNTs were covered by thin COF layers with a thickness of about 5 nm (Fig. 12e). Interestingly, the capacity of COF@CNTs gradually increased from 362 to 1536 mA h g−1 at 100 mA g−1 after 320 cycles (Fig. 12d). The activation process and high capacity were attributed to (1) the 14-electron redox mechanism involving the reaction between Li+ ion and COF skeleton (one Li+ per CN linkage and six Li+ per benzene ring); (2) the expansion of the interlamellar space in COFs by ion insertion during the lithiation/delithiation process.
Besides, the formation and transformation of radical intermediates during the lithiation/delithiation process were observed. Moreover, stabilizing the radical intermediates is an efficient method to improve electrochemical performance.188,192 β-Ketoenamine linked DAAQ-COF was employed as the anode in sodium-ion batteries (SIBs) and it resulted in C–O˙ and α-C radical intermediates upon charging and discharging.188 It was found that reducing the thickness of COFs could promote the stability of radical intermediates as well as their contributive capacity, owing to the restriction of interlayer electron self-exchange behavior and the increase of available active sites. The COF with a thickness of 4–12 nm exhibited a high capacity of 420 mA h g−1 at 100 mA g−1 and outstanding cycling stability with 99% capacity retention over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 5 A g−1.
000 cycles at 5 A g−1.
Despite the fact that very high capacity (>1500 mA h g−1) can be achieved by using COF-based anodes, their high voltage, long activation process, and high cost are still greatly limiting their real application.
3.1.3.1 Solid electrolyte. Since liquid electrolytes suffer from serious safety issues including electrolyte leakage, flammability, and dendrite formation, solid electrolytes have been considered with great interest.11,40,201,202 However, the processability and mechanical strength of inorganic electrolytes and the ionic conductivity of polymeric solid electrolytes still cannot fulfill the demands for practical application. For example, poly(ethylene oxide) (PEO) has been accepted as a promising candidate for solid state electrolytes in the past few years, but its further application has been hindered by limited ionic conductivity (ionic conductivity can up to 10−4 S cm−1 under 65–78 °C) and generally a low t+ value. Regarding inorganic ceramic materials (e.g., Al2O3 and Li7La3Zr2O12), although they have been widely studied owing to their outstanding conductivity (ionic conductivity ranges from 10−5 to 10−3 S cm−1 at room temperature), the poor machinability and the often instability at electrodes limit their practical application.
The basic mechanism for Li+ ion transport is the Grotthuss (hopping) mechanism, in which the ion can hop from sites to sites. COFs may provide the following opportunities: (1) open channels and well-defined pore structures create directional ion conduction pathways; (2) hopping ability is promoted by introducing functional groups and/or guest molecules with hopping sites for ions; (3) versatile structural design features the selective adsorption of counter ions, which can facilitate the migration of target ions.61,153,203–210
There are two effective strategies to increase Li-ion conductivity: (1) building an ionic skeleton to facilitate the dissociation of ion pairs and promote target ionic migration; (2) introducing soft and flexible building units or polymer chains in the framework to increase hopping sites.
Feng et al. successfully prepared an anionic 3D COF (CD-COF-Li) with Li+ as the counterion by linking soft and flexible building blocks, and investigated its ion conduction behavior as a quasi-solid electrolyte.203 With the encapsulation of a small amount of ethylene carbonate (EC)/dimethyl carbonate (DMC) and LiPF6 in the channels, the resultant composite exhibited a conductivity of up to 2.7 × 10−3 S cm−1 at 30 °C with a low activation energy of 0.26 eV. Besides, the comparison between neutral framework and cationic/anionic framework was investigated for further demonstrating the promotion effect of the ionic framework.61,153,205,208,209 It is worth mentioning that the organic solvents that can greatly increase ionic conductivity are inevitably used in the solid electrolyte fabrication process, but not all the works clearly clarify whether the solvent molecules are residual or not.
Poly(ethylene oxide) (PEG)-based polymers with oxygen atoms as Li+ ion hopping sites are representative polymeric solid-state electrolytes; however, the existence of the crystalline area restricts their chain oscillation and ion conduction ability. Anchoring oligo(ethylene oxide) chains to the pore walls or accommodating PEG in COFs provides chances to suppress the oxyalkyl chain crystallization and accelerate ion conduction in the channels.204–207 A polyelectrolyte COF, TPB-BMTP-COF, was loaded with LiClO4 through a solution diffusion method.204 The Li-ion conductivity of Li+@TPB-BMTP-COF was up to 1.66 × 10−4 S cm−1 at 80 °C, which is 30 times and 731 times higher than that of Li+@TPB-DMTP-COF (methoxy groups on the edge phenyl units) and Li+@TPB-TP-COF (bare pore walls), respectively. Li+@TPB-BMTP-COF possessed the lowest activation energy of 0.87 eV among these three electrolytes. The obviously increased conductivity and decreased activation energy of Li+@TPB-BMTP-COF could be attributed to the advantage that the covalently bonded dense oligo(ethylene oxide) chains can complex with lithium ions and form a polyelectrolyte interface in the channels, thus accelerating the dissociation of the ionic compounds and facilitating the ion hopping for Li-ion transport between the neighboring hopping sites.
Without the complex monomer synthetic procedure, Feng and Wang et al. directly encapsulated low-molecular-weight polyethylene glycol (PEG, Mw = 800) and lithium salt into the channels of COFs to prepare all-solid-state electrolytes without any solvent residue (Fig. 13a).205 The results showed that PEG in the confined space can indeed significantly boost the ion conduction rate by orders of magnitude. PEG-Li+@EB-COF-ClO4 showed a high ion conductivity, reaching 1.78 × 10−3 S cm−1 at 120 °C. Interestingly, PEG 800 has a phase transition from the elastic to viscous state at around 30 °C, but after the incorporation of PEG into the channels, the differential scanning calorimetry (DSC) curve did not show any endothermic/exothermic peaks from −10 to 120 °C. It indicated a possibility that the incorporation of PEG chains in the channels could inhibit the formation of crystalline regions of PEG, and endowed a wide operating temperature range (Fig. 13b).
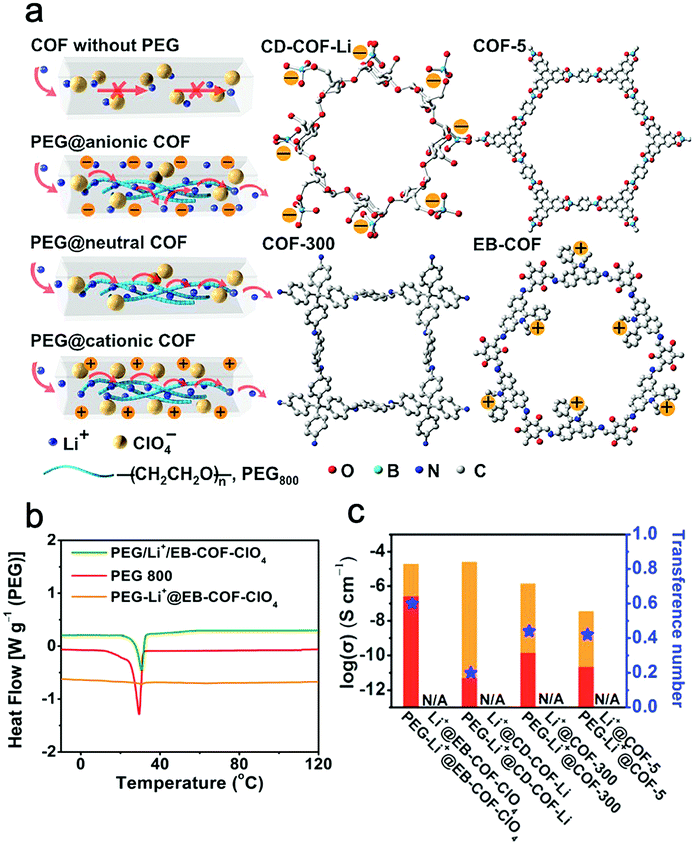 | ||
| Fig. 13 (a) Schematic for Li+ transport in neutral or ionic COFs, and chemical structures of CD-COF, COF-5, COF-300, and EB-COF. (b) DSC curves of PEG, PEG-Li+@EB-COF-ClO4, and PEG/Li+/EB-COF-ClO4 (mechanical mixture sample). (c) The contribution of Li+ conduction (red area) and anion conduction (orange area). Reproduced from ref. 205 with permission from American Chemical Society, copyright 2019. | ||
Notably, t+ is another important factor for electrolytes that depends on the proportion of the target ions to the total ions carried through the electrolyte. During the charge–discharge process, Li+ and its counter anions migrate oppositely, while anions are blocked by the anode and accumulated on it. The accumulation of anions leads to a concentration gradient and polarization problem, which will cause increased internal impedance and the occurrence of inevitable side reaction. Thus, improving the value of t+ close to unity is important for cell performance.
Feng and Wang et al. chose two neutral COFs (COF-5, COF-300), a cationic COF (EB-COF-ClO4), and an anionic COF (CD-COF-Li) with the inclusion of PEG and LiClO4 to explore their t+ (Fig. 13a).205 Among these four PEG-included COFs, PEG-Li+@EB-COF-ClO4 showed the highest t+ of 0.60, while PEG-Li+@CD-COF-Li only possessed a low t+ of 0.2 (Fig. 13c). It is attributed to the anionic skeleton that can fix Li+ to restrain its mobility, but the cationic skeleton can trap anions and promote Li+ migration. To further improve t+, a single-ion conducting strategy was provided. Lee and Zhang et al. reported a series of crystalline imidazolate-containing diamine (1-H, 1-CH3, 1-CF3) iCOFs (Li-ImCOFs) as quasi-solid electrolytes.208 Ion conductivities of 7.2 × 10−3, 5.3 × 10−3, and 8.0 × 10−5 S cm−1 were achieved at room temperature for CF3-Li-ImCOF, H-Li-ImCOF, and CH3-Li-ImCOF (with ∼20% propylene carbonate) with t+ values of 0.81, 0.88, and 0.93, respectively. These high t+ values are owing to the fact that, without further addition of Li salt, the anionic groups were fixed in COFs’ skeleton and immobilized, thus providing higher Li+ mobility. It is noted that the ion conductivity obtained via the single ion conducting strategy in all-solid-state electrolytes needs to be further improved.209
In addition to the Li+ ion conduction, COFs can also serve as proton conductors in the proton exchange membrane fuel cells (PEMFCs).211–219 Nafion-based electrolytes used as proton conductors are strongly dependent on humidity and are deactivated above 80 °C. The current strategies for developing COF-based proton conductors rely on encapsulating proton carriers in the channel,212,213 constructing an ionic skeleton,214 covalently decorating pendant acid groups, or combining a proton-conductive polymer to form a composite conductor.215–217 Nitrogenous molecules (e.g., imidazole molecules, pyridines), acidic molecules (e.g., H3PO4, polyoxometalates), water molecules, and pendant sulfonic acid groups are usually considered as favorable proton carriers encapsulated in COFs’ channels or fixed on the walls to enhance the integral proton conductivity.
Jiang et al. endowed TPB-DMTP-COF with high proton conductivity by loading proton carriers (triazole or imidazole) in it (Fig. 14a).212 The im@TPB-DMTP-COF exhibited a conductivity of 4.37 × 10−3 S cm−1 at 130 °C with 155% imidazole loading. Besides, Li and Zang et al. applied ethidium bromide (EB) based cationic 2D COFs with a series of counterions (EB-COF:X, X = F, Cl, Br, I, and PW12O403−) for proton conductors.214 Since hydrophilic PW12O403− possesses a good water retention ability, it may attract water clusters and form interconnected hydrogen bonding networks throughout the channels. Therefore, EB-COF:PW12 with an abundant proton pathway showed a proton conductivity of 3.32 × 10−3 S cm−1 under 97% RH at 25 °C.
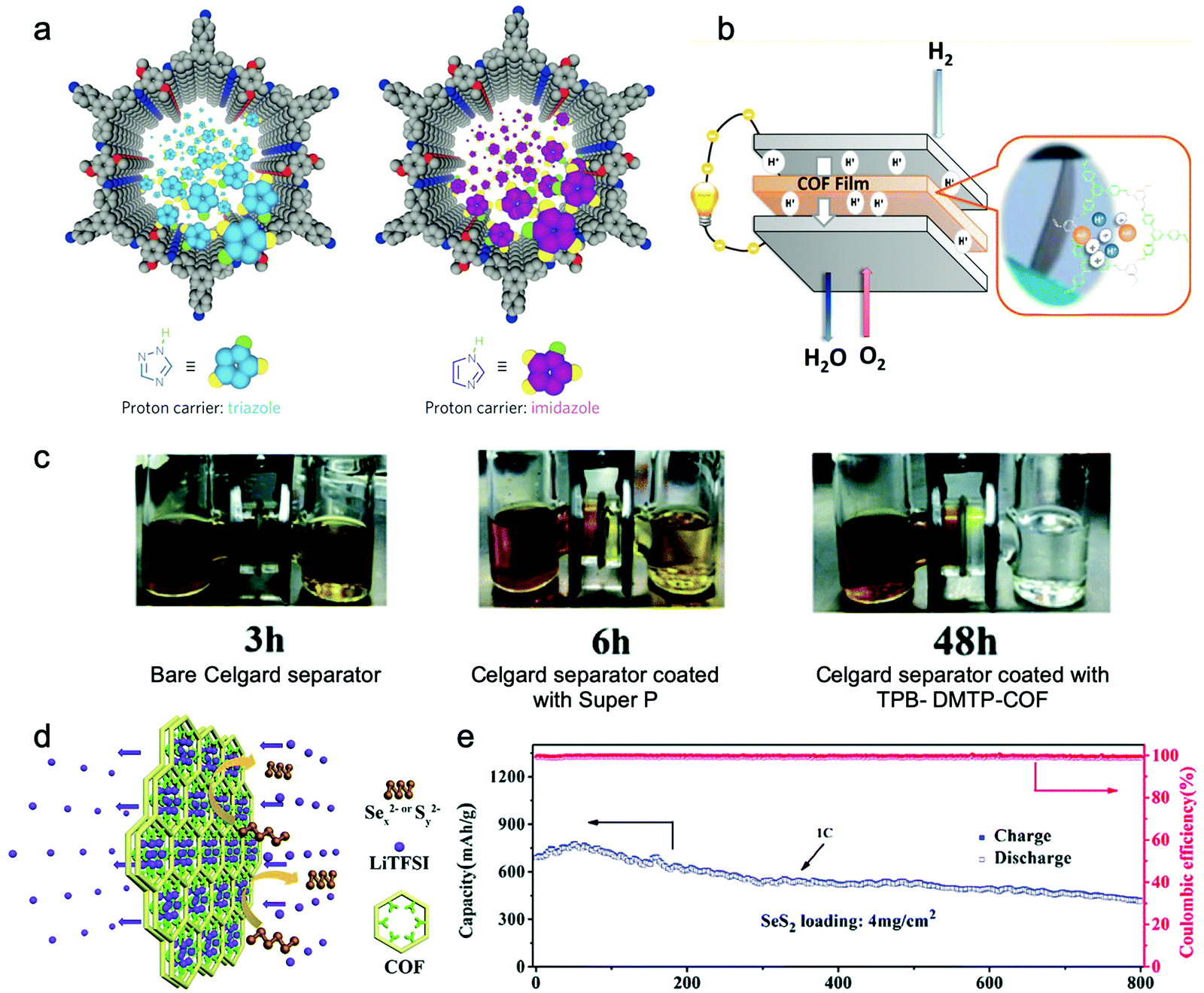 | ||
| Fig. 14 (a) Schematic for proton carriers in COFs’ channels. Reproduced from ref. 212 with permission from Nature Publishing Group, copyright 2016. (b) Schematic of the PEMFCs with COF films as membrane electrode assemblies (MEA). Reproduced from ref. 211 with permission from American Chemical Society, copyright 2017. (c) Visual experiments displaying permeation of polysulfides with different separators. (d) Schematic illustration of the usage of the TPB-DMTP-COF separator. (e) Cycling performance of the Li–SeS2 cell with the TPB-DMTP-COF modified separator at 1C. Reproduced from ref. 222 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Remarkably, COF-based proton conductors were applied to construct a PEMFC (Fig. 14b).211,220 For example, the RT-COF-1AcB film prepared from 1,3,5-tris(4-aminophenyl)benzene (TAPB) (58) and 1,3,5-benzenetricarbaldehyde (BTCA) (23) possessed a proton conductivity of 5.25 × 10−4 S cm−1 at 313 K under 100% RH. When assembled in a PEMFC, it exhibited an optimal balanced performance with high conductivity (1.1 × 10−2 S cm−1 at 323 K) and low H2 fuel crossover, leading to a maximum power density (12.95 mW cm−2) and a maximum current density (53.1 mA cm−2).
Up to now, the reported COFs and their composites could achieve a Li+ conductivity of 10−3 S cm−1 at room temperature and a high ion transference number (>0.9). However, in most cases, COFs were pressed into pellets for the ion conduction measurements. The ability to form self-standing films for COFs makes them good candidates as flexible solid-state electrolytes but this remains largely unexplored.
