
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQSAR without borders
Eugene N.
Muratov
 ab,
Jürgen
Bajorath
c,
Robert P.
Sheridan
d,
Igor V.
Tetko
e,
Dmitry
Filimonov
f,
Vladimir
Poroikov
f,
Tudor I.
Oprea
ghi,
Igor I.
Baskin
jk,
Alexandre
Varnek
j,
Adrian
Roitberg
l,
Olexandr
Isayev
a,
Stefano
Curtalolo
m,
Denis
Fourches
n,
Yoram
Cohen
o,
Alan
Aspuru-Guzik
p,
David A.
Winkler
qrst,
Dimitris
Agrafiotis
u,
Artem
Cherkasov
*v and
Alexander
Tropsha
*a
ab,
Jürgen
Bajorath
c,
Robert P.
Sheridan
d,
Igor V.
Tetko
e,
Dmitry
Filimonov
f,
Vladimir
Poroikov
f,
Tudor I.
Oprea
ghi,
Igor I.
Baskin
jk,
Alexandre
Varnek
j,
Adrian
Roitberg
l,
Olexandr
Isayev
a,
Stefano
Curtalolo
m,
Denis
Fourches
n,
Yoram
Cohen
o,
Alan
Aspuru-Guzik
p,
David A.
Winkler
qrst,
Dimitris
Agrafiotis
u,
Artem
Cherkasov
*v and
Alexander
Tropsha
*a
aUNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC, USA. E-mail: alex_tropsha@unc.edu
bDepartment of Pharmaceutical Sciences, Federal University of Paraiba, Joao Pessoa, PB, Brazil
cDepartment of Life Science Informatics, University of Bonn, Bonn, Germany
dMerck & Co. Inc., Kenilworth, NJ, USA
eInstitute of Structural Biology, Helmholtz Zentrum München – Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH) and BIGCHEM GmbH, Neuherberg, Germany
fInstitute of Biomedical Chemistry, Moscow, Russia
gDepartment of Internal Medicine and UNM Comprehensive Cancer Center, University of New Mexico, Albuquerque, NM, USA
hDepartment of Rheumatology, Gothenburg University, Sweden
iNovo Nordisk Foundation Center for Protein Research, University of Copenhagen, Denmark
jDepartment of Chemistry, University of Strasbourg, Strasbourg, France
kFaculty of Physics, M. V. Lomonosov Moscow State University, Moscow, Russia
lDepartment of Chemistry, University of Florida, Gainesville, FL, USA
mMaterials Science, Center for Autonomous Materials Design, Duke University, Durham, NC, USA
nDepartment of Chemistry, North Carolina State University, Raleigh, NC, USA
oInstitute of The Environment and Sustainability, University of California, Los Angeles, CA, USA
pDepartment of Chemistry, University of Toronto, Toronto, ON, Canada
qMonash Institute of Pharmaceutical Sciences, Monash University, Melbourne, VIC, Australia
rLa Trobe Institute for Molecular Science, La Trobe University, Bundoora, Australia
sCSIRO Manufacturing, Clayton, Australia
tSchool of Pharmacy, University of Nottingham, Nottingham, UK
uNovartis Institutes for BioMedical Research (NIBR), Cambridge, MA, USA
vVancouver Prostate Centre, University of British Columbia, Vancouver, BC, Canada. E-mail: acherkasov@prostatecentre.com
First published on 1st May 2020
Abstract
Prediction of chemical bioactivity and physical properties has been one of the most important applications of statistical and more recently, machine learning and artificial intelligence methods in chemical sciences. This field of research, broadly known as quantitative structure–activity relationships (QSAR) modeling, has developed many important algorithms and has found a broad range of applications in physical organic and medicinal chemistry in the past 55+ years. This Perspective summarizes recent technological advances in QSAR modeling but it also highlights the applicability of algorithms, modeling methods, and validation practices developed in QSAR to a wide range of research areas outside of traditional QSAR boundaries including synthesis planning, nanotechnology, materials science, biomaterials, and clinical informatics. As modern research methods generate rapidly increasing amounts of data, the knowledge of robust data-driven modelling methods professed within the QSAR field can become essential for scientists working both within and outside of chemical research. We hope that this contribution highlighting the generalizable components of QSAR modeling will serve to address this challenge.