3.1.3.2 Separator and interlayer. As mentioned in Section 3.1.1.2, one of the most effective strategies to suppress the shuttle effect of phase-changing electrodes is to fabricate a composite electrode by impregnating redox-active components (e.g., S and Se) in a porous host. Although this leads to a remarkable promotion in their electrochemical performance, the energy density is inevitably decreased because the host materials make up a mass percentage. To block the shuttle effect without sacrificing the redox-active component loadings in cathode composites, introducing a cation (e.g. Li+ and Na+) selective separator/interlayer between the electrodes that inhibits the passage of soluble intermediates (e.g., PSs) is developed. Compared with the conventional materials used in interlayers/separators including polymers, graphene-based materials, carbon materials, and metal oxides, 2D COFs with pre-designable and well-defined channels in nanometer-scale are more beneficial for selective passage of cations.221–224
Wang and Hu et al. prepared a COF-rGO double-layer film by depositing dispersed COF-1 onto graphene through filtration and applied it as an ion-sieving separator in both organic sodium-ion batteries and Li–S batteries.221 The cell assembled with the COF-rGO membrane showed an excellent initial capacity of 1386.9 mA h g−1 which slowly decreased to 1169.4 mA h g−1 after 50 cycles at 0.1C, while the capacity of the cells without an ionic sieve decreased from 869.9 mA h g−1 to 519.1 mA h g−1 after 50 cycles. Together with permeation measurements that demonstrated that the COF-rGO membrane could efficiently block the passage of polysulfides, these results proved that the improvement in the electrochemical performance was originated from the inhibition of the shuttle of polysulfides by the ionic sieve membrane.
Besides, a separator coated with TPB-DMTP-COF was proved to preferentially adsorb LiTFSI species into its cavities by strong hydrogen-bonding interactions, which created a narrower cavity size and further blocked the transportation of polysulfide/polyselenide species in the electrolyte and enhanced the Li+ ion conductivity at the same time (Fig. 14d).222 After separating the LiTFSI solution and LiTFSI/Li2S6 solution in the H-tube by the TPB-DMTP-COF-coated separator, the LiTFSI solution side remained colorless after 48 h, which indicated its effective inhibition for the transport of Li2S6 species (Fig. 14c). Meanwhile, a three-times increment in the lithium ion migration coefficient was achieved after using the TPB-DMTP-COF-coated separator. At a SeS2 loading of 2 mg cm−2, a specific capacity of 844.6 mA h g−1 was achieved at 0.5C. After increasing the SeS2 loading to 4 mg cm−2, a specific capacity of 684 mA h g−1 at 1C was achieved, and a capacity of 416.3 mA h g−1 was retained after 800 cycles (Fig. 14e).
Lee and Kwak et al. presented a strategy to construct a “microporous COF net on mesoporous CNT net” hybrid architecture and fabricated a self-standing film as the interlayer (NN interlayer) between sulfur cathode and separator to capture Li2Sx.223 Since the high conductivity and the hierarchical structure including mesopores and micropores of the NN interlayer were beneficial for electron and ion transportation as well as PS capture, the cell equipped with the NN interlayer exhibited a capacity retention of 84% after 300 cycles at 2.0C, but 15% for the cell without the interlayer.
3.2 Supercapacitor
Supercapacitors (SCs) possess high power density, high coulombic efficiency, long cycling life, and fast charge–discharge rate. Traditional SCs store electrical energy based on the surface charging mechanism through electrochemical double-layer capacitors (EDLCs) that attract anions and cations from the electrolyte to opposite electrodes’ internal surfaces after applying an external electrical field. The capacity of EDLCs is related to the available surface sites of the electrode materials, where surface area, porosity, thickness of the double layer, dielectric constant, and ion species in the electrolyte all have impacts.6,9 The representative electrode materials like porous carbon-based active materials for EDLCs have a high specific surface area, good chemical and electrical stability, and good conductivity. Another crucial class of supercapacitors based on reversible surface redox reactions, namely faradaic supercapacitors (FS) or pseudocapacitors, have shown at least an order of magnitude improvement in energy densities. The pseudocapacitance of the electrode materials strongly relies on the pore size and distribution, porosity, and redox-active species. Accordingly, combining the surface charging mechanism and the reversible surface redox reactions can significantly increase the energy density of supercapacitors. COFs with inherently large surface area (abundant adsorption sites), well-defined channels and long-range ordered arrangement (idealized ion transport pathways within the electrode–electrolyte interface), and redox-active building blocks (excellent redox activity) can contribute to both EDLCs and pseudocapacitors. COF-based SCs have been extensively developed in recent years. There are three ways for COFs to apply as electrodes in SCs: (1) synthesizing a redox-active skeleton by directly introducing or post-synthetically functionalizing with reversible redox response units; (2) constructing COF-based composites with conductive materials by inclusion or in situ polymerization of conductive polymers in their nanochannels to enhance both electrical conductivity and processability; (3) pyrolyzing COFs to form porous carbon materials to enhance conductivity and capacitance.Although high charge carrier transportation rates are achieved in the nano-scaled region of 2D COFs with high periodicity, the electron conductivity of COFs is still limited due to the existence of disordered area and particle boundaries. Therefore, pristine COFs are usually mixed with or grown on conductive materials to afford SCs. The contribution of redox-active groups was illustrated by comparing DAAQ-TFP COF possessing reversible redox-active units 2,6-diaminoanthraquinone (DAAQ) (57) (SBET = 435 m2 g−1) and DAB-TFP COF without redox-active groups (SBET = 365 m2 g−1).225 Not surprisingly, DAAQ-TFP COF provided a capacitance of 48 ± 10 F g−1 with a current density of 0.1 A g−1 and the capacitance remained at 40 ± 9 F g−1 after more than 5000 cycles, while DAB-TFP COF showed a double-layer capacitance behavior that delivered a lower capacitance of 15 ± 6 F g−1. However, only 2.5% of the DAAQ moieties in DAAQ-TFP COF were utilized to participate in the electrode reaction, which may be attributed to the inferior conductivity originated from randomly oriented and polycrystalline DAAQ-TFP COF particles. Yang and Guo et al. presented that the conductivity of COFs could be improved by enhancing their nitrogen content. A triazine-based COF, PDC-MA-COF with a nitrogen content of 47.87%, showed an electronic conductivity of 3.34 × 10−2 S cm−1.227 The PDC-MA-COF exhibited a capacitance of 335 F g−1 at 1 A g−1 and approximately 19.71% triazine units were accessed during the electrochemical process.
An alternative approach to introducing redox-active groups to COFs is post-synthetic modification. Jiang et al. prepared radical COFs by post functionalizing the channel wall of imine-linked COFs with TEMPO via click chemistry.228 [HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C]X%-NiP-COFs (X = 0, 50, and 100) assembled by 5,10,15,20-tetrakis(4′-tetraphenylamino) porphyrin (NiP) (62) with different molar ratios of 2,5-bis(2-propynyloxy)terephthalaldehyde (BPTA) (7) and 2,5-dimethoxyterephthalaldehyde (DMTA) (3) were grafted with 4-azido-2,2,6,6-tetramethyl-1-piperidinyloxy to yield [TEMPO]50%-NiP-COF and [TEMPO]100%-NiP-COF (Fig. 15a and b). After decorating TEMPO on the walls, a pair of reversible peaks appeared in CV curves. [TEMPO]100%-NiP-COF exhibited a capacitance of 167 F g−1 at 100 mA g−1, similar to redox-active MOFs (Fig. 15c).
C]X%-NiP-COFs (X = 0, 50, and 100) assembled by 5,10,15,20-tetrakis(4′-tetraphenylamino) porphyrin (NiP) (62) with different molar ratios of 2,5-bis(2-propynyloxy)terephthalaldehyde (BPTA) (7) and 2,5-dimethoxyterephthalaldehyde (DMTA) (3) were grafted with 4-azido-2,2,6,6-tetramethyl-1-piperidinyloxy to yield [TEMPO]50%-NiP-COF and [TEMPO]100%-NiP-COF (Fig. 15a and b). After decorating TEMPO on the walls, a pair of reversible peaks appeared in CV curves. [TEMPO]100%-NiP-COF exhibited a capacitance of 167 F g−1 at 100 mA g−1, similar to redox-active MOFs (Fig. 15c).
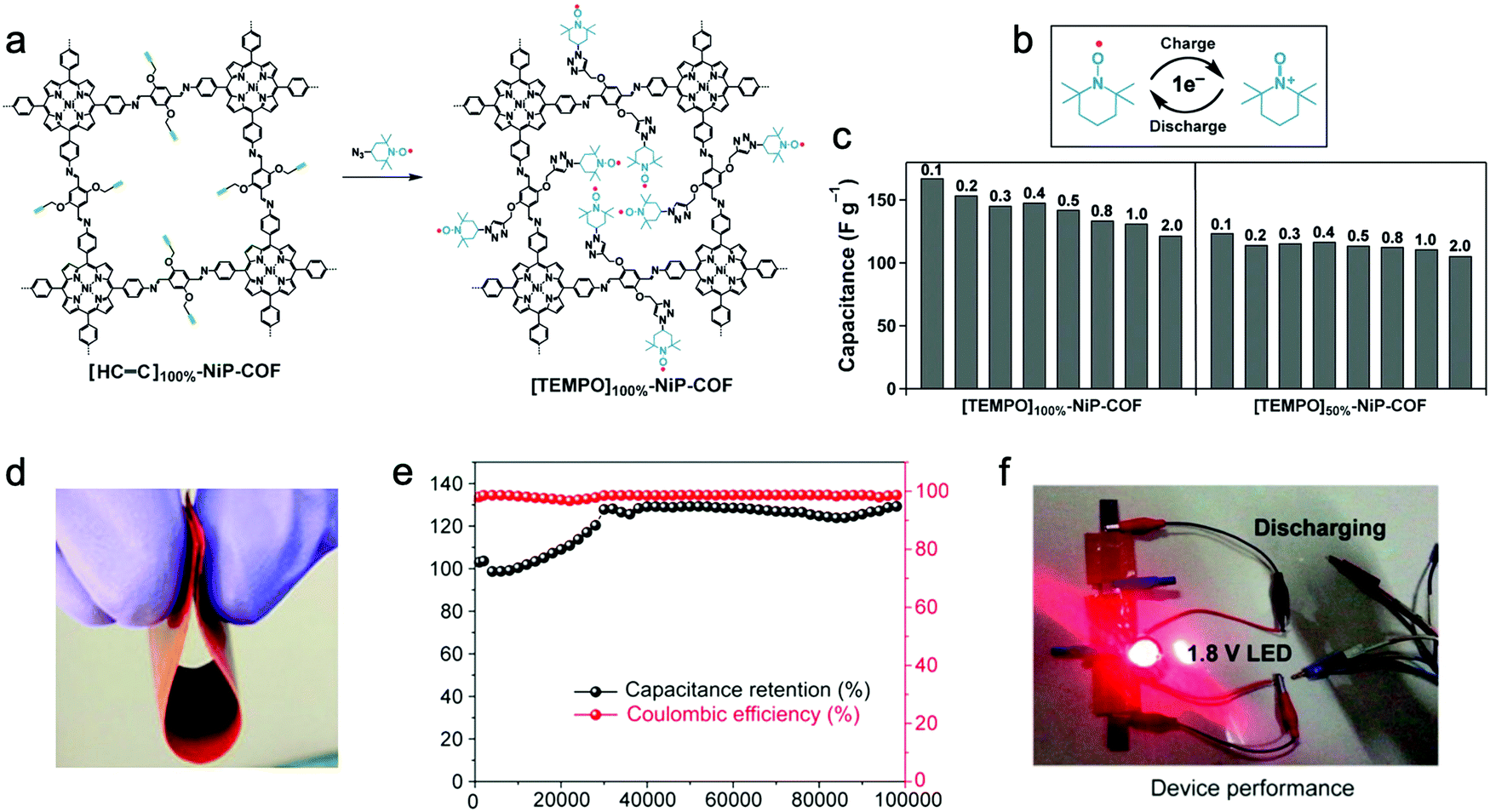 | ||
| Fig. 15 (a) Schematic illustration of the synthesis of a radical COF ([TEMPO]100%-NiP-COF) in which TEMPO radicals are anchored on the walls. (b) The redox reaction involves one electron of TEMPO radicals during the charge–discharge process. (c) Capacitance of radical COFs at various current density (on the top). Reproduced from ref. 228 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, copyright 2015. (d) Photo of the self-standing and flexible thin sheet. (e) Cycling performance of TpOMe-DAQ with 1 cm2 area exposed. (f) Photo of the enkindling experiment: a 1.8 V LED was lighted by three solid-state-devices connected with each other. Reproduced from ref. 229 with permission from American Chemical Society, copyright 2018. | ||
Recently, researchers found that the existence of interlayer hydrogen bonds can effectively enhance 2D COFs’ electrochemical stability.226,227,229 In the charge–discharge process of TpPa-(OH)2 synthesized from 1,3,5-triformylpholoroglucinol (Tp) (24) and 2,5-dihydroxy-1,4-phenyldiamine [Pa-(OH)2] (44), a concerted electron–proton transfer reaction occurred.226 TpPa-(OH)2 exhibited a capacitance of 416 F g−1 at 0.5 A g−1, while high accessibility (43%) of redox-active hydroquinone units was achieved. The capacitance remained at 66% after 10000 cycles, attributed to the fact that the H-bonding between the carbonyl oxygen of the benzoquinone functionality and its neighboring amine functionality inhibited the decomposition of benzoquinone.
Constructing conductive COFs is a promising method for designing high-performance COF-based supercapacitors. Gu, Li, and Yan et al. built a Ni-COF containing Ni(II)-Salphen units. The Ni-COF possessed an electrical conductivity up to 1.3 × 10−2 S cm−1 and showed a high specific capacitance of 1257 F g−1 at 1 A g−1 with an outstanding cycling performance in that the capacitance remained at 94% after 10000 cycles. An excellent energy density of 130 W h kg−1 at a power density of 839 W kg−1 was achieved after assembling in a two-electrode configuration. In contrast, the COF without Ni2+ (Ni0-COF) exhibited an electrical conductivity of 8.4 × 10−6 S cm−1, and a much lower capacitance of 184 F g−1 at 1 A g−1. The great promotion of electrical conductivity may be attributed to the strong π-conjugation and square-planar Ni(II) coordination, which further improved the electrochemical performance.254
In the above-mentioned examples, pristine COFs are usually mixed with carbon black before measuring their capacitance. Preparing highly oriented COF films is able to effectively alleviate the influence of the grain boundaries and random orientation on the conductivity. A β-ketoenamine-linked 2D COF was used to fabricate crystalline and oriented thin films on the Au electrode surface with adjustable thickness.249 The vertical orientation along with the stacking direction with respect to the substrate was determined by grazing incidence X-ray diffraction (GIXD). A much higher electrochemical accessibility (83%) of anthraquinone groups in DAAQ-TFP COF thin films was achieved compared to that of the randomly oriented microcrystalline COF powder (3%). A 7.5-fold increase in areal capacitance was observed from powder (0.4 mF cm−2 at 150 μA cm−2) to films (3.0 mF cm−2 at 150 μA cm−2). Besides, Banerjee et al. assembled and tested solid-state supercapacitors based on free-standing COF thin sheets.229,230 For example, an imine-based COF, TpOMe-DAQ, was fabricated as uniform and continuous thin sheets with a centimeter scale and a thickness of about 200 μm (Fig. 15d).229 The pristine COF sheets exhibited an outstanding areal capacitance of up to 1600 mF cm−2 (gravimetric capacitance 169 F g−1). Interestingly, an increment of the initial capacitance was obtained in the first 30000 cycles under a current density of 10 mA cm−2, and the resultant capacitance did not decay in the following 70000 cycles (Fig. 15e). The activation process may be related to the increasing number of redox-active centers that became available to electrolytes during the cycling progress. The symmetric solid-state supercapacitor device was assembled with two 1 cm2 pristine COF thin sheets and a 2 M aq. H2SO4/PVA gel (poly(vinyl alcohol)) as an electrolyte. The device showed a high areal capacitance of 84 mF cm−2 (8.8 F g−1) and delivered energy and power densities of ∼2.9 μW h cm−2 and ∼61.8 μW cm−2, respectively. Using these three solid-state devices connected in a series could light up a 1.8 V LED for 20 s (Fig. 15f).
Researchers also grew COFs on CNTs or graphene to enhance the composites’ conductivity.243,244 For instance, two β-ketoenamine-based 2D COF hybrid films with conductive carbon nanofiber (CNF) were prepared by including CNF into the COF precursor matrix.243 The intermolecular π⋯π interaction between the π electrons in COF backbones and the sp2 graphene carbons of CNF built efficient electron transfer pathways from COFs to the matrix. An increment of 109-fold in the electrical conductivity between COF-CNF hybrids (0.25 × 10−3 S cm−1) and pristine COFs was found. The COF-CNF hybrids delivered a high areal capacitance of 464 mF cm−2 at 0.25 mA cm−2, much higher than that of the mixture of COF and CNF (6.25 mF cm−2), owing to the feasible long channel electronic movement through the π⋯π interaction. Integrating the COF-CNF-based supercapacitors with a highly efficient perovskite solar cell achieved a power conversion efficiency of 16.7% and an areal capacitance of 42.0 mF cm−2 for the 300 s photocharging at a current density of 0.25 mA cm−2.
Besides, incorporating conductive polymers into the channels was proved to be a feasible strategy to enhance the electron transfer ability of COF-based composites.242,245 As an example, a redox-active AQ-COF as a scaffold infiltrated with conductive poly(3,4-ethylenedioxythiophene) (PEDOT) was prepared by a facile in situ solid-state polymerization method.242 The resultant PEDOT@AQ-COF nanocomposite showed an excellent electrical conductivity of 1.1 S cm−1, which was 10 orders of magnitude higher than that of AQ-COF. In PEDOT@AQ-COF, the redox-active groups can directly receive the electrons from PEDOT chains encapsulated in the channels, providing a rapid electronic exchange between the working electrode and redox-active groups. Thus, PEDOT@AQ-COF showed a peak current density of 27 A g−1 at a sweep rate of 5 mV s−1 in CV measurements, while AQ-COF/PEDOT (the mixture of AQ-COF and PEDOT) showed only 1.8 A g−1. The specific capacitance was up to 1663 F g−1 at 1 A g−1 for the all-organic PEDOT@AQ-COF. Besides, it also showed a comparable rate performance with a capacitance of 998 F g−1 at 500 A g−1 and was available to operate over 10000 cycles at 50 A g−1 without capacitance decay.
Hao et al. pyrolyzed a series of C3-symmetric COFs under ionothermal conditions. The resultant porous carbons showed a typical double-layer capacitor behavior.259 Three Schiff-base BTT-COFs with different pore sizes (BTT-DADP COF, BTT-DAB COF, and BTT-TAB COF) were synthesized and treated in melt ZnCl2 at 700 °C for 20 h, leading to the formation of porous carbon materials. The surface area was largely increased after pyrolyzing, which may be attributed to the additive ZnCl2 that provided the padding and supported the pore structures during thermal decomposition. The specific capacitance of BTT-DADP COF-700 assembled symmetric supercapacitor was more than 100 F g−1 at 1 A g−1, and the capacitance remained at 77.5% of the initial after 10000 cycles at 10 A g−1. A similar strategy was implemented by Awaga and Wu et al.253 The multiple heteroatom-doped carbons (ONC-T1s) with ultra-high specific surface area were pyrolysed from AQ-COF impregnated with K2CO3, in which ONC-T1-850 (SBET = 1518 m2 g−1) showed a typical EDLC behavior, with a high specific capacitance of 1171 F g−1 at 1 A g−1.
In addition to directly pyrolyzing pristine COFs to produce porous carbon materials, carbonizing the hybrid COF materials was also investigated. Talyzin et al. grew COF-1 perpendicularly to the surface of graphene (v-COF-GO) via covalent bonding.112 The vertically oriented COF-1 may provide an interconnected pore network to facilitate ion diffusion. After carbonization in molten salts, the orientation of the resultant boron-doped carbon nanostructures (v-CNS-RGO) still remained, and v-CNS-RGO showed a good specific capacitance and excellent cycling stability.
With the rapid development of emergency multifunctional electronics, thinner and lighter supercapacitors with high volumetric power density are needed. The electrode materials are of vital importance. The volumetric energy density of a supercapacitor is positively correlated with its volumetric capacitance (equal to gravimetric capacitance multiplied by bulk density). However, there is a trade-off effect between the gravimetric capacitance and bulk density. For electrode materials, proper surface area with accessible pore volume for ions and high active site density can greatly promote the ion adsorption, which is beneficial for gravimetric capacitance but generally results in a relatively low bulk density. To achieve an ideal supercapacitor electrode, the materials are required to balance the density, porosity, and pore size, instead of only pursuing a high surface area. Furthermore, the capacitor performance is also influenced by other factors, such as electronic conductivity (amount of conductive additive, and electrochemical accessibility of COFs' redox-active sites), loading amount of active materials (volume fraction), and electrolyte system (working voltage). It is a challenge to well balance these parameters to fabricate an outstanding energy-storage device.
We have summarized the performances of supercapacitors based on COFs and carbonized COFs in Tables 3 and 4, respectively. Some of them were compared in the Ragone plot (Fig. 16),264 possessing a decent energy density with an acceptable power density. Even few devices showed an energy density higher than 5 W h kg−1 at a power density greater than 1 kW kg−1 under a working voltage larger than 1 V, which can fulfill the basic requirements for the industry devices.
| Electrode | Specific capacitance | Current density | Working voltage (V) | Retention (cycle) | S BET (m2 g−1) | Ref. |
|---|---|---|---|---|---|---|
| In three-electrode configuration | ||||||
| NIBDZ | 88.4 F g−1 | 0.5 A g−1 | 0–1.0 | 93.61% (5000) | 120 | 233 |
| TPT-DAHQ COF | 256 F g−1 | 0.5 A g−1 | 0.1–1.0 | 98.8% (1750) | 1855 | 232 |
| PDC-MA-COF | 355 F g−1 | 1.0 A g−1 | 0–1.5 | 78% (9000) | 748.2 | 227 |
| DAB-TFP-COF | 15 ± 6 F g−1 | 0.1 A g−1 | −1.0 to 0.25 | — | 365 | 225 |
| DAAQ-TFP-COF | 48 ± 10 F g−1 | 83.3 (5000) | 435 | |||
| TpOMe-DAQ | 169 F g−1 | 3.3 mA cm−2 | −0.5 to 0.5 | 65% (50000) |
1531 | 229 |
| [TEMPO]100%-NiP-COF | 167 F g−1 | 0.1 A g−1 | 0–0.8 | — | 5.2 | 228 |
| [TEMPO]50%-NiP-COF | 124 F g−1 | 100 | 264 | |||
| TPA-COF-1 | 51.3 F g−1 | 0.2 A g−1 | 0.2–0.7 | — | 714 | 234 |
| TPA-COF-2 | 14.4 F g−1 | 478 | ||||
| TPA-COF-3 | 5.1 F g−1 | 557 | ||||
| TPA-COF-4 | 2.4 F g−1 | 1132 | ||||
| TFP-NDA-COF | 348 F g−1 | 0.5 A g−1 | 0–1.0 | 75% (8000) | 353 | 235 |
| Dq2Da1Tp COF thin sheet | 122 F g−1 | 1.56 mA cm−2 | −0.7 to 0.3 | — | 1004 | 230 |
| DqTpCOF thin sheet | 154 F g−1 | — | 940 | |||
| Dq1Da1Tp thin sheet | 111 F g−1 | — | 804 | |||
| Car-TPA COF | 13.6 F g−1 | 0.2 A g−1 | 0–0.6 | — | 1334 | 236 |
| Car-TPP COF | 14.5 F g−1 | 743 | ||||
| Car-TPT COF | 17.4 F g−1 | 721 | ||||
| TpBD-(OH)2 | 90 F g−1 | 0.5 A g−1 | −0.2 to 0.5 | — | 197 | 226 |
| TpPa-(OH)2 | 416 F g−1 | 66% (10000) |
369 | |||
| TDFP-1 | 418 F g−1 | 0.5 A g−1 | 0–1.0 | 95% (1000) | 651 | 237 |
| TpPa-COF@PANI | 95 F g−1 | 0.2 A g−1 | 0–0.7 | 83% (30000) |
574.4 | 238 |
| SWCNTs-TpPa-COFs | 153 F g−1 | 0.5 A g−1 | −0.2 to 0.8 | >2000 | 235.5 | 239 |
| COFDAAQ-BTA-3DG | 31 mF cm−2 | 0.5 mA cm−2 | −1.05 to 0.4 | 24.0% (2000) | — | 240 |
| COFs/NH2-rGO | 533 F g−1 | 0.2 A g−1 | 0–0.5 | 79% (1000) | — | 241 |
| PEDOT@AQ-COF | 1663 F g−1 | 1 A g−1 | −0.2 to 0.6 | >10000 |
131 | 242 |
| DqDaTp-CNF | 364 mF cm−2 | 0.25 mA cm−2 | −0.5 to 0.5 | — | 532 | 243 |
| g-C34N6-COF/CNT MSC | 13.1 mF cm−2 | 2 mA cm−2 | 0–0.8 | 93.1% (5000) | 1003 | 244 |
| PEDOT/DAAQ-TFP | 350 mF/cm3 | — | −0.3 to 0.6 | >10000 | — | 245 |
| NH2-f-MWCNT@COFTTA–DHTA | 127.5 F g−1 | 0.4 A g−1 | 0–0.8 | 96% (1000) | 1157 | 246 |
| Fe3O4/COF-5d | 112 F g−1 | 0.5 A g−1 | −0.1 to 0.7 | 76% (2000) | 872 | 247 |
| NiNWs@TpPa-COFs | 426 F g−1 | 2 A g−1 | −0.2 to 0.6 | >2500 | 596.7 | 248 |
| DAAQ-TFP thin films | 3 mF cm−2 | 0.15 mA cm−2 | −2.0 to 0.5 | 93% (5000) | — | 249 |
| TaPa-Py COF | 209 F g−1 | 0.5 A g−1 | −0.3 to 0.4 | — | 687 | 250 |
| DAB-TFP COF | 98 F g−1 | 0.5 A g−1 | −0.3 to 0.4 | — | 385 | |
| TPPDA-TPTPE COF | 237.1 F g−1 | 2 A g−1 | −0.92 to 0.18 | 86.2 (5000) | 1067 | 251 |
| TPPDA-TPPyr COF | 188.7 F g−1 | 85.6 (5000) | 1020 | |||
| PG-BBT | 724 F g−1 | 1 A g−1 | 0–0.6 | 96% (10000) |
507 | 252 |
| ONC-T1-700 | 768 F g−1 | 1 A g−1 | 0–0.6 | 98.3% (10000) |
3451 | 253 |
| ONC-T1-850 | 1711 F g−1 | 101.5% (10000) |
1518 | |||
| Ni-COF | 1257 F g−1 | 1 A g−1 | 0–0.6 | 94% (10000) |
362 | 254 |
| In two-electrode configuration | ||||||
| PDC-MA-COF//AC | 94 F g−1 | 1.0 A g−1 | 0–1.5 | 88% (20000) | 748.2 | 227 |
| CT-Dq1Da1TpCOF//CT-Dq1Da1TpCOF | 8.5 mF cm−2 | 0.39 mA cm−2 | 0–1.0 | 78% (7000) | 804 | 230 |
| CT-DqTpCOF//CT-DqTpCOF | 12 mF cm−2 | 0.39 mA cm−2 | 80% (2500) | 940 | ||
| TpPa-(OH)2//TpPa-(OH)2 | 214 ± 1 F g−1 | 0.2 A g−1 | 0–0.7 | 88% (10000) | 369 | 226 |
| DqDaTp-CNF//DqDaTp-CNF | 167 F g−1 | 0.5 mA cm−2 | 0–1 | 76% (4500) | 532 | 243 |
| PEDOT/DAAQ-TFP//AC | 197 F g−1 | — | −0.9 to 0 | — | — | 245 |
| TaPa-Py COF//TaPa-Py COF | 102 F g−1 | 0.5 A g−1 | 0–0.8 | 92% (6000) | 687 | 250 |
| DAB-TFP COF//DAB-TFP COF | 42.7 F g−1 | — | 385 | |||
| e-JUC-510//e-JUC-510 | 4.17 mF cm−2 | 1000 mV s−1 | 0–1.3 | — | 666 | 255 |
| e-JUC-511//e-JUC-511 | 5.46 mF cm−2 | 100% (10000) | 416.6 | |||
| e-JUC-512//e-JUC-512 | 5.85 mF cm−2 | 100% (10000) | 336 | |||
| PG-BBT//AC | 220 F g−1 | 1 A g−1 | 0–1.5 | — | 507 | 252 |
| ONC-T1-700/CNT//ONC-T1-700/CNT | 480 F g−1 | 1 A g−1 | 0–0.6 | — | 3451 | 253 |
| ONC-T1-850/CNT//ONC-T1-850/CNT | 700 F g−1 | — | 1518 | |||
| Ni-COF//AC | 417 F g−1 | 1 A g−1 | 0–1.5 | — | 362 | 254 |
| TpOMe-DAQ//TpOMe-DAQ | 84 mF cm−2 | 0.25 mA cm−2 | 0–1 | >70% (30000) | 1531 | 229 |
| Electrode | Specific capacitance | Current density | Working voltage (V) | Retention (cycle) | S BET (m2 g−1) | Ref. |
|---|---|---|---|---|---|---|
| In three-electrode configuration | ||||||
| TAPT-DHTA-COF0.1@PPZS900 | 411 F g−1 | 0.5 A g−1 | −1.0 to 0 | 100% (10000) |
456 | 256 |
| (N)G2 | 460 F g−1 | 1 A g−1 | −1.0 to 0 | 90% (10000) |
1147 | 257 |
| Carbonized ACOF1 | 234 F g−1 | 1 A g−1 | −1.0 to 0 | — | 1596 | 258 |
| In two-electrode configuration | ||||||
| BTT-DADP COF-700//BTT-DADP COF-700 | >100 F g−1 | 1 A g−1 | 0–3.5 | 77.5% (10000) |
2288 | 259 |
| v-CNS-RGO//v-CNS-RGO | >160 F g−1 | 1 A g−1 | −0.5 to 0.5 | 100% (3000) | 700 | 112 |
| (N)G2//(N)G2 | 175 F g−1 | 0.2 A g−1 | 0–1.0 | — | 1147 | 257 |
 | ||
| Fig. 16 The specific power vs. specific energy for EES devices delineated in the Ragone plot. Reproduced from ref. 264 with permission from American Chemical Society, copyright 2018. The electrochemical performance of COF-based supercapacitors has been reported: (★): ref. 252; (▲): ref. 226; (●): ref. 227; (■): ref. 250. | ||
4 Electrochemical energy conversion
For catalysts used in electrochemical energy conversion, possessing catalytic sites with high activity and selectivity, high electrical conductivity for electron transport and path for rapid mass transport is highly desired. In addition, chemical stability towards water and long-term stability upon catalytic cycles also should be taken into consideration.Many porous frameworks including amorphous porous organic polymers,265,266 metal–organic frameworks (MOFs),267,268 COFs,267 and zeolite269,270 have been reported to possess electrocatalytic activity and have led to great achievements. They have attracted great interest as electrocatalytic materials because of their large surface area and permanent porosity, which are beneficial for charge/mass transport and full contact between substrate and active sites. Compared with other host materials, COFs have several advantages: (1) in addition to the highly ordered structural feature, the active sites can be incorporated into either backbones or side chains with atomic precision, facilitating electrocatalytic mechanism study; (2) the chemical and physical environment around the active sites is able to be easily modulated, resulting in the fine-tuning of their catalytic activity; (3) their chemical stability can be achieved by choosing proper linkages; (4) 2D COF NSs with conjugated electron structure may provide sufficient conductivity; (5) the rich choices in the linkers and linkages lead to tremendous opportunities for the designing of high performance electrocatalysts. Moreover, despite boroxine and boronate linkage-based COFs are sensitive to moisture, most of the developed linkages (e.g., imine, hydrazine, azine, ketene, imide, squaraine, triazine, and ethylenic linkages) are chemically stable in water.56,271 The introduction of tautomerization, hydrogen bonding, and interlayer interactions in the COFs can further improve the stability of imine linkage-based COFs in acidic and basic solutions. The polyarylether-based COFs are able to withstand harsh chemical environments including strong acids and bases, as well as oxidation and reduction conditions,272 the stability of which outperformed most of the known crystalline porous materials.
To solve the conductivity problem of bulk COF materials in energy storage application, two general strategies are usually employed: the first one is to physically mix COFs with conductive supporting materials (e.g., graphene and CNT) or directly grow COFs on them to prepare COF-based composites; the second one is treating COFs at high temperature to pyrolyze them into heteroatom-doped porous carbon. The former approach can retain the ordered structures of COFs and atomically distributed metal ions, which is beneficial for the maximal utilization of catalytic atoms and the realization of high selectivity and activity; the latter approach is easier to achieve high conductivity and robust chemical stability but suffers from the agglomeration of metal ions.
In this section, the recent advances related to the application of COFs in electrocatalysis, including oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and CO2 reduction reaction (CO2RR), are presented.
4.1 ORR
The oxygen reduction reaction (ORR, O2 + 4H+ + 4e− → 2H2O), as a rate-determining step (RDS), plays a vital role in energy storage and conversion devices, including metal–air batteries and fuel cells.273–276Fuel cells are electrochemical power generation devices that directly convert chemical energy into electrical energy. In proton exchange membrane fuel cells, hydrogen is oxidized at the anode to form protons and electrons, and electrons flow from the anode to the cathode to participate in the ORR, while the protons combine with the reduced oxygen species to form H2O on the cathode in the electrolyte. They are considered as one of the new clean and efficient power generation technologies in the 21st century.
Metal–air batteries have attracted great interest around the world due to their high theoretical energy density (approximately 2–10 times higher than that of lithium-ion batteries).277 The negative electrodes of metal–air batteries, combining the features of traditional batteries and fuel cells, are made of metals as in the case of traditional batteries. The oxygen supplied by air is continuously pumped into the cathode as the reactant to conduct ORR, whose efficiency greatly influences the battery voltage and energy density. Among the various kinds of metal–air batteries, Zn–air batteries exhibit great potential for future energy applications.