 Artem Cherkasov | Artem Cherkasov is a Professor of Medicine at the University of British Columbia (Vancouver, Canada) and a Director of Therapeutics Development at Vancouver Prostate Centre. Research interests include computer-aided drug discovery (CADD), QSAR modeling, drug reprofiling and development of new cancer therapies. Dr Cherkasov co-authored more than 200 research papers, 80 patent filings and several book chapters. During his tenure at the UBC, Dr Cherkasov has been a principal applicant or co-applicant on a number of successful grants totaling over 70M dollars, and licensed 6 drug candidates to big pharma companies, major international venture funds and spin off companies. |
 Alexander Tropsha | Alexander Tropsha, PhD is K. H. Lee Distinguished Professor and Associate Dean for Data and Data Science at the UNC Eshelman School of Pharmacy (ranked #1 in the country by US News & World Report), UNC-Chapel Hill. Prof. Tropsha obtained his PhD in Chemical Enzymology in 1986 from Moscow State University, Russia and came to UNC-Chapel Hill in 1989 as a postdoctoral fellow. He joined the School of Pharmacy in 1991 as an Assistant Professor and became full proffesor in 2002. His research interests are in the areas of Computer-Assisted Drug Design, Computational Toxicology, Cheminformatics, (Nano)Materials Informatics, and Structural Bioinformatics. His has authored or co-authored more than 230 peer-reviewed research papers, reviews and book chapters and co-edited two monographs. He is an Associate Editor of the ACS Journal of Chemical Information and Modeling. His research has been supported by multiple grants from the NIH, NSF, EPA, DOD, foundations, and private companies. |
Introduction
Quantitative structure–activity relationship (QSAR) modeling is a well-established computational approach to chemical data analysis. QSAR models are developed by establishing empirical, linear or non-linear relationships between values of chemical descriptors computed from molecular structure and experimentally measured properties or bioactivities of those molecules, followed by application of these models to predict or design novel chemicals with desired properties.Historically, QSAR modeling has been largely applied to computer-aided drug discovery. Many papers, reviews, and book chapters describing the methods and applications of QSAR modeling have appeared in the scientific literature since the seminal publication by Hansch et al. in 19621 that effectively pioneered the field. More than five years ago, some of the contributors to this paper co-authored a comprehensive review of QSAR modeling,2 where we discussed the evolution of methods and best practices of QSAR. Since then, the field has grown and evolved substantially. The Web of Science core collection lists more than 5600 papers on QSAR published within last five years, a substantial fraction of the ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 papers that have been published on this subject since 1962. Many publications have advanced the traditional areas of QSAR modeling3 such as prediction of biological activities and ADME/Tox properties, building on successful use of QSAR modeling in chemical, agrochemical, pharmaceutical,4 and cosmetic industries.5 However, new and interesting directions and application areas have also emerged, such as process chemistry6,7 and (retro)synthetic route prediction and optimization.8 Thus, models have become an integral component of the drug discovery process, providing substantial guidance in planning experiments.4,9
000 papers that have been published on this subject since 1962. Many publications have advanced the traditional areas of QSAR modeling3 such as prediction of biological activities and ADME/Tox properties, building on successful use of QSAR modeling in chemical, agrochemical, pharmaceutical,4 and cosmetic industries.5 However, new and interesting directions and application areas have also emerged, such as process chemistry6,7 and (retro)synthetic route prediction and optimization.8 Thus, models have become an integral component of the drug discovery process, providing substantial guidance in planning experiments.4,9
In cheminformatics molecules are represented by mathematical descriptors that encode molecular structures and properties. Multivariate statistical methods or machine learning are employed to establish relationships between descriptors and a target property, such as molecular bioactivity. It is easy to see that analogous representations can be generated for many types of data where objects are represented by their features, and the general objective is to predict object properties (endpoints) from these features. For instance, in clinical data, the objects would be patients, the features would be clinical or pharmacological biomarkers characteristic of the patients, and the target property would be the any health outcomes such as the rate of patient survival.