At present, Pt is considered to be the best ORR electrocatalyst in fuel cells.278 Nevertheless, its scarcity, high cost, poor long-term stability, as well as low tolerance to the fuel crossover effect are main obstacles.279 Among the benchmark performers of non-precious metal catalysts (NPMC) that display high activity and stability in catalysis, metal/nitrogen/carbon (M/N/C, M = Fe, Co, Ni, etc.) nanohybrids prepared by pyrolysis of precursors have been demonstrated to be one of the most promising candidates to replace Pt-based catalysts.280,281
Direct pyrolysis of COFs always leads to the formation of an uncontrollable 3D carbon structure with the loss of the 2D structure and inherent porous feature, even though the precursors possess layered 2D structures. Accounting for these problems, Jiang and co-workers proposed a strategy based on template carbonization, which successfully converted a conventional COF (TAPT-DHTA-COF) into carbon sheets (PA@TAPT-DHTA-COF1000) with high conductivity, hierarchical pores, and sufficient catalytic edges.282 They inserted phytic acid (PA) as a template as well as directing agent into the channels and layers during pyrolysis of 2D COF at 1000 °C under N2. The multi-fold functions of PA included stripping the layered 2D COFs into several small-sized carbon plates, guiding the conversion of 2D COFs to 2D carbon when it covered the surface of COFs, triggering the formation of 2D porous carbon structure during its network decomposition, and finally, supplying the carbon sheets with phosphorus. For ORR in aqueous KOH solutions (0.1 M), the resulting PA@TAPT-DHTA-COF1000 reached an E0 value of 0.921 V (V vs. RHE), an E1/2 value of 0.751 V (V vs. RHE) and a limit current density of 6.5 mA cm−2. They further improved its ORR activity by pyrolyzing it at 900 °C under NH3 to form PA@TAPT-DHTA-COF1000NH3, whose ORR catalytic performance (E0, 0.941 V; E1/2, 0.831 V; jlimit, 7.2 mA cm−2) was superior to that of Pt/C (E0, 0.911 V; E1/2, 0.781 V; jlimit, 6.0 mA cm−2).
In 2019, Thomas's group reported a “salt-mediated technique” for COFs’ growth with p-toluenesulfonic acid (PTSA) as a molecular organizer during bipyridine-COF (TpBpy) synthesis and silica nanoparticles as hard templates to facilitate the formation of mesoporous structures (Fig. 17a).130 Iron species were introduced into the system coordinated with bipyridine moieties to produce SiO2@TpBpy-Fe, which then formed Fe–Nx sites after a subsequent pyrolysis process followed by the treatment with HCl and NaOH.288 Electrocatalytic studies showed that the resultant mC-TpBpy-Fe exhibited almost comparable values (E0 of 0.920 V, E1/2 of 0.845 V) to those of commercial Pt/C under basic conditions (Fig. 17b and c). More importantly, it showed long-term durability and good tolerance to methanol during the catalytic reaction and a competitive discharge performance when applied in a Zn–air battery.
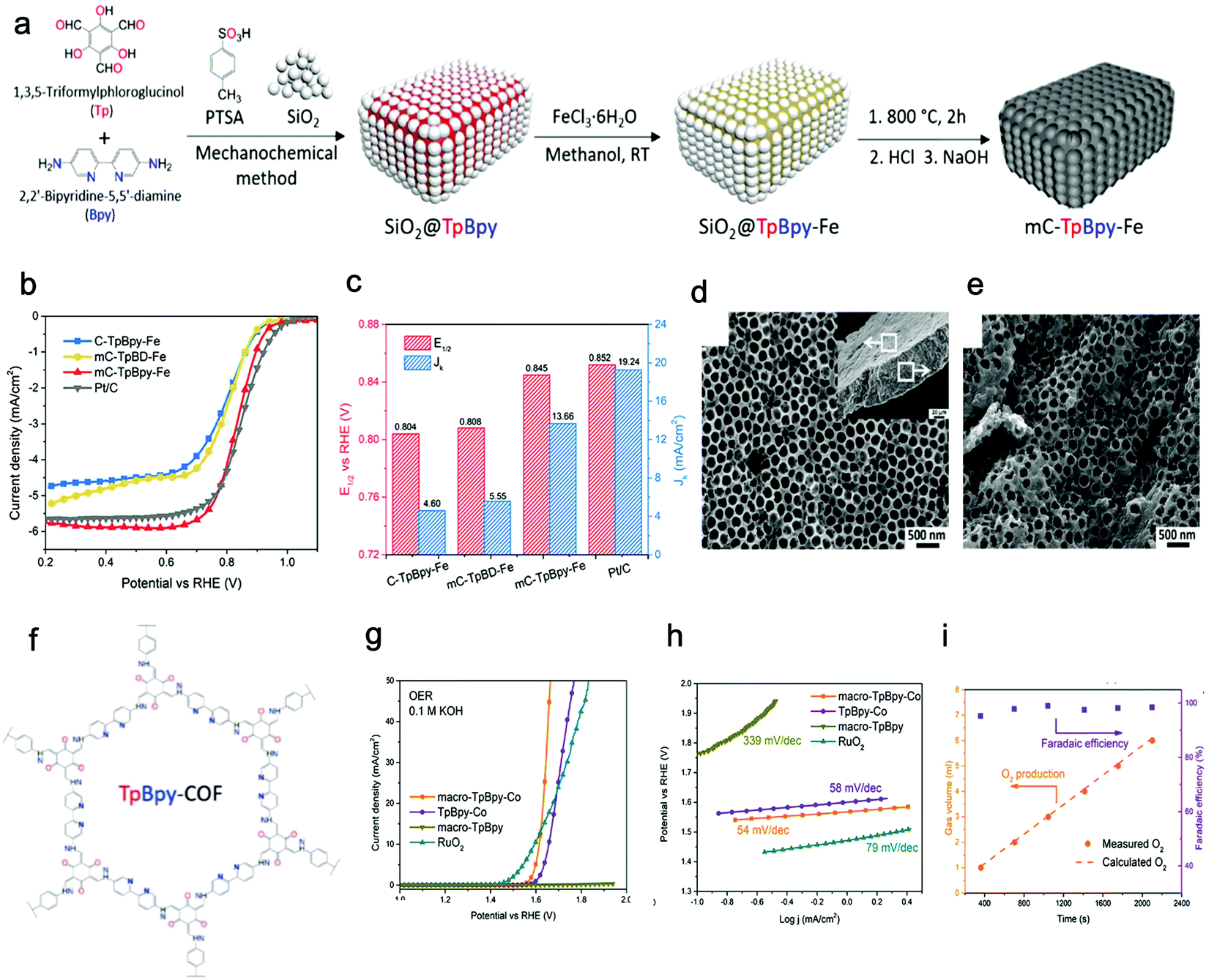 | ||
| Fig. 17 (a) Synthesis of mC-TpBpy-Fe by PTSA-assisted mechanical milling using SiO2 nanoparticles as the hard template. (b) LSV curves of mC-TpBpy-Fe, mC-TpBD-Fe, C-TpBpy-Fe, and Pt/C in O2− saturated KOH solution. (c) Corresponding columnar charts of these samples’ half-wave potential and kinetic current (at 0.8 V). Reproduced from ref. 288 with permission from American Chemical Society, copyright 2019. (d) and (e) Top-view and cross-sectional SEM images of macro-TpBpy, respectively. (f) Chemical structure of macro-TpBpy. (g) OER polarization curves. (h) Tafel plots of macro-TpBpy, TpBpy-Co, macro-TpBpy-Co, and RuO2 catalysts. (i) Faradaic efficiency of macro-TpBpy-Co for oxygen evolution. Reproduced from ref. 300 with permission from American Chemical Society, copyright 2019. | ||
Single-atom catalysts (SACs) take advantage of maximal atom utilization and high catalytic activity. However, the isolated metal atoms in the materials tend to agglomerate under elevated temperatures due to their thermodynamic instability.152,289,290 Therefore, the process for preparing a single-atom catalyst by pyrolysis requires fine adjustment of the carbonization temperature and the reduction of metal ion loading. Recently, Peng, Shi, and Huo et al. synthesized SACs with good electrocatalytic activity via a pyrolysis-free synthetic method.289 They first prepared a fully closed π-conjugated COF (COFBTC) via benzene-1,2,4,5-tetracarbonitrile polymerization and then assembled it with the graphene matrix via intermolecular interaction. The as-synthesized pfSAC-Fe catalysts showed excellent electrocatalytic performance for ORR. When the ratio of COFBTC was increased, the E1/2 value changed from 0.88 V to 0.91 V. Besides, the optimized catalyst, pfSAC-Fe-0.2, showed a high kinetic current density Jk of 25.86 mA cm−2, a low Tafel slope of 31.7 mV and good methanol tolerance, which were superior to those of commercial Pt/C (E1/2, 0.86 V; Jk, 6.49 mA cm−2). The pfSAC-Fe driven Zn–air battery exhibited a specific capacity of 732 mA h g−1 at 100 mA cm−2 with 89.3% zinc utilization and a power density of 123.43 mW cm−2 (which was higher than that of the battery driven by Pt/C (113.81 mW cm−2)). Moreover, it exhibited a remarkable long cycling life over 300 hours with less than 0.1% decrease, whereas the performance of the Pt/C-based battery decreased within 20 hours.
4.2 OER
OER as an anodic reaction occurs in water splitting and in the charging processes of rechargeable metal–air batteries. As seen from the reaction pathways of OER, H2O is oxidized to O2 accompanied with 4e− transfer. OER can operate in either acidic or alkaline solutions, and the corresponding processes are 2H2O → O2 + 4H+ + 4e− and 4OH− → O2 + 2H2O + 4e−, respectively.Unfortunately, OER suffers from sluggish kinetics owing to the large energy barriers in the complex procedures including breaking four O–H bonds, removing four electrons from H2O molecules, and forming energy-intensive O–O bonds. RuO2 and Ir-based materials are considered to be benchmarked electrocatalysts for OER. However, limited storage, high cost, and poor availability of these noble metal-based electrocatalysts hamper their wide commercial usage. Thus, there is an upsurge in attempts to develop effective, low-cost, and stable catalysts for oxidizing water with minimum energy.
In order to better evaluate and compare the properties of OER electrocatalysts, a series of criteria have been established, involving η10 (the overpotential needed to generate a catalytic current density of 10 mA cm−2), j0 (the value of OER current density), Tafel slope, the yield of oxygen, turnover frequency (TOF), and faradaic efficiency (FE). Besides, since OER involves the generation and liberation of oxygen, the problem arising from the leaching of the catalyst leading to suboptimal long-term stability should be taken into account. Moreover, the nonconducting gas phase will hinder the exposure of active sites and cause the current density to decrease.
As an alternative candidate, transition metal-based materials became the subject of comprehensive research on OER electrocatalysts. So far, Fe/Co/Ni/Mo/Zr/V-based materials (either free-standing or supported on other materials)291 have been explored for catalyzing OER, among which cobalt-based catalysts became one of the hot spots. In the case of free-standing transition metal-based catalysts, a number of them suffered from high overpotential and sluggish kinetics in the electrochemical study. On the one hand, there still exist lots of molecular water-oxidizing complexes (WOCs) that will be eventually dissociated into a homogeneous phase during catalysis.292,293 On the other hand, carbon materials294 and metal foam295,296 as platforms to support metal nanoparticles or clusters as catalytic active materials also confront several problems, such as complicated synthetic procedures.297,298 COFs equipped with some inherent virtues appear satisfactory for preparing electrocatalysts.
In 2016, Banerjee and Kurungot et al. designed Co-TpBpy via coordinating Co species to the bipyridine sites on the TpBpy-COF synthesized by the Schiff-base condensation of Tp (24) and 2,2′-bipyridyl 5,5′-diamine (Bpy) (50).299 The resulting Co-TpBpy retained high accessible surface area (450 m2 g−1) and porosity. The electrochemical properties measured in 0.1 M aqueous phosphate buffer (pH = 7) showed an overpotential of 400 mV at a current density of 1 mA cm−2, a TOF of 0.23 s−1, a Tafel slope of 59 mV dec−1, an FE of 95%, and good cycling stability. Further, Thomas et al. introduced polystyrene spheres as hard templates and PTSA during the preparation process to enable the formation of TpBpy-COF with a hierarchical pore structure (Fig. 17d–f).300 Macro-TpBpy-Co exhibited an outstanding performance as a OER catalyst with an overpotential of 380 mV at a current density of 10 mA cm−2, a Tafel slope of 54 mV dec−1, and an FE of 98% (Fig. 17g–i). It should be noted that the TOF value of Macro-TpBpy-Co was much higher than that of microporous TpBpy-Co and RuO2. The good electrocatalytic properties were attributed to the homogeneous and continuous macroporous structure, which could facilitate the transportation of O2 gas bubbles and thereby promote the reaction. It was proved that the Co ions coordinated with pyridine not only worked as the active sites but also increased the conductivity of the catalyst.
Additionally, COFs have been utilized as supports for metal nanoparticles (NPs). The size of NPs and the electronic interaction between the NPs and the host COFs both played important roles in enhancing their catalytic activity. For instance, IISERP-COF2 was prepared by using nonplanar building units with tetrahedral sp3 nitrogen, which introduced flexibility in COF skeletons to generate mesopores capable of confining the in situ formation of CoxNiy(OH)2 NPs down to about 2 nm size without any end-capping reagent.301 Owing to the localized electronic interactions between the NPs and the COF skeleton, the resultant nanocomposite exhibited an excellent performance toward the OER process with an overpotential of 258 mV at 10 mA cm−2 in 0.1 M KOH, which was close to the record of about 200 mV for noble-metal-free electrocatalysts. Also, it possessed a Tafel slope of 38.9 mV dec−1, indicating outstanding kinetics. Similarly, metallic Ni3N NPs were loaded in a 2D benzimidazole-based COF (IISERP-COF3) with donor–acceptor nodes and face-to-face stacked layers of conjugated π-components that were crucial to generate aligned conduction pathways.302 The resulting IISERP-COF3_Ni3N possessed a low overpotential of 230 mV at 10 mA cm−2, a TOF value of 0.52 s−1, an FE of 0.98 at 1 mA cm−2, a Tafel slope of 79 mV dec−1, and an O2 evolution rate of 230 mmol h−1 g−1.
To enhance the conductivity, COFs have also been pyrolyzed into carbon hosts for OER reaction. Kurungot and Vaidhyanathan et al. developed a soft pyrolysis approach to prepare two composites consisting of small RuO2 NPs and carbon derived from COFs (Fig. 18a and b). They explained that the preparation of RuO2 nanoparticles usually requires surfactant assistance and high-temperature annealing under oxidizing conditions. However, when the temperature was higher than 500 °C, the size of RuO2 NPs would increase significantly. Therefore, they controlled the temperature to about 400 °C and finally obtained RuO2@C composite materials by the soft pyrolysis method. The two resultant composites exhibited good OER performance with a η10 of 210 and 217 mV for composite-I and composite-II, respectively (Fig. 18c and d).303 It is worth noting that most of the small-sized NPs were dispersed within the interlayers of the pyrolyzed COF, only leaving fewer on the surface to form more accessible active sites.
 | ||
| Fig. 18 (a and b) Schematic illustration of the reaction of IISERP-COF1 and IISERP-COF6. (c and d) LSV curves (left) and Tafel slopes (right) of IISERP-COF1 and IISERP-COF6. Reproduced from ref. 303 with permission from American Chemical Society, copyright 2019. (e) Synthetic scheme of COF (SB-PORPy). (f) Polarization curves for the hydrogen production on GC, Vulcan, and SB-PORPy-COF electrodes. (g) Tafel slope of the SB-PORPy-COF electrode. Reproduced from ref. 308 with permission from American Chemical Society, copyright 2017. | ||
There are other strategies for producing efficient COF-based electrocatalysts such as introducing transition metal species into macrocyclic clusters (e.g., porphyrin and phthalocyanine) and blending metal-porphyrin with other conductive materials (e.g., metal foam and CNTs).118,304 For instance, Chen and Zhang et al. developed a metalloporphyrin-based GDY (graphdiyne) analogue (Co-PDY nanosheets) synthesized via Glaser–Hay coupling reaction on copper foam.118 During the catalytic process, the cobalt centers of metalloporphyrin acted as electrocatalytic active sites, while rapid electron transfer occurred inside the skeleton and the expanded pore structure accelerated the small molecules’ diffusion. These three factors endow Co-PDY with superb OER catalytic performance (low overpotential of 270 mV at 10 mA cm−2, Tafel slope of 99 mV dec−1, and long-term cycling stability) (Fig. 19a–e), which was superior to that of commercial catalysts such as RuO2 (η10 of 370 mV), IrO2 (η10 of 340 mV), Ru/C (20%) (η10 of 390 mV), and Ir/C (20%) (η10 of 380 mV).
 | ||
| Fig. 19 (a) Schematic illustration of the reaction of Co-PDY. (b) SEM images of Co-PDY/CF before (left) and after (right) the continuous electrocatalysis test for 10 h. (c) TEM images of Co-PDY/CF before (left) and after (right) the continuous electrocatalysis test for 10 h. (d) LSV curves of Co-PDY/CF, PDY/CF, and CF. (e) Tafel slope of Co-PDY/CF, PDY/CF, and CF. Reproduced from ref. 118 with permission from the Royal Society of Chemistry, copyright 2019. | ||
4.3 HER
Hydrogen, as a kind of clean fuel with high energy density, has been deemed to be one of the most promising alternative candidates to replace fossil fuels. At present, hydrogen is mostly produced by steam reforming of hydrocarbons. Besides, heating coal at high temperature without oxygen or partially burning coal with steam can also generate hydrogen; however, these techniques are with a high energy cost and are tedious because hydrogen needs to be separated from the mixture gases of H2, CO, CO2, etc.305 The electrolysis of water is an ideal method to produce high-purity H2via HER. The procedure of electrocatalytic HER is a multi-step reaction involving adsorption, reduction, and desorption processes, including a Volmer step (H+ + e → Hads) and a Tafel step (Hads + H+ + e → H2) or a Heyrovsky step (2Hads → H2). To date, Pt-based materials show the best electrocatalytic performance toward HER but scarcity and high cost limit their large-scale practical applications. The transition-metal-based catalysts applied for electrocatalytic HER still suffer from the corrosion of the metal or leaching of metal ions and easier deactivation, which are the major obstacles for their commercial utilization.306,307 Thus, tremendous efforts have been devoted to investigating efficient metal-free electrocatalysts for HER. Recently, metal-free COF-based materials have been reported to show good HER activity.308,309For example, SB-PORPy-COF prepared by assembling porphyrin and pyrene units via the Schiff-base reaction was applied for HER (Fig. 18e).308 It possessed high surface area (∼869 m2 g−1), permanent microporosity, and excellent stability, allowing it to achieve electrocatalytic properties with an onset potential of 50 mV, a low overpotential of 380 mV at 5 mA cm−2, and a Tafel slope of 116 mV dec−1 (Fig. 18f and g). Recently, free-standing 2D conjugated COF films were also applied to electrocatalyze HER. Li's group reported that graphene-like 2DCCOF1 with homogeneous pores was prepared by Suzuki polymerization on the interface of water and toluene at 2 °C.309 To explore potential applications in electrocatalysis, they horizontally deposited the 2DCCOF1 film on a copper electrode and an electrochemical study showed that the 2DCCOF1 coated electrode was active for catalyzing HER with an overpotential of 541 mV at 10 mA cm−2 and a Tafel slope of 130 mV dec−1. However, the performance of the COF-based catalysts mentioned above did not exceed the performance of MoS2 nanosheets (benchmarked Pt-free HER catalyst) with an overpotential of 187 mV at 10 mA cm−2 and a Tafel slope of 43 mV.310
4.4 CO2 reduction
The massive emission of CO2 caused by the consumption of fossil fuels and industrial production has become a serious environmental issue, leading to worldwide extensive research on efficient and eco-friendly technologies for converting CO2 into other value-added carbon products.311 Indeed, extensive approaches have been attempted to facilitate this transformation, among which the electrolytic approach emerged as a promising route to reduce CO2. It benefits from the following aspects: (1) using water as reaction media is in favor of promoting proton and electron transfer; (2) since the final products can be used as fuels and chemical feedstocks and the electrolyte can be adequately recycled, zero-emission of pollutants in the overall process is realized; (3) the electricity used can be originated from renewable resources including wind, solar, geothermal energy, etc.312–314Typically, the electrochemical reduction (ECR) of CO2 involves multiple steps to different products, accompanying 2-, 4-, 6-, 8-, 12-, 14-, or 18-electron reaction pathways. There are three major steps for heterogeneous catalysis: (1) chemical adsorption of CO2 on active sites of electrocatalysts; (2) electron transfer and/or proton migration to cleave C–O bonds and/or form C–H bonds; (3) configuration rearrangement of products (e.g., CO, HCOO− or HCOOH, CH4, C2H4, C2H5OH, and CH3OH) followed by desorption from the electrode surface and dissolution in the electrolyte.315
There still remain several challenges to achieve efficient conversion of CO2: (1) a large overpotential is required to produce the CO2˙− intermediate due to the high energy barrier for its formation; (2) sluggish kinetics, interference of competitive off-pathway reduction of water as well as the limited soluble and mass transfer ability of CO2 lead to a low conversion efficiency; (3) the feedstocks produced in the process are mixed together and separating them is a high-cost process; (4) catalytic active sites of electrocatalysts may be blocked and destroyed by impurities and by-products in the reaction system, resulting in a limited lifetime.313 Consequently, the electrocatalysts for CO2 reduction should be designed with high activity and selectivity.
Many investigations have elaborated that COF-based materials are capable of acting as electrocatalysts for CO2 reduction.
Metalloporphyrins/phthalocyanines, as well as related complexes, are highly promising as CO2 conversion electrocatalysts.316–321 Yaghi and Chang et al. incorporated cobalt porphyrin units into COFs and the resulting COF-366-Co promoted carbon monoxide evolution in a neutral CO2 saturated solution at −0.67 V with an overpotential of −0.55 V.39 COF-366-Co exhibited a high FE (90%) and turnover number (up to 290000) without decomposition over 24 hours. Furthermore, they developed a COF-366-Co analogue, namely COF-367-Co, by replacing 1,4-benzenedicarboxaldehyde (BDA) with BPDA as the strut. Cyclic voltammetry studies showed that COF-367-Co displayed improved catalytic performance with an onset potential of −0.40 V and an increased FE (91%) at −0.67 V. They explained that the larger pores in expanded COF-367-Co enabled higher CO2 adsorption and provided more possibilities for CO2 to contact active sites, finally resulting in enhancement in activity and selectivity.
In most cases, COFs are synthesized via solvothermal methods to form powders, followed by drop-casting onto electrode surfaces for electrocatalysis. However, there is only a small quantity of COF reaction centers remaining electrochemically accessible and electroactive. Besides, the low aqueous solubility, and confined diffusion and adsorption of CO2 within the COF channels hinder the CO2 reduction process. Following this, many researchers have attempted to directly grow COFs onto the surface of electrodes to prepare thin films.39,320,322 A series of oriented COF films were prepared on HOPG and their electrocatalytic activity toward CO2 reduction was investigated.322 The resulting COF-366-Co film exhibited apparently improved catalytic ability with an FE of 87%. In addition to porphyrin and phthalocyanine macrocycles, tricarbonyl rhenium(I) complexes had quite a high catalytic activity. It was evidenced that COF-2,2′-bpy-Re containing Re-2,2′-bpy fragments could work as an electrocatalyst for CO2 reduction.323 COF-Re_Co and COF-Re_Fe, containing both metalloporphyrin and metal bipyridine fragments, were prepared and showed a certain degree of CO2 reduction activity.319
Metal-free COF based electrocatalysts for CO2 reduction324–326 have been reported recently. Deng's group324 reduced the imine linkage in a 3D COF (COF-300) to form more stable COF-300-AR, which exhibited a good electrocatalytic performance for CO2 reduction with a good CO conversion efficiency. They elaborated that the obvious activity of COF-300-AR for CO2 reduction could be attributed to the porosity of COFs, which facilitated the diffusion of CO2 molecules to the electrode, and after that, amine groups close to the electrode surface promoted the conversion of CO2 into carbamate intermediates.
5 Summary and outlook
In this review, we have summarized the recent progress in the synthetic methods of COF NSs and thin films, along with the potential applications of COFs in batteries, capacitors, and electrocatalysts.5.1 Synthetic strategy of COF NSs and thin films
To date, several approaches to design COF NSs and thin films with desired structure, chemical property, and thickness have been proposed, which are of great importance to investigate their properties for diverse applications. The synthetic strategies are classified into “bottom-up” and “top-down” approaches, both of which have their own merits and limitations.In the case of the bottom-up strategy, solvothermal synthesis has received a considerable amount of attention because of its easy operation and broad applicability. When using the solvothermal method, the type and ratio of the selected solvents as well as the heating temperature have a significant influence on successfully controlling the morphology and periodicity of COF NSs and films. Additionally, the heated solvents under high pressure may promote the progress of the reaction and provide a unique environment for the formation of products. However, this method has a relatively insufficient ability to control the thickness of COF films and a certain number of unreacted monomers and oligomers are present in the obtained products.
Considering the on-surface synthesis, this method has been illustrated to be effective for synthesizing single-layered nanomaterials. It is essential to select the appropriate substrates. Specifically, well-defined single-crystal surfaces are capable of providing an ideal atomically flat surface to guide the growth of NSs and thin films in two dimensions and support the resulting products for further applications. Moreover, the appropriate interactions between the substrates and monomers can assist the dispersion of precursors on the surface, in favor of avoiding the formation of multi-layer structures and facilitating the formation of high-quality COF NSs and thin films. The commonly used substrates include graphene, HOPG, and metal substrates (Au, Ag, Cu, etc.). It is worth mentioning that metal substrates may catalyze the coupling reaction on the surface to confine the formation of highly ordered sCOFs. However, most of the syntheses were conducted under UHV conditions, resulting in unexpected irreversible cross-linking and giving rise to the formation of disordered nanostructures. To improve the surface coverage and reduce the degree of disorder, manipulating the kinetic process by controlling the feed rate of building units may provide a solution. On the other hand, introducing a small amount of water in a closed system to maintain the reaction balance and enhance reversibility is favourable for obtaining uniform sCOFs with few defects. Additionally, directly polymerizing at room temperature by the solid–liquid interface method is able to prepare sCOFs with limited monomers of small molecular sizes. However, the surface coverage and periodicity of sCOFs still require further improvement and the as-formed sCOFs are difficult to be peeled off from the substrates.
The interface formed between liquid and liquid or air can also provide a confined space for the growth of COF NSs and thin films. Different monomers (sometimes involving catalysts) are dissolved in one or two (compatible or incompatible) solvents. Under certain conditions of temperature and pressure, the precursors polymerize at the interface to form relatively large-sized COF films and NSs. However, this approach seems to be not efficient enough to produce well-ordered and high-quality thin COF films, and it is difficult to control the number of COF layers by this means as well.
For the top-down strategy, it is a method of stripping bulk COFs into single-/multi-layered COF NSs by introducing external (sometimes by internal) forces or solvents rather than growing them directly on selected substrates or interface. Compared to the bottom-up approach, this method is easier for scale-up but suffers from the difficulties in controlling the thickness, homogeneity, and quality of COF NSs. We elaborated on the top-down strategy involving mechanical exfoliation, solvent-assisted exfoliation, chemical exfoliation, and self-exfoliation as well as the difference among them that can be attributed to distinct peel strength. The mechanical exfoliation of bulk COFs via ball milling or grinding is a conventional exfoliation technique that applies mechanical forces to break the van der Waals interaction between the layers of bulk COF materials without destroying the covalent bonds of the frameworks; then, single- or few-layered 2D COF nanosheets are produced. Moreover, solvent-assisted exfoliation under sonication conditions is also widely used for COF exfoliation. In contrast, the self-exfoliation approach is more likely to depend on internal forces among the building blocks to strip the multi-layered COFs into the single or few-layered structure. Several iCOFs are exfoliated by mixing with solvents in a self-exfoliation method.
Although great achievements have been made, some obstacles need to be overcome in future development. (1) Quality: it is difficult to precisely control the degree of periodicity, crystal domain area, molecular orientation, and defect of the COF films and NSs. Thus, insightful mechanism studies on the growth of COFs based on advanced in situ characterization techniques (e.g., TEM, XPS, and Raman spectroscopy) are required; (2) Diversity: in different approaches, due to the solubility, activity, and volatility of the monomers as well as the reversibility of the reactions only limited number of COF films and NSs have been prepared. Therefore, it is desired to develop more facile preparation methods with improved applicability; (3) Electrochemical stability: despite the fact that most of the COFs are chemically robust and are even stable against harsh chemical environments, their electrochemical stability still requires further deep investigation; (4) Scale-up: only up to centimeter-sized COF films and NSs with low yield have been prepared and the reaction conditions always require a large amount of organic solvent, high vacuum, ultra-regular surface, and long production period. Besides, the structure–function relationships of COF films and NSs still remain to be largely explored.
5.2 COFs for energy storage and conversion
Within the last few years, research on COF-based electrochemical energy storage has emerged rapidly and become diversified. As a concluding remark, the research directions focused on EES are summarized:(1) As the electrode: COFs are positioned to address the challenge in organic electrode materials with a controllable degree of porosity and more stable frameworks (e.g., various organic solvents, and strong acid or base). The following issues should be considered to achieve higher performance: enhancing the density of redox-active sites; increasing or decreasing the voltage plateau for cathode and anode, respectively; increasing the electron conductivity to fully utilize their redox-active sites; balancing the density and porosity by structural optimization to utilize their pores and active sites efficiently; widening the operation potential windows by using organic electrolytes or ionic liquid electrolytes in supercapacitors; charge–discharge mechanism is unclear (especially for anode), and theoretical and experimental studies need to be further investigated.
(2) As the host for the phase-changing component: the prerequisites for COFs to act as a host in phase changing cathode-based batteries, such as Li–S batteries, are high specific surface area and large pore volume, leading to high active component loading in composites. Besides, the introduction of specific functional groups can provide interactions to restrain the migration of soluble intermediates out of the pores and further inhibit the shuttle effect.
(3) As an electrolyte: the ionic skeleton can effectively promote ion-pair dissociation and facilitate ion migration, and the transference number of target ions can be improved by using different strategies. COFs incorporated with flexible chains provide a chance to solve the crystallization and phase transition problems in polymeric electrolytes, leading to a wide usage temperature range. However, when targeting all-solid-state electrodes for portable devices, it is urgently needed to develop solvent-free flexible COF films with improved ion conductivity and transference number. Moreover, besides the Li+ ion, the conduction behaviors of other ions, such as Na+, K+, Mg2+, Zn2+, and Al3+, have remained unexplored.
(4) As dramatically developed materials, COFs are still very young in the EES fields. Owing to their ordered structures, designable pore size, modifiable skeletons, and high specific surfaces, they have huge potential for developing higher theoretical gravimetric energy density EES systems. Recently, many new fields of EES were investigated, such as aqueous zinc ion batteries,327 Li–CO2 batteries,183,328 and artificial SEI layers for use in anodes.329
COFs combine the merits of both molecular and heterogeneous catalysts, and many inherent advantages make them promising candidates for preparing efficient electrocatalysts. Since the activity, selectivity, and stability of a catalyst are the main factors to be evaluated, we provide a summary and outlook of the development of COF-based electrocatalysts from these aspects.
The selectivity of an electrocatalyst is mainly determined by the active sites and their surroundings, while the activity is governed by the types, electronic structures, and dispersity of active sites, as well as electron/substrate accessibility.
Multi-electron macrocycles (e.g., porphyrin and phthalocyanine), metal ions (e.g., Fe, Co, Ni, Ir, Ru, and even dual ions), and metallic nanoparticles (e.g., RuO, Ni3N) as active sites have been introduced onto the skeleton or into the channels via building block design, post-synthetic modification, or in situ generation. The unique structural features of COFs ensure the atomic distribution of active sites like macrocycles and metal ions, while the predesignable and well-aligned channels can confine the formation of metallic nanoparticles in nano-sized diameters. Inherent electron conduction channels existing in the 2D COFs with π–π electron cloud overlap between the adjacent layers as well as the conjugated skeletons in both 2D and 3D COFs. To further alleviate the conductivity loss of bulk COFs caused by interfacial boundaries and disordered area, several efforts have been made, including physically mixing COFs with conductive components (e.g., carbon black), growing COFs on conductive materials (e.g., CNTs and graphene), preparing highly oriented thin COF films on electrodes, exfoliating bulk COFs into NSs, and pyrolyzing them into porous carbons. The large pore diameters, open and straight channels, and high porosity of COFs are favorable for mass transport; their pore structures and environment have been further modulated, and hierarchical structures have been prepared to facilitate reactant and product diffusion. Functional building units with electron-donating or -withdrawing properties have been introduced to tune their electronic structures.
Therefore, ideally, in a COF-catalyzed electrochemical reaction, favorite products can be obtained with high purity and low energy consumption. However, up to now, there still remains a large room to increase the selectivity and activity of COF-based electrochemical reactions, including ORR, HER, OER, CO2 reduction, and N2 fixation.330 In addition, although the stability of COFs and their performance are almost satisfactory for electrochemical reaction, partial decomposition of the COF skeletons and leaching of metal ions have been found in acidic or alkaline test solutions.
To overcome these challenges, the following efforts are suggested: (1) thoroughly investigating their structure–function relationships via precise molecular design; (2) carefully tuning the surrounding environment of active sites that affects the adsorption, bond cleavage and formation, and desorption process; (3) exploring the reaction pathways and their influencing factors via experimental works and theoretical calculations; (4) preparing COFs and their NSs with high periodicity, fewer defects, and high orientation to promote their mass transport and charge transfer process; (6) increasing the loading and accessibility of active sites; (7) verifying their structure evolution after electrochemical processes; (8) increasing their stability under electrocatalytic conditions; (9) developing scalable production methods to reduce their cost.