Regardless of the nature of the data, the same machine learning (ML) approaches can be used universally to analyze and process data in any domain. Furthermore, despite differences in the information content and meaning of the data, different research fields share similar data handling routines. These often replicate the workflows and protocols already created, evaluated, and used in QSAR. Indeed, the general data cycle associated with QSAR projects (Fig. 1) can be easily adopted for similar data-analytical investigations in other fields. To further illustrate this point, Table 1 provides a collection of recent references describing studies in diverse research areas that cite some or many concepts from QSAR. Examples include fields as diverse as climatology,10 urban engineering,11 student admissions,12 remote sensing13 and clinical informatics (discussed in one of the sections of this contribution). Importantly, QSAR modeling was among research fields that relatively early highlighted such subjects as the importance of data curation,14 rigorous validation of developed models,15 and data reproducibility,16 that have recently become a significant concern to the general scientific community.17
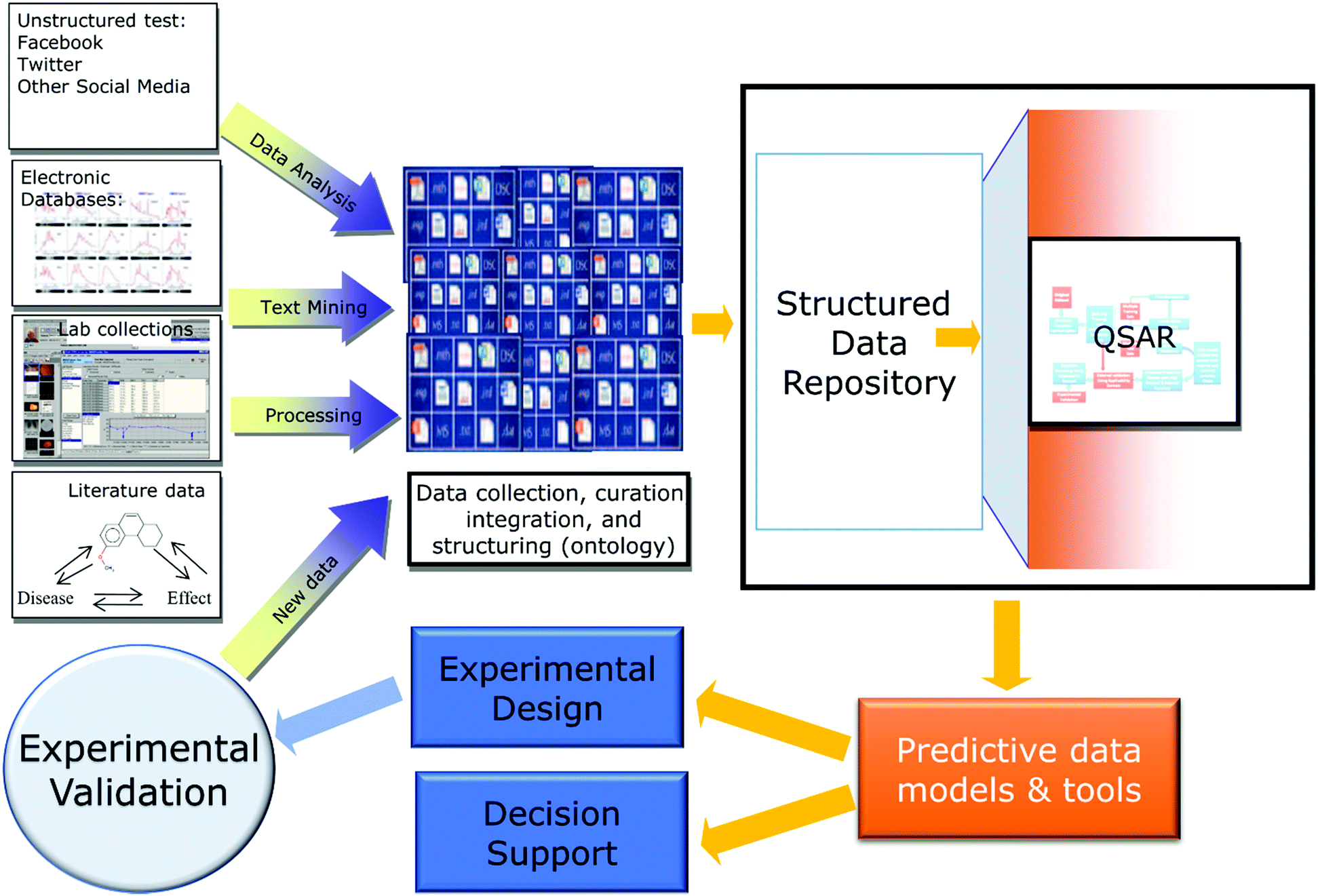 | ||
| Fig. 1 Data cycle associated with QSAR modeling projects. | ||
| Cited paper | Title | Journal | Year/ref. |
|---|---|---|---|
| 315 | Sensory analysis of red wines: Discrimination by adaptive fuzzy partition | Journal of Sensory Studies | 2008/318 |
| 15 | Improved wheat yield and production forecasting with a moisture stress index, AVHRR and MODIS data | Crop and Pasture Science | 2009/319 |
| 15 | Use of genetic algorithm and neural network approaches for risk factor selection: A case study of West Nile virus dynamics in an urban environment | Computers Environment and Urban Systems | 2010/11 |
| 15 | Whole cell-catalyzed transesterification of waste vegetable oil | Global Change Biology Bioenergy | 2010/320 |
| 15 | New Ground-Motion Prediction Equations Using Multi Expression Programing | Journal of Earthquake Engineering | 2011/321 |
| 322 | Qualitocracy: A Data Quality Collaborative Framework Applied to Citizen Science | IEEE Conference Proceedings | 2012/323 |
| 15 | Gene expression programming as a basis for new generation of electricity demand prediction models | Computers and Industrial Engineering | 2014/324 |
| 315 | Development of a model for quality evaluation of litchi fruit | Computers and Electronics in Agriculture | 2014/325 |