Abbreviations
| COFs | Covalent organic frameworks |
| EES | Electrochemical energy storage |
| EEC | Electrochemical energy conversion |
| 2D | Two dimensional |
| 3D | Three dimensional |
| NSs | Nanosheets |
| CTFs | Covalent triazine-based frameworks |
| CORFs | Covalent organic radical frameworks |
| FT-IR | Fourier Transform Infrared |
| OPA | o-Phthalaldehyde |
| DMF | N,N-Dimethylformamide |
| THF | Tetrahydrofuran |
| PXRD | Powder X-ray diffraction |
| AFM | Atomic Force Microscope |
| DLS | Dynamic light scattering |
| HRTEM | High-resolution transmission electron microscopy |
| TEM | Transmission electron microscopy |
| iCOFs | Ionic covalent organic frameworks |
| Py | 4,4′,4′′,4′′′-(Pyrene-1,3,6,8-tetrayl)tetraaniline |
| SAED | Selected-area electron diffraction |
| TP | 1,3,5-Triformylphloroglucinol |
| DANTB | 2,7-Bis((E)-benzylideneamino)benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone |
| Tb | 1,3,5-Triformylbenzene |
| CNTs | Carbon nanotubes |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxyl |
| LSBs | Lithium–sulfur batteries |
| PSs | Lithium polysulfides |
| BOC | Boron/oxygen co-doped porous carbon |
| XPS | X-ray photoelectron spectroscopy |
| 1H-NMR | 1H nuclear magnetic resonance |
| IR | Infrared spectra |
| LIBs | Lithium-ion batteries |
| SIBs | Sodium-ion batteries |
| δ i | Ionic conductivity |
| t + | Transference number (of Li+) |
| γ-CD | γ-Cyclodextrin |
| EC | Ethylene carbonate |
| DMC | Dimethyl carbonate |
| PEG | Poly(ethylene oxide) |
| TPB | 1,3,5-Tri(4-aminophenyl)benzene |
| BMTP | 2,5-Bis((2-methoxyethoxy)methoxy)terephthalaldehyde |
| DSC | Differential scanning calorimetry |
| PEMFC | Proton exchange membrane fuel cells |
| EB | Ethidium bromide |
| TAPB | 1,3,5-Tris(4-aminophenyl)benzene |
| BTCA | 1,3,5-Benzenetricarbaldehyde |
| RH | Relative humidity |
| SCs | Supercapacitors |
| EDLCs | Electrochemical double-layer capacitors |
| FS | Faradaic supercapacitors |
| DAAQ | 2,6-Diaminoanthraquinone |
| PDC | 1,4-Piperazinedicarboxaldehyde |
| MA | Melamine |
| NiP | 5,10,15,20-Tetrakis(4′-tetraphenylamino) porphyrin |
| BPTA | 2,5-Bis(2-propynyloxy)terephthalaldehyde |
| DMTA | 2,5-Dimethoxyterephthalaldehyde |
| PVA | Poly(vinyl alcohol) |
| TFP | 1,3,5-Triformylphloroglucinol |
| GIXD | Grazing incidence X-ray diffraction |
| CNF | Conductive carbon nanofiber |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| BTT | Benzotrithiophene tricarbaldehyde |
| HOPG | Highly oriented pyrolytic graphite |
| sCOFs | Single-layered covalent organic frameworks |
| STM | Scanning tunneling microscopy |
| UHV | Ultrahigh vacuum |
| LAP | Laminar assembly polymerization |
| BDBA | 1,4-Benzenediboronic acid |
| HHTP | 2,3,6,7,10,11-Hexahydroxytriphenylene |
| TMC | Benzene-1,3,5-tricarbonyl trichloride |
| DFT | Density functional theory |
| 4ATTF | Tetrathiafulvalene equipped with four benzaldehyde groups |
| PPDA | p-Phenylenediamine |
| SLG | Single-layer graphene |
| TBA | 2,4,6-Trimethylbenzaldehyde |
| BII | Buffering interlayer interface |
| PTSA | Amine-p-toluene sulfonic acid |
| TTA | 4,4′,4′′-(1,3,5-Triazine-2,4,6-triyl)trianiline |
| DHTA | 2,5-Dihydroxyterethaldehyde |
| ORR | Oxygen reduction reaction |
| HER | Hydrogen evolution reaction |
| OER | Oxygen evolution reaction |
| CO2RR | CO2 reduction reaction |
| RDS | Rate-determining step |
| NPMC | Non-precious metal catalysts |
| PTM | Polychlorotriphenylmethyl |
| PA | Phytic acid |
| SAC | Single-atom catalysts |
| η 10 | Overpotential (at a current density of 10 mA cm−2) |
| j 0 | Current density |
| TOF | Turnover frequency |
| FE | Faradaic efficiency |
| WOCs | Water-oxidizing complexes |
| Bpy | 2,2′-Bipyridyl 5,5′-diamine |
| NPs | Nanoparticles |
| BDA | 1,4-Benzenedicarboxaldehyde |
| ECR | Electrochemical reduction |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21922502, 21674012, 21625102, 21971017) and Beijing Institute of Technology Research Fund Program.Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- M. R. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 12647 CrossRef PubMed.
- C. X. Zu and H. Li, Energy Environ. Sci., 2011, 4, 2614–2624 RSC.
- W. Xia, A. Mahmood, R. Q. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- Y. G. Wang, Y. F. Song and Y. Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- L. Wang, Y. Z. Han, X. Feng, J. W. Zhou, P. F. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- F. X. Wang, X. W. Wu, X. H. Yuan, Z. C. Liu, Y. Zhang, L. J. Fu, Y. S. Zhu, Q. M. Zhou, Y. P. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC.
- H. Y. Jin, C. X. Guo, X. Liu, J. L. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- X. B. Cheng, C. Z. Zhao, Y. X. Yao, H. Liu and Q. Zhang, Chem, 2019, 5, 74–96 CAS.
- N. Chen, H. Q. Zhang, L. Li, R. J. Chen and S. J. Guo, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- Z. B. Liang, C. Qu, W. H. Guo, R. Q. Zou and Q. Xu, Adv. Mater., 2018, 30, 1702891 CrossRef PubMed.
- Q. R. Fang, Z. B. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. H. Wang, S. L. Qiu and Y. S. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- X. Feng, X. S. Ding and D. L. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed.
- G. Zhang, M. Tsujimoto, D. Packwood, N. T. Duong, Y. Nishiyama, K. Kadota, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2018, 140, 2602–2609 CrossRef CAS PubMed.
- Z. F. Pang, T. Y. Zhou, R. R. Liang, Q. Y. Qi and X. Zhao, Chem. Sci., 2017, 8, 3866–3870 RSC.
- R. W. Tilford, S. J. Mugavero, 3rd, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741–2746 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. L. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- X. Y. Chen, K. Y. Geng, R. Y. Liu, K. T. Tan, Y. F. Gong, Z. P. Li, S. S. Tao, Q. H. Jiang and D. L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 2–44 CrossRef.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 2–30 CrossRef.
- Y. F. Zeng, R. Q. Zou and Y. L. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- N. Huang, X. Chen, R. Krishna and D. L. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- J. T. Yu, Z. Chen, J. L. Sun, Z. T. Huang and Q. Y. Zheng, J. Mater. Chem., 2012, 22, 5369–5373 RSC.
- S. S. Yuan, X. Li, J. Y. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- C. Zhang, B. H. Wu, M. Q. Ma, Z. K. Wang and Z. K. Xu, Chem. Soc. Rev., 2019, 48, 3811–3841 RSC.
- X. Q. Zou and G. S. Zhu, Adv. Mater., 2018, 30, 1700750 CrossRef PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepulveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, I. X. F. X. Llabres, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- X. Chen, M. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Q. Guo, L. L. Liu, S. Irle and D. L. Jiang, J. Am. Chem. Soc., 2015, 137, 3241–3247 CrossRef CAS PubMed.
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Doblinger, S. Herbert, K. Peters, D. Bohm, P. Knochel, D. D. Medina, D. Fattakhova-Rohlfing and T. Bein, J. Am. Chem. Soc., 2018, 140, 2085–2092 CrossRef CAS PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. B. Zhao, A. R. Paris, D. Y. Kim, P. D. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- S. L. Wang, L. Ma, Q. Y. Wang, P. P. Shao, D. Ma, S. Yuan, P. Lei, P. F. Li, X. Feng and B. Wang, J. Mater. Chem. C, 2018, 6, 5369–5374 RSC.
- L. Ma, X. Feng, S. Wang and B. Wang, Mater. Chem. Front., 2017, 1, 2474–2486 RSC.
- Y. P. Song, Q. Sun, B. Aguila and S. Q. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- P. P. Shao, J. Li, F. Chen, L. Ma, Q. B. Li, M. X. Zhang, J. W. Zhou, A. X. Yin, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2018, 57, 16501–16505 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- T. Q. Ma, K. A. Kapustin, S. X. Yin, L. Liang, Z. Y. Zhou, J. Niu, L. H. Li, Y. Y. Wang, J. Su, J. Li, X. G. Wang, W. D. Wang, W. Wang, J. L. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. H. Chen and D. L. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O’Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- S. C. Yan, X. Guan, H. Li, D. H. Li, M. Xue, Y. S. Yan, V. Valtchev, S. L. Qiu and Q. R. Fang, J. Am. Chem. Soc., 2019, 141, 2920–2924 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- K. Geng, T. He, R. Liu, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00550.
- S. Patwardhan, A. A. Kocherzhenko, F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. C, 2011, 115, 11768–11772 CrossRef CAS.
- X. Feng, L. Liu, Y. Honsho, A. Saeki, S. Seki, S. Irle, Y. Dong, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2618–2622 CrossRef CAS PubMed.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. P. Dong and D. L. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- S. Wan, F. G. Andara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. F. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed.
- C. Y. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef PubMed.
- J. W. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- X. Gao, Y. Dong, S. W. Li, J. W. Zhou, L. Wang and B. Wang, Electrochem. Energy Rev., 2020, 3, 81–126 CrossRef.
- B. N. Zheng, X. D. Lin, X. C. Zhang, D. C. Wu and K. Matyjaszewski, Adv. Funct. Mater., 2019 DOI:10.1002/adfm.201907006.
- S. J. Kim, H. T. Wang and Y. M. Lee, Angew. Chem., Int. Ed., 2019, 58, 17512–17527 CrossRef CAS PubMed.
- H. Wang, Z. T. Zeng, P. Xu, L. S. Li, G. M. Zeng, R. Xiao, Z. Y. Tang, D. L. Huang, L. Tang, C. Lai, D. N. Jiang, Y. Liu, H. Yi, L. Qin, S. J. Ye, X. Y. Ren and W. W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- R. H. Dong, T. Zhang and X. L. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- P. T. Xiao and Y. X. Xu, J. Mater. Chem. A, 2018, 6, 21676–21695 RSC.
- Z. K. Peng, H. M. Ding, R. F. Chen, C. Gao and C. Wang, Acta Chim. Sin., 2019, 77, 681–689 CrossRef.
- W. Zheng, C. S. Tsang, L. Y. S. Lee and K. Y. Wong, Mater. Today Chem., 2019, 12, 34–60 CrossRef CAS.
- S. B. Alahakoon, C. M. Thompson, G. Occhialini and R. A. Smaldone, ChemSusChem, 2017, 10, 2116–2129 CrossRef CAS PubMed.
- Y. Zhong, B. R. Cheng, C. Park, A. Ray, S. Brown, F. Mujid, J. U. Lee, H. Zhou, J. Suh, K. H. Lee, A. J. Mannix, K. Kang, S. J. Sibener, D. A. Muller and J. Park, Science, 2019, 366, 1379–1384 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456–4458 RSC.
- M. Bieri, M. T. Nguyen, O. Gröning, J. M. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669–16676 CrossRef CAS PubMed.
- N. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678–6679 CrossRef CAS PubMed.
- S. Schlögl, T. Sirtl, J. Eichhorn, W. M. Heckl and M. Lackinger, Chem. Commun., 2011, 47, 12355–12357 RSC.
- O. Ourdjini, R. Pawlak, M. Abel, S. Clair, L. Chen, N. Bergeon, M. Sassi, V. Oison, J. M. Debierre, R. Coratger and L. Porte, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 125421 CrossRef.
- T. Faury, S. Clair, M. Abel, F. Dumur, D. Gigmes and L. Porte, J. Phys. Chem. C, 2012, 116, 4819–4823 CrossRef CAS.
- S. Spitzer, A. Rastgoo-Lahrood, K. Macknapp, V. Ritter, S. Sotier, W. M. Heckl and M. Lackinger, Chem. Commun., 2017, 53, 5147–5150 RSC.
- A. C. Marele, R. Mas-Ballesté, L. Terracciano, J. Rodríguez-Fernández, I. Berlanga, S. S. Alexandre, R. Otero, J. M. Gallego, F. Zamora and J. M. Gómez-Rodríguez, Chem. Commun., 2012, 48, 6779–6781 RSC.
- B. Yang, J. Björk, H. P. Lin, X. Q. Zhang, H. M. Zhang, Y. Y. Li, J. Fan, Q. Li and L. F. Chi, J. Am. Chem. Soc., 2015, 137, 4904–4907 CrossRef CAS PubMed.
- C. Chen, T. Joshi, H. F. Li, A. D. Chavez, Z. Pedramrazi, P. N. Liu, H. Li, W. R. Dichtel, J. L. Bredas and M. F. Crommie, ACS Nano, 2018, 12, 385–391 CrossRef CAS PubMed.
- S. Weigelt, C. Busse, C. Bombis, M. M. Knudsen, K. V. Gothelf, E. Lægsgaard, F. Besenbacher and T. R. Linderoth, Angew. Chem., Int. Ed., 2008, 47, 4406–4410 CrossRef CAS PubMed.
- Z. M. Gong, B. Yang, H. P. Lin, Y. Y. Tang, Z. Y. Tang, J. J. Zhang, H. M. Zhang, Y. Y. Li, Y. S. Xie, Q. Li and L. F. Chi, ACS Nano, 2016, 10, 4228–4235 CrossRef CAS PubMed.
- T. Joshi, C. Chen, H. F. Li, C. S. Diercks, G. Q. Wang, P. J. Waller, H. Li, J. L. Bredas, O. M. Yaghi and M. F. Crommie, Adv. Mater., 2019, 31, 1805941 CrossRef PubMed.
- J. C. Russell, M. O. Blunt, J. M. Garfitt, D. J. Scurr, M. Alexander, N. R. Champness and P. H. Beton, J. Am. Chem. Soc., 2011, 133, 4220–4223 CrossRef CAS PubMed.
- C. Z. Guan, D. Wang and L. J. Wan, Chem. Commun., 2012, 48, 2943–2945 RSC.
- R. Tanoue, R. Higuchi, N. Enoki, Y. Miyasato, S. Uemura, N. Kimizuka, A. Z. Stieg, J. K. Gimzewski and M. Kunitake, ACS Nano, 2011, 5, 3923–3929 CrossRef CAS PubMed.
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed.
- J. Y. Yue, X. H. Liu, B. Sun and D. Wang, Chem. Commun., 2015, 51, 14318–14321 RSC.
- W. L. Dong, L. Wang, H. M. Ding, L. Zhao, D. Wang, C. Wang and L. J. Wan, Langmuir, 2015, 31, 11755–11759 CrossRef CAS PubMed.
- J. Y. Yue, M. Markoulides, A. C. Regan, S. Y. Li, N. Chronakis, A. Gourdon, T. Chen, H. J. Yan and D. Wang, Chem. Commun., 2017, 53, 428–431 RSC.
- Y. X. Yu, J. B. Lin, Y. Wang, Q. D. Zeng and S. B. Lei, Chem. Commun., 2016, 52, 6609–6612 RSC.
- L. Yu, Z. B. Li and D. Wang, Chem. Commun., 2016, 52, 13771–13774 RSC.
- Y. P. Mo, X. H. Liu and D. Wang, ACS Nano, 2017, 11, 11694–11700 CrossRef CAS PubMed.
- L. M. Wang, J. Y. Yue, X. Y. Cao and D. Wang, Langmuir, 2019, 35, 6333–6339 CrossRef CAS PubMed.
- X. H. Liu, C. Z. Guan, S. Y. Ding, W. Wang, H. J. Yan, D. Wang and L. J. Wan, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS PubMed.
- J. Y. Yue, Y. P. Mo, S. Y. Li, W. L. Dong, T. Chen and D. Wang, Chem. Sci., 2017, 8, 2169–2174 RSC.
- C. X. Wang, J. L. Chen, C. H. Shu, K. J. Shi and P. N. Liu, Phys. Chem. Chem. Phys., 2019, 21, 13222–13229 RSC.
- D. Cui, J. M. MacLeod, M. Ebrahimi, D. F. Perepichka and F. Rosei, Chem. Commun., 2015, 51, 16510–16513 RSC.
- D. L. Cui, J. M. MacLeod and F. Rosei, Small, 2019, 15, 1903294 CrossRef CAS PubMed.
- X. H. Liu, C. Z. Guan, Q. N. Zheng, D. Wang and L. J. Wan, J. Chem. Phys., 2015, 142, 101905 CrossRef PubMed.
- J. F. Dienstmaier, A. M. Gigler, A. J. Goetz, P. Knochel, T. Bein, A. Lyapin, S. Reichlmaier, W. M. Heckl and M. Lackinger, ACS Nano, 2011, 5, 9737–9745 CrossRef CAS PubMed.
- L. R. Xu, X. Zhou, Y. X. Yu, W. Q. Tian, J. Ma and S. B. Lei, ACS Nano, 2013, 7, 8066–8073 CrossRef CAS PubMed.
- C. Lu, Y. Li, L. M. Wang, H. J. Yan, L. Chen and D. Wang, Chem. Commun., 2019, 55, 1326–1329 RSC.
- L. R. Xu, X. Zhou, W. Q. Tian, T. Gao, Y. F. Zhang, S. B. Lei and Z. F. Liu, Angew. Chem., Int. Ed., 2014, 53, 9564–9568 CrossRef CAS PubMed.
- Q. T. Fan, C. C. Wang, Y. Han, J. F. Zhu, W. Hieringer, J. Kuttner, G. Hilt and J. M. Gottfried, Angew. Chem., Int. Ed., 2013, 52, 4668–4672 CrossRef CAS PubMed.
- X. H. Liu, Y. P. Mo, J. Y. Yue, Q. N. Zheng, H. J. Yan, D. Wang and L. J. Wan, Small, 2014, 10, 4934–4939 CrossRef CAS PubMed.
- C. L. Tan, X. H. Cao, X. J. Wu, Q. Y. He, J. Yang, X. Zhang, J. Z. Chen, W. Zhao, S. K. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- J. Sun, A. Klechikov, C. Moise, M. Prodana, M. Enachescu and A. V. Talyzin, Angew. Chem., Int. Ed., 2018, 57, 1034–1038 CrossRef CAS PubMed.
- M. S. Kim, W. J. Lee, S. M. Paek and J. K. Park, ACS Appl. Mater. Interfaces, 2018, 10, 32102–32111 CrossRef CAS PubMed.
- S. Park, M. S. Kim, W. Jang, J. K. Park and D. H. Wang, Nanoscale, 2018, 10, 4708–4717 RSC.
- B. Sun, C. H. Zhu, Y. Liu, C. Wang, L. J. Wan and D. Wang, Chem. Mater., 2017, 29, 4367–4374 CrossRef CAS.
- M. L. Xu, L. Y. Wang, Y. Xie, Y. H. Song and L. Wang, Sens. Actuators, B, 2019, 281, 1009–1015 CrossRef CAS.
- S. Y. Kim and H. C. Choi, Chem. Commun., 2019, 2, 60 CrossRef.
- H. Huang, F. M. Li, Y. Zhang and Y. Chen, J. Mater. Chem. A, 2019, 7, 5575–5582 RSC.
- W. B. Liu, X. K. Li, C. M. Wang, H. H. Pan, W. P. Liu, K. Wang, Q. D. Zeng, R. M. Wang and J. Z. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed.
- X. D. Chen, H. Zhang, C. G. Ci, W. W. Sun and Y. Wang, ACS Nano, 2019, 13, 3600–3607 CrossRef CAS PubMed.
- W. Y. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. H. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 CrossRef CAS PubMed.
- J. I. Feldblyum, C. H. McCreery, S. C. Andrews, T. Kurosawa, E. J. Santos, V. Duong, L. Fang, A. L. Ayzner and Z. N. Bao, Chem. Commun., 2015, 51, 13894–13897 RSC.
- S. Y. Kim, H. Lim, J. Lee and H. C. Choi, Langmuir, 2018, 34, 8731–8738 CrossRef CAS PubMed.
- T. Zhang, H. Y. Qi, Z. Q. Liao, Y. D. Horev, L. A. Panes-Ruiz, P. S. Petkov, Z. Zhang, R. Shivhare, P. P. Zhang, K. J. Liu, V. Bezugly, S. H. Liu, Z. K. Zheng, S. Mannsfeld, T. Heine, G. Cuniberti, H. Haick, E. Zschech, U. Kaiser, R. H. Dong and X. L. Feng, Nat. Commun., 2019, 10, 4225 CrossRef PubMed.
- K. J. Liu, H. Y. Qi, R. H. Dong, R. Shivhare, M. Addicoat, T. Zhang, H. Sahabudeen, T. Heine, S. Mannsfel, U. Kaiser, Z. K. Zheng and X. L. Feng, Nat. Chem., 2019, 11, 994–1000 CrossRef CAS PubMed.
- H. Sahabudeen, H. Y. Qi, M. Ballabio, M. Položij, S. Olthof, R. Shivhare, Y. Jing, S. Park, K. J. Liu, T. Zhang, J. Ma, B. Rellinghaus, S. Mannsfeld, T. Heine, M. Bonn, E. Cμnovas, Z. K. Zheng, U. Kaiser, R. H. Dong and X. L. Feng, Angew. Chem., Int. Ed., 2020, 59, 2–11 CrossRef PubMed.
- S. Park, Z. Q. Liao, B. Ibarlucea, H. Y. Qi, H. H. Lin, D. Becker, J. Melidonie, T. Zhang, H. Sahabudeen, L. Baraban, C. Baek, Z. K. Zheng, E. Zschech, A. Fery, T. Heine, U. Kaiser, G. Cuniberti, R. H. Dong and X. L. Feng, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201916595.
- Y. Li, M. C. Zhang, X. H. Guo, R. Wen, X. Li, X. F. Li, S. J. Li and L. J. Ma, Nanoscale Horiz., 2018, 3, 205–212 RSC.
- K. Dey, M. Pal, K. C. Rout, H. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- Z. F. Wang, Q. Yu, Y. B. Huang, H. D. An, Y. Zhao, Y. F. Feng, X. Li, X. L. Shi, J. J. Liang, F. S. Pan, P. Cheng, Y. Chen, S. Q. Ma and Z. J. Zhang, ACS Cent. Sci., 2019, 5, 1352–1359 CrossRef CAS PubMed.
- J. J. Liu, W. Zan, K. Li, Y. Yang, F. X. Bu and Y. X. Xu, J. Am. Chem. Soc., 2017, 139, 11666–11669 CrossRef CAS PubMed.
- Q. Hao, C. Q. Zhao, B. Sun, C. Lu, J. Liu, M. J. Liu, L. J. Wan and D. Wang, J. Am. Chem. Soc., 2018, 140, 12152–12158 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- W. R. Cui, C. R. Zhang, W. Jiang, R. P. Liang, S. H. Wen, D. Peng and J. D. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 9408–9415 CrossRef CAS.
- G. H. Liu, Z. Y. Jiang, H. Yang, C. D. Li, H. J. Wang, M. D. Wang, Y. M. Song, H. Wu and F. S. Pan, J. Membr. Sci., 2019, 572, 557–566 CrossRef CAS.
- S. Wang, Q. Y. Wang, P. P. Shao, Y. Z. Han, X. Gao, L. Ma, S. Yuan, X. J. Ma, J. W. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- X. W. Mu, J. Zhan, X. M. Feng, B. H. Yuan, S. L. Qiu, L. Song and Y. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 23017–23026 CrossRef CAS PubMed.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed.
- Y. W. Peng, Y. Huang, Y. H. Zhu, B. Chen, L. Y. Wang, Z. C. Lai, Z. C. Zhang, M. T. Zhao, C. L. Tan, N. L. Yang, F. W. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef CAS PubMed.
- J. Q. Dong, X. Li, S. B. Peh, Y. Yuan, D. Y. X. Wang, D. D. Ji, S. J. Peng, G. L. Liu, S. M. Ying, D. Q. Yuan, J. W. Jiang, S. Ramakrishna and D. Zhao, Chem. Mater., 2018, 31, 146–160 CrossRef.
- K. Wang, Z. Zhang, L. Lin, J. Chen, K. Hao, H. Y. Tian and X. S. Chen, Chem. Mater., 2019, 31, 3313–3323 CrossRef CAS.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- P. Wang, F. Zhou, C. Zhang, S. Y. Yin, L. L. Teng, L. L. Chen, X. X. Hu, H. W. Liu, X. Yin and X. B. Zhang, Chem. Sci., 2018, 9, 8402–8408 RSC.
- S. J. Gan, X. N. Tong, Y. Zhang, J. H. Wu, Y. Q. Hu and A. H. Yuan, Adv. Funct. Mater., 2019, 29, 1902757 CrossRef CAS.
- W. R. Cui, C. R. Zhang, W. Jiang, R. P. Liang and J. D. Qiu, ACS Appl. Nano Mater., 2019, 2, 5342–5349 CrossRef CAS.
- C. L. Zhang, S. M. Zhang, Y. H. Yan, F. Xia, A. N. Huang and Y. Z. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed.
- G. Das, F. Benyettou, S. K. Sharma, S. K. Sharama, T. Prakasam, F. Gandara, V. A. de la Pena-O'Shea, N. Saleh, R. Pasricha, R. Jagannathan, M. A. Olson and A. Trabolsi, Chem. Sci., 2018, 9, 8382–8387 RSC.
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed.
- N. Zhang, T. S. Wang, X. Wu, C. Jiang, F. Chen, W. Bai and R. Bai, RSC Adv., 2018, 8, 3803–3808 RSC.
- P. Peng, L. Shi, F. Huo, S. J. Zhang, C. X. Mi, Y. H. Cheng and Z. H. Xiang, ACS Nano, 2019, 13, 878–884 CrossRef CAS PubMed.
- H. W. Chen, H. Y. Tu, C. J. Hu, Y. Liu, D. R. Dong, Y. F. Sun, Y. F. Dai, S. L. Wang, H. Qian, Z. Y. Lin and L. W. Chen, J. Am. Chem. Soc., 2018, 140, 896–899 CrossRef CAS PubMed.
- A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., Int. Ed., 2018, 57, 8443–8447 CrossRef CAS PubMed.
- L. L. Wang, C. Zeng, H. Xu, P. C. Yin, D. C. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. G. Ma, Chem. Sci., 2019, 10, 1023–1028 RSC.
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604–15608 CrossRef CAS PubMed.
- Z. Y. Fan, K. Nomura, M. S. Zhu, X. X. Li, J. W. Xue, T. Majima and Y. Osakada, Chem. Commun., 2019, 2, 55 CrossRef.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Diaz Diaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef CAS PubMed.
- M. D. Wang, F. S. Pan, H. Yang, Y. Cao, H. J. Wang, Y. M. Song, Z. Lu, M. Z. Sun, H. Wu and Z. Y. Jiang, J. Mater. Chem. A, 2019, 7, 9912–9923 RSC.
- Y. G. Zhang, M. X. Tan, H. Li, Y. G. Zheng, S. J. Gao, H. Zhang and J. Y. Ying, Chem. Commun., 2011, 47, 7365–7367 RSC.
- D. W. Burke, C. Sun, I. Castano, N. C. Flanders, A. M. Evans, E. Vitaku, D. C. McLeod, R. H. Lambeth, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Angew. Chem., Int. Ed., 2020, 59, 5165 CrossRef CAS PubMed.
- X. J. Zhan, Z. Chen and Q. C. Zhang, J. Mater. Chem. A, 2017, 5, 14463–14479 RSC.
- M. Y. Wang, H. Guo, R. Xue, Q. Li, H. Liu, N. Wu, W. Q. Yao and W. Yang, ChemElectroChem, 2019, 6, 2984 CrossRef CAS.
- D. H. Yang, Z. Q. Yao, D. H. Wu, Y. H. Zhang, Z. Y. Zhou and X. H. Bu, J. Mater. Chem. A, 2016, 4, 18621–18627 RSC.
- J. Q. Lv, Y. X. Tan, J. F. Xie, R. Yang, M. X. Yu, S. S. Sun, M. D. Li, D. Q. Yuan and Y. B. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS PubMed.
- S. Q. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. B. Sheng, X. D. Zhuang, E. Brunner, T. Heine, R. Berger and X. L. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS PubMed.
- F. Xu, S. B. Jin, H. Zhong, D. C. Wu, X. Q. Yang, X. Chen, H. Wei, R. W. Fu and D. L. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS PubMed.
- Z. Q. Luo, L. J. Liu, J. X. Ning, K. X. Lei, Y. Lu, F. J. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed.
- G. Wang, N. Chandrasekhar, B. P. Biswal, D. Becker, S. Paasch, E. Brunner, M. Addicoat, M. H. Yu, R. Berger and X. L. Feng, Adv. Mater., 2019, 31, 1901478 CrossRef PubMed.
- R. Fang, S. Zhao, Z. Sun, D. W. Wang, H. M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- Z. Cheng, H. Pan, H. Zhong, Z. Xiao, X. Li and R. Wang, Adv. Funct. Mater., 2018, 28, 1707597 CrossRef.
- H. P. Liao, H. M. Ding, B. J. Li, X. P. Ai and C. Wang, J. Mater. Chem. A, 2014, 2, 8854–8858 RSC.
- H. P. Liao, H. M. Wang, H. M. Ding, X. S. Meng, H. Xu, B. S. Wang, X. P. Ai and C. Wang, J. Mater. Chem. A, 2016, 4, 7416–7421 RSC.
- Y. Meng, G. Q. Lin, H. M. Ding, H. P. Liao and C. Wang, J. Mater. Chem. A, 2018, 6, 17186–17191 RSC.
- Z. A. Ghazi, L. Y. Zhu, H. Wang, A. Naeem, A. M. Khattak, B. Liang, N. A. Khan, Z. X. Wei, L. S. Li and Z. Y. Tang, Adv. Energy Mater., 2016, 6, 1601250 CrossRef.
- X. D. Chen, Y. J. Xu, F. H. Du and Y. Wang, Small Methods, 2019, 3, 1900338 CrossRef CAS.
- Z. C. Xiao, D. B. Kong, Q. Song, S. K. Zhou, Y. B. Zhang, A. Badshah, J. X. Liang and L. J. Zhi, Nano Energy, 2018, 46, 365–371 CrossRef CAS.
- Q. Jiang, Y. S. Li, X. X. Zhao, P. X. Xiong, X. Yu, Y. H. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC.
- D. G. Wang, N. Li, Y. M. Hu, S. Wan, M. Song, G. Yu, Y. H. Jin, W. F. Wei, K. Han, G. C. Kuang and W. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 42233–42240 CrossRef CAS PubMed.
- F. Xu, S. H. Yang, X. Chen, Q. H. Liu, H. J. Li, H. Q. Wang, B. Q. Wei and D. L. Jiang, Chem. Sci., 2019, 10, 6001–6006 RSC.
- L. Y. Bai, Q. Gao and Y. L. Zhao, J. Mater. Chem. A, 2016, 4, 14106–14110 RSC.
- S. Huang, D. Chen, C. Meng, S. Wang, S. Ren, D. Han, M. Xiao, L. Sun and Y. Meng, Small, 2019, 15, 1904830 CrossRef CAS PubMed.
- S. Haldar, K. Roy, S. Nandi, D. Chakraborty, D. Puthusseri, Y. Gawli, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2018, 8, 1702170 CrossRef.
- Z. D. Lei, X. D. Chen, W. W. Sun, Y. Zhang and Y. Wang, Adv. Energy Mater., 2019, 9, 1801010 CrossRef.
- J. J. Liu, P. B. Lyu, Y. Zhang, P. Nachtigall and Y. X. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef PubMed.
- B. C. Patra, S. K. Das, A. Ghosh, K. A. Raj, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655–16663 RSC.
- S. Gu, S. F. Wu, L. J. Cao, M. C. Li, N. Qin, J. Zhu, Z. Q. Wang, Y. Z. Li, Z. Q. Li, J. J. Chen and Z. G. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- Z. D. Lei, Q. S. Yang, S. Y. Guo, W. W. Sun, H. Liu, L. P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef.
- X. D. Chen, Y. S. Li, L. Wang, Y. Xu, A. Nie, Q. Q. Li, F. Wu, W. W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31, 1901640 CrossRef.
- S. Feng, H. Xu, C. Zhang, Y. Chen, J. H. Zeng, D. L. Jiang and J. X. Jiang, Chem. Commun., 2017, 53, 11334–11337 RSC.
- J. S. Wu, S. F. Wu, M. C. Li, H. Phan, D. G. Wang, T. S. Herng, J. Ding and Z. G. Lu, Angew. Chem., Int. Ed., 2018, 57, 8007–8011 CrossRef.
- Z. Wang, Y. Li, P. Liu, Q. Qi, F. Zhang, G. Lu, X. Zhao and X. Huang, Nanoscale, 2019, 11, 5330–5335 RSC.
- C. J. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. J. Xu, D. S. Li, S. Zhang and Q. Zhang, ChemSusChem, 2019, 12, 1–8 CrossRef.
- E. Vitaku, C. N. Gannett, K. L. Carpenter, L. Shen, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2020, 142, 16–20 CrossRef CAS PubMed.
- X. F. Yang, B. Dong, H. Z. Zhang, R. Ge, Y. A. Gao and H. M. Zhang, RSC Adv., 2015, 5, 86137–86143 RSC.
- X. Zhang, Z. Wang, L. Yao, Y. Y. Mai, J. Q. Liu, X. L. Hua and H. Wei, Mater. Lett., 2018, 213, 143–147 CrossRef CAS.
- D. G. Wang, Y. Wang, M. Song, G. C. Kuang and K. Han, Chem. Commun., 2019, 55, 13247–13250 RSC.
- J. Y. Wang, L. P. Si, Q. Wei, X. J. Hong, L. G. Lin, X. Li, J. Y. Chen, P. B. Wen and Y. P. Cai, J. Energy Chem., 2019, 28, 54–60 CrossRef.
- Z. Yang, C. Peng, R. Meng, L. Zu, Y. Feng, B. Chen, Y. Mi, C. Zhang and J. Yang, ACS Cent. Sci., 2019, 5, 1876–1883 CrossRef CAS PubMed.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- Q. Zhao, S. Stalin, C. Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- Y. Y. Zhang, J. Y. Duan, D. Ma, P. F. Li, S. W. Li, H. W. Li, J. W. Zhou, X. J. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed.
- Q. Xu, S. S. Tao, Q. H. Jiang and D. L. Jiang, J. Am. Chem. Soc., 2018, 140, 7429–7432 CrossRef CAS PubMed.
- Z. B. Guo, Y. Y. Zhang, Y. Dong, J. Li, S. W. Li, P. P. Shao, X. Feng and B. Wang, J. Am. Chem. Soc., 2019, 141, 1923–1927 CrossRef CAS PubMed.
- Z. Xie, B. Wang, Z. F. Yang, X. Yang, X. Yu, G. L. Xing, Y. H. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 58, 15742–15746 CrossRef CAS PubMed.
- G. Zhang, Y. L. Hong, Y. Nishiyama, S. Y. Bai, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2018, 141, 1227–1234 CrossRef PubMed.
- Y. M. Hu, N. Dunlap, S. Wan, S. L. Lu, S. F. Huang, L. Sellinger, M. Ortiz, Y. H. Jin, S. H. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518–7525 CrossRef CAS PubMed.
- K. H. Jeong, S. Park, G. Y. Jung, S. H. Kim, Y. H. Lee, S. K. Kwak and S. Y. Lee, J. Am. Chem. Soc., 2019, 141, 5880–5885 CrossRef CAS PubMed.
- D. A. Vazquez-Molina, G. S. Mohammad-Pour, C. Lee, M. W. Logan, X. F. Duan, J. K. Harper and F. J. Uribe-Romo, J. Am. Chem. Soc., 2016, 138, 9767–9770 CrossRef CAS PubMed.
- C. Montoro, D. Rodriguez-San-Miguel, E. Polo, R. Escudero-Cid, M. L. Ruiz-Gonzalez, J. A. R. Navarro, P. Ocon and F. Zamora, J. Am. Chem. Soc., 2017, 139, 10079–10086 CrossRef CAS PubMed.