| 15 and 315 | Good practices in LIBS analysis: Review and advices | Spectrochimica Acta Part B-Atomic Spectroscopy | 2014/326 |
| 327 | Characterization of Softwood and Hardwood LignoBoost Kraft Lignins with Emphasis on their Antioxidant Activity | BioResources | 2014/328 |
| 315 | Gene expression models for prediction of dam breach parameters | Journal of Hydroinformatics | 2014/329 |
| 315 | An entrainment model for non-uniform sediment | Earth Surface Processes and Landforms | 2015/330 |
| 15 | Indirect estimation of the ultimate bearing capacity of shallow foundations resting on rock masses | International Journal of Rock Mechanics and Mining Sciences | 2015/331 |
| 15 | A novel protocol for assessment of aboveground biomass in rangeland environments | Rangeland Journal | 2015/332 |
| 15 | Statistical Modeling of Soil Moisture, Integrating Satellite Remote-Sensing (SAR) and Ground-Based Data | Remote Sensing | 2015/13 |
| 315 | Testing and Prediction of Material Compatibility of Biofuel Candidates with Elastomeric Materials | International Journal of Fuels and Lubricants | 2015/333 |
| 315 | Regression Algorithms in Hyperspectral Data Analysis for Meat Quality Detection and Evaluation | Comprehensive Reviews in Food Science and Food Safety | 2016/334 |
| 315 | Evolutionary patterns and physicochemical properties explain macroinvertebrate sensitivity to heavy metals | Ecological Applications | 2016/335 |
| 315 | Restricted attention to social cues in schizophrenia patients | European Archives of Psychiatry and Clinical Neuroscience | 2016/336 |
| 322 | Molecular descriptor data explain market prices of a large commercial chemical compound library | Scientific Reports | 2016/337 |
| 15 | A hybrid intelligent fuzzy predictive model with simulation for supplier evaluation and selection | Expert Systems with Applications | 2016/338 |
| 315 | Development of a stage-dependent prognostic model to predict psychosis in ultra-high-risk patients seeking treatment for co-morbid psychiatric disorders | Psychological Medicine | 2016/339 |
| 315 | Prediction of Timing of Watermain Failure Using Gene Expression Models | Water Resources Management | 2016/340 |
| 15 | A new approach for modeling of flow number of asphalt mixtures | Archives of Civil and Mechanical Engineering | 2017/341 |
| 15 | Next generation prediction model for daily solar radiation on horizontal surface using a hybrid neural network and simulated annealing method | Energy Conversion and Management | 2017/342 |
| 322 | Computer-Assisted Decision Support for Student Admissions Based on their Predicted Academic Performance | Journal of American Pharmaceutical Education | 2017/12 |
| 315 | Predicting Bond Strength between FRP Plates and Concrete Substrate: Applications of GMDH and MNLR Approaches | Journal of Advanced Concrete Technology | 2017/343 |
| 15 | Gene Expression Programming Approach to Cost Estimation Formulation for Utility Projects | Journal of Civil Engineering and Management | 2017/344 |
| 315 | Prediction of flow duration curves for ungauged basins | Journal of Hydrology | 2017/345 |
| 15 | Maize [Zea Mays (L.)] crop-nutrient response functions extrapolation for Sub-Saharan Africa | Nutrient Cycling in Agroecosystems | 2017/346 |
| 15 | Performance assessment of existing models to predict brittle failure modes of steel-to-timber connections loaded parallel-to-grain with dowel-type fasteners | Engineering Structures | 2018/347 |
| 315 | A comparative study on groundwater spring potential analysis based on statistical index, index of entropy and certainty factors models | Geocarto International | 2018/348 |
| 349 | Environmental factors influencing snowfall and snowfall prediction in the Tianshan Mountains, Northwest China | Journal of Arid Land | 2018/350 |
| 15 and 315 | Prediction of riprap stone size under overtopping flow using data-driven models | International Journal of River Basin Management | 2018/351 |
| 15 | Forecasting experiments of a dynamical–statistical model of the sea surface temperature anomaly field based on the improved self-memorization principle | Ocean Science | 2018/10 |
| 315 | Expressed emotion as a predictor of the first psychotic episode – Results of the European prediction of psychosis study | Schizophrenia Research | 2018/352 |
Here we integrate contributions from some of the leading experts in QSAR modeling that illustrate the breadth and generality of modern data processing and modeling practices in the field and highlight the applicability of these methods outside of the traditional borders of the field.18 The contributors have worked both on methodology and applications of QSAR modeling for most of their professional life. Some of the co-authors have pivoted their research into other areas where QSAR-like approaches have not been used before, illustrating the main theme of this paper by their own careers. We engaged other scientists who work in areas where data modeling was not common but who have started using QSAR-like methods in their research. We are confident that many fields that employ statistical modeling approaches will benefit significantly from the experience accumulated within the QSAR community in the last 55 years.
We start this contribution by discussing fundamental concepts of QSAR, such as chemical similarity. We describe the impact of recent advances, such as deep learning (DL), on traditional areas of QSAR modeling, such as drug discovery and development and chemical safety prediction. We then reflect how the complexity of algorithms and the size, diversity, and complexity of chemical bioactivity data have grown. We also illustrate how modern computational methods are capable of modeling multiple bioactivity endpoints simultaneously, addressing the issue of multi-objective optimization. We then extend traditional boundaries of QSAR by summarizing recent, exciting developments in organic synthesis planning and retrosynthetic pathway prediction, advances in robotic chemistry, and applications of machine learning to quantum chemistry. Finally, to further illustrate the breadth of applicability of modern QSAR approaches, we discuss their use in materials and nanomaterials science, regenerative medicine, and health care. Throughout the discussion, we identify methodological similarities between drug discovery approaches and those employed in other areas. We further propose that experience and best practice of data curation, model development, and validation accumulated by the QSAR community provides valuable guidance for many areas where statistical and machine learning data modeling is applied.
This broad, platform applicability of QSAR algorithms and protocols across all data-rich areas of modern science underpins the appeal of QSAR as a robust, predictive data analysis and modelling tool. We advise contemporary chemists to become familiar with the major computational approaches discussed in this contribution. To this end, borrowing from a recent “In the Pipeline” blog by Derek Lowe,19 “it is not that machines are going to replace chemists. It's that the chemists who use machines will replace those that don’t”! We hope that this paper will stimulate experimental scientists to consider deeper integration of computational methods and models into their research projects, to consider how the data they generate will be modelled when planning experiments and will serve as useful reference for computational chemists as well.
Clearly, QSAR modeling is an established and useful computational chemistry approach. However, many practitioners still consider it limited to modeling and prediction of chemical bioactivities and/or properties. One aim of this Perspective is to outline the opportunities presented by recent and emerging developments in artificial intelligence (AI), machine learning (ML) and other approaches to modeling Big Data within the traditional QSAR modeling. However, our prime objective is to emphasize the impact that QSAR methods and approaches have, or will shortly have, on many modern data-driven areas of molecular research beyond traditional QSAR areas. We called this paper QSAR without borders, to emphasize the plausible impact that many data modeling approaches developed and practiced by the QSAR community may have on many areas of the scientific pursuit.
Chemical similarity
Classical QSAR is defined by linear (regression) models derived from a set of small molecules sharing the same (target-specific) biological activity. A QSAR model predicts changes in potency as a function of structural modifications.1,20 The evolution of QSAR modeling from linear to more complex machine learning models addressing non-linear relationships between chemical structure and bioactivity was discussed in a paper co-written by one of the founders of classical QSAR, Prof. Toshio Fujita in 2016.20 Chemical bioactivity data employed in model development are generally derived from investigations of analog series from medicinal chemistry. These sets of compounds usually share a common core structure (scaffold) and carry different substituents (R-groups) at one or more sites. Descriptor-based linear regression models then predict potency of newly designed analogs to further extend such congeneric series, a fundamental task of classical QSAR. This prediction scheme is provides a useful guide to compound design and synthesis, making QSAR one of the most popular predictive approaches in medicinal chemistry since its seminal development.1QSAR modeling is based upon the premise that structurally similar compounds exhibit similar biological effects, often referred to as the similarity-property principle (SPP). The SPP postulates a conceptual link between molecular similarity and biological activity and implies that gradual changes in compound structure are accompanied by gradual changes in potency, which provides a rationale for the derivation of linear QSAR models. In congeneric series, analogs share the same core, which renders them similar. R-group replacements result in incremental changes in structure and ensuing potency variations should be predictable. The applicability domain of these predictions is defined by the SPP and requires the presence of “SAR continuity”,21 as illustrated in Fig. 2.
 | ||
| Fig. 2 SAR characteristics of cyclooxygenase-2 inhibitors. Substitutions are highlighted. For each compound, the pIC50 value is reported. | ||