- H. Xu, S. S. Tao and D. L. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef CAS PubMed.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- H. P. Ma, B. L. Liu, B. Li, L. M. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. S. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed.
- S. Chandra, T. Kundu, K. Dey, M. Addicoat, T. Heine and R. Banerjee, Chem. Mater., 2016, 28, 1489–1494 CrossRef CAS.
- Y. W. Peng, G. D. Xu, Z. G. Hu, Y. D. Cheng, C. L. Chi, D. Q. Yuan, H. S. Cheng and D. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 18505–18512 CrossRef CAS PubMed.
- M. Z. A. Aykanat and K. A. Mirica, Chem. Mater., 2018, 31, 819–825 Search PubMed.
- H. Zhong, Z. H. Fu, J. M. Taylor, G. Xu and R. H. Wang, Adv. Funct. Mater., 2017, 27, 1701465 CrossRef.
- W. F. Kong, W. Jia, R. Wang, Y. F. Gong, C. C. Wang, P. Y. Wu and J. Guo, Chem. Commun., 2019, 55, 75–78 RSC.
- H. S. Sasmal, H. B. Aiyappa, S. N. Bhange, S. Karak, A. Halder, S. Kurungot and R. Banerjee, Angew. Chem., Int. Ed., 2018, 57, 10894–10898 CrossRef CAS PubMed.
- C. Jiang, M. Tang, S. L. Zhu, J. D. Zhang, Y. C. Wu, Y. Chen, C. Xia, C. L. Wang and W. P. Hu, Angew. Chem., Int. Ed., 2018, 57, 16072–16076 CrossRef CAS PubMed.
- Y. Yang, X. J. Hong, C. L. Song, L. H. Li, Y. X. Zheng, D. D. Zhou, M. Zhang, Y. P. Cai and H. X. Wang, J. Mater. Chem. A, 2019, 7, 16323–16329 RSC.
- J. Yoo, S. J. Cho, G. Y. Jung, S. H. Kim, K. H. Choi, J. H. Kim, C. K. Lee, S. K. Kwak and S. Y. Lee, Nano Lett., 2016, 16, 3292–3300 CrossRef CAS PubMed.
- Q. Xu, K. L. Zhang, J. Qian, Y. Guo, X. K. Song, H. L. Pan, D. Wang and X. P. Li, ACS Appl. Energy Mater., 2019, 2, 5793–5798 CrossRef.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS.
- S. Chandra, D. Roy Chowdhury, M. Addicoat, T. Heine, A. Paul and R. Banerjee, Chem. Mater., 2017, 29, 2074–2080 CrossRef CAS.
- L. Li, F. Lu, R. Xue, B. L. Ma, Q. Li, N. Wu, H. Liu, W. Q. Yao, H. Guo and W. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 26355–26363 CrossRef CAS PubMed.
- F. Xu, H. Xu, X. Chen, D. C. Wu, Y. Wu, H. Liu, C. Gu, R. W. Fu and D. L. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS PubMed.
- A. Halder, M. Ghosh, M. A. Khayum, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS PubMed.
- M. A. Khayum, V. Vijayakumar, S. Karak, S. Kandambeth, M. Bhadra, K. Suresh, N. Acharambath, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2018, 10, 28139–28146 CrossRef PubMed.
- W. Liu, M. Ulaganathan, I. Abdelwahab, X. Luo, Z. X. Chen, S. J. Rong Tan, X. W. Wang, Y. P. Liu, D. C. Geng, Y. Bao, J. Y. Chen and K. P. Loh, ACS Nano, 2017, 12, 852–860 CrossRef PubMed.
- A. F. M. El-Mahdy, Y. H. Hung, T. H. Mansoure, H. H. Yu, T. Chen and S. W. Kuo, Chem. – Asian J., 2019, 14, 1429–1435 CrossRef CAS.
- A. Roy, S. Mondal, A. Halder, A. Banerjee, D. Ghoshal, A. Paul and S. Malik, Eur. Polym. J., 2017, 93, 448–457 CrossRef CAS.
- A. F. M. El-Mahdy, C.-H. Kuo, A. Alshehri, C. Young, Y. Yamauchi, J. Kim and S.-W. Kuo, J. Mater. Chem. A, 2018, 6, 19532–19541 RSC.
- S. K. Das, K. Bhunia, A. Mallick, A. Pradhan, D. Pradhan and A. Bhaumik, Microporous Mesoporous Mater., 2018, 266, 109–116 CrossRef CAS.
- A. F. M. El-Mahdy, C. Young, J. Kim, J. You, Y. Yamauchi and S. W. Kuo, ACS Appl. Mater. Interfaces, 2019, 11, 9343–9354 CrossRef CAS PubMed.
- P. Bhanja, K. Bhunia, S. K. Das, D. Pradhan, R. Kimura, Y. Hijikata, S. Irle and A. Bhaumik, ChemSusChem, 2017, 10, 921–929 CrossRef CAS.
- S. Liu, L. Yao, Y. Lu, X. Hua, J. Liu, Z. Yang, H. Wei and Y. Mai, Mater. Lett., 2019, 236, 354–357 CrossRef CAS.
- Y. Han, Q. Zhang, N. T. Hu, X. Zhang, Y. Y. Mai, J. Q. Liu, X. L. Hua and H. Wei, Chin. Chem. Lett., 2017, 28, 2269–2273 CrossRef CAS.
- Z. Q. Zha, L. R. Xu, Z. K. Wang, X. G. Li, Q. M. Pan, P. A. Hu and S. B. Lei, ACS Appl. Mater. Interfaces, 2015, 7, 17837–17843 CrossRef CAS PubMed.
- P. Y. Wang, Q. Wu, L. F. Han, S. Wang, S. M. Fang, Z. H. Zhang and S. M. Sun, RSC Adv., 2015, 5, 27290–27294 RSC.
- Y. Wu, D. W. Yan, Z. Y. Zhang, M. M. Matsushita and K. Awaga, ACS Appl. Mater. Interfaces, 2019, 11, 7661–7665 CrossRef CAS PubMed.
- A. K. Mohammed, V. Vijayakumar, A. Halder, M. Ghosh, M. Addicoat, U. Bansode, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2019, 11, 30828–30837 CrossRef CAS PubMed.
- J. S. Xu, Y. F. He, S. Bi, M. Wang, P. Yang, D. Q. Wu, J. J. Wang and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12065–12069 CrossRef CAS PubMed.
- C. R. Mulzer, L. X. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed.
- B. Sun, J. Liu, A. M. Cao, W. G. Song and D. Wang, Chem. Commun., 2017, 53, 6303–6306 RSC.
- Y. Z. Liao, J. H. Li and A. Thomas, ACS Macro Lett., 2017, 6, 1444–1450 CrossRef CAS.
- Y. Han, N. Hu, S. Liu, Z. Hou, J. Liu, X. Hua, Z. Yang, L. Wei, L. Wang and H. Wei, Nanotechnology, 2017, 28, 33LT01 CrossRef PubMed.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodriguez-Calero, R. P. Bisbey, H. D. Abruna and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS.
- A. M. Khattak, Z. A. Ghazi, B. Liang, N. A. Khan, A. Iqbal, L. S. Li and Z. Y. Tang, J. Mater. Chem. A, 2016, 4, 16312–16317 RSC.
- A. F. M. El-Mahdy, M. G. Mohamed, T. H. Mansoure, H. H. Yu, T. Chen and S. W. Kuo, Chem. Commun., 2019, 55, 14890–14893 RSC.
- T. Li, X. D. Yan, Y. Liu, W. D. Zhang, Q. T. Fu, H. Y. Zhu, Z. J. Li and Z. G. Gu, Polym. Chem., 2020, 11, 47–52 RSC.
- D. W. Yan, Y. Wu, R. Kitauraa and K. Awaga, J. Mater. Chem. A, 2019, 7, 26829–26837 RSC.
- T. Li, W. D. Zhang, Y. Liu, Y. X. Li, C. K. Cheng, H. Y. Zhu, X. D. Yan, Z. J. Li and Z. G. Gu, J. Mater. Chem. A, 2019, 7, 19676–19681 RSC.
- Y. Yusran, H. Li, X. Guan, D. Li, L. Tang, M. Xue, Z. Zhuang, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, Adv. Mater., 2020, 32, 1907289 CrossRef CAS PubMed.
- Q. Xu, Y. P. Tang, L. P. Zhai, Q. H. Chen and D. L. Jiang, Chem. Commun., 2017, 53, 11690–11693 RSC.
- J. Romero, D. Rodriguez-San-Miguel, A. Ribera, R. Mas-Ballesté, T. F. Otero, I. Manet, F. Licio, G. Abellán, F. Zamora and E. Coronado, J. Mater. Chem. A, 2017, 5, 4343–4351 RSC.
- G. Kim, J. Yang, N. Nakashima and T. Shiraki, Chem. – Eur. J., 2017, 23, 17504–17510 CrossRef CAS PubMed.
- H. T. Wei, J. D. Ning, X. Cao, X. H. Li and L. Hao, J. Am. Chem. Soc., 2018, 140, 11618–11622 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- M. Kim, P. Puthiaraj, Y. J. Qian, Y. Kim, S. Jang, S. Hwang, E. Na, W. S. Ahn and S. E. Shim, Electrochim. Acta, 2018, 284, 98–107 CrossRef CAS.
- Y. Y. Lei, X. M. Liang, L. Yang, P. Jiang, Z. Y. Lei, S. S. Wu and J. W. Feng, J. Mater. Chem. A, 2020, 8, 4376–4385 RSC.
- Y. Q. Li, X. T. Xu, S. J. Hou, J. Q. Ma, T. Lu, J. C. Wang, Y. F. Yao and L. K. Pan, Chem. Commun., 2018, 54, 14009–14012 RSC.
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS.
- X. X. Lin, P. Peng, J. N. Guo and Z. H. Xiang, Chem. Eng. J., 2019, 358, 427–434 CrossRef CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- H. Zhu, D. X. Liu, D. T. Zou and J. Y. Zhang, J. Mater. Chem. A, 2018, 6, 6130–6154 RSC.
- W. G. Tu, Y. Xu, S. M. Yin and R. Xu, Adv. Mater., 2018, 30, 1707582 CrossRef PubMed.
- X. Meng and F. S. Xiao, Chem. Rev., 2014, 114, 1521–1543 CrossRef CAS PubMed.
- K. L. Yeung and W. Han, Catal. Today, 2014, 236, 182–205 CrossRef CAS.
- S. J. Lyle, P. J. Waller and O. M. Yaghi, Trends Chem., 2019, 1, 172–184 CrossRef.
- X. Y. Guan, H. Li, Y. C. Ma, M. Xue, Q. R. Fang, Y. S. Yan, V. Valtchev and S. L. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS PubMed.
- M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- Y. J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- Y. G. Li and H. J. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- A. C. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- M. Zhou, H. L. Wang and S. J. Guo, Chem. Soc. Rev., 2016, 45, 1273–1307 RSC.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- Q. Xu, Y. P. Tang, X. B. Zhang, Y. Oshima, Q. H. Chen and D. L. Jiang, Adv. Mater., 2018, 30, 1706330 CrossRef PubMed.
- Q. P. Lin, X. H. Bu, A. G. Kong, C. Y. Mao, F. Bu and P. Y. Feng, Adv. Mater., 2015, 27, 3431–3436 CrossRef CAS.
- Q. Zuo, G. Z. Cheng and W. Luo, Dalton Trans., 2017, 46, 9344–9348 RSC.
- S. B. Ren, J. Wang and X. H. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 25875–25880 CrossRef CAS PubMed.
- W. J. Ma, P. Yu, T. Ohsaka and L. Q. Mao, Electrochem. Commun., 2015, 52, 53–57 CrossRef CAS.
- X. H. He, Q. He, Y. C. Deng, M. Peng, H. Y. Chen, Y. Zhang, S. Y. Yao, M. T. Zhang, D. Q. Xiao, D. Ma, B. H. Ge and H. B. Ji, Nat. Commun., 2019, 10, 3663 CrossRef.
- X. J. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Y. Ye, G. Y. Tian, J. Schmidt and A. Thomas, Chem. Mater., 2019, 31, 3274–3280 CrossRef CAS.
- P. Peng, L. Shi, F. Huo, C. X. Mi, X. H. Wu, S. J. Zhang and Z. H. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef.
- W. P. Liu, C. M. Wang, L. J. Zhang, H. H. Pan, W. B. Liu, J. Chen, D. J. Yang, Y. J. Xiang, K. Wang, J. Z. Jiang and X. D. Yao, J. Mater. Chem. A, 2019, 7, 3112–3119 RSC.
- Q. Zhao, Z. H. Yan, C. C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- Y. Hou, M. Qiu, T. Zhang, J. Ma, S. Liu, X. Zhuang, C. Yuan and X. Feng, Adv. Mater., 2017, 29, 1604480 CrossRef PubMed.
- M. Shalom, D. Ressnig, X. F. Yang, G. Clavel, T. P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2015, 3, 8171–8177 RSC.
- X. Y. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- J. R. Galán-Mascarós, ChemElectroChem, 2015, 2, 37–50 CrossRef.
- B. Zhang, X. L. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. L. Han, J. X. Xu, M. Liu, L. R. Zheng, F. P. García de Arquer, C. T. Dinh, F. J. Fan, M. J. Yuan, E. Yassitepe, N. Chen, T. Regier, P. F. Liu, Y. H. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. G. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- X. J. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- D. Mullangi, V. Dhavale, S. Shalini, S. Nandi, S. Collins, T. Woo, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1600110 CrossRef.
- S. Nandi, S. K. Singh, D. Mullangi, R. Illathvalappil, L. George, C. P. Vinod, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1601189 CrossRef.
- D. Chakraborty, S. Nandi, R. Illathvalappil, D. Mullangi, R. Maity, S. K. Singh, S. Haldar, C. P. Vinod, S. Kurungot and R. Vaidhyanathan, ACS Omega, 2019, 4, 13465–13473 CrossRef CAS.
- H. X. Jia, Z. J. Sun, D. C. Jiang and P. W. Du, Chem. Mater., 2015, 27, 4586–4593 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- Y. Yan, T. He, B. Zhao, K. Qi, H. F. Liu and B. Y. Xia, J. Mater. Chem. A, 2018, 6, 15905–15926 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS.
- S. Bhunia, S. K. Das, R. Jana, S. C. Peter, S. Bhattacharya, M. Addicoat, A. Bhaumik and A. Pradhan, ACS Appl. Mater. Interfaces, 2017, 9, 23843–23851 CrossRef CAS.
- D. Zhou, X. Y. Tan, H. M. Wu, L. H. Tian and M. Li, Angew. Chem., Int. Ed., 2019, 58, 1376–1381 CrossRef CAS.
- M. A. Lukowski, A. S. Danie, F. Meng, A. Forticaux, L. S. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS.
- R. Schlogl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- H. L. Wu, M. Zeng, X. Zhu, C. C. Tian, B. B. Mei, Y. Song, X. L. Du, Z. Jiang, C. G. Xia and S. Dai, ChemElectroChem, 2018, 5, 2717–2721 CrossRef CAS.
- X. Zhang, H. Y. Liu, J. Q. Qin, H. S. Han, C. X. Qiu, S. Y. Zhang, X. Y. Hao, W. Liu and Y. J. Song, Chem. Commun., 2019, 55, 5659–5662 RSC.
- M. H. Zhu, D. T. Yang, R. Q. Ye, J. Zeng, N. Corbin and K. Manthiram, Catal. Sci. Technol., 2019, 9, 974–980 RSC.
- E. M. Johnson, R. Haiges and S. C. Marinescu, ACS Appl. Mater. Interfaces, 2018, 10, 37919–37927 CrossRef CAS PubMed.
- P. L. Cheung, S. K. Lee and C. P. Kubiak, Chem. Mater., 2019, 31, 1908–1919 CrossRef CAS.
- C. L. Yao, J. C. Li, W. Gao and Q. Jiang, Chem. – Eur. J., 2018, 24, 11051–11058 CrossRef CAS PubMed.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- D. A. Popov, J. M. Luna, N. M. Orchanian, R. Haiges, C. A. Downes and S. C. Marinescu, Dalton Trans., 2018, 47, 17450–17460 RSC.
- H. Y. Liu, J. Chu, Z. L. Yin, X. Cai, L. Zhuang and H. X. Deng, Chem, 2018, 4, 1696–1709 CAS.
- X. Zhu, C. C. Tian, H. H. Wu, Y. Y. He, L. He, H. Wang, X. D. Zhuang, H. L. Liu, C. G. Xia and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 43588–43594 CrossRef CAS PubMed.
- Y. H. Wang, J. X. Chen, G. X. Wang, Y. Li and Z. H. Wen, Angew. Chem., Int. Ed., 2018, 57, 13120–13124 CrossRef CAS PubMed.
- M. A. Khayum, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC.
- X. Li, H. Wang, Z. Chen, H. S. Xu, W. Yu, C. Liu, X. Wang, K. Zhang, K. Xie and K. P. Loh, Adv. Mater., 2019, 31, 1905879 CrossRef CAS PubMed.
- D. D. Chen, S. Huang, L. Zhong, S. J. Wang, M. Xiao, D. M. Han and Y. Z. Meng, Adv. Funct. Mater., 2019, 30, 1907717 CrossRef.
- S. Liu, M. F. Wang, T. Qian, H. Q. Ji, J. Liu and C. L. Yan, Nat. Commun., 2019, 10, 3898 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |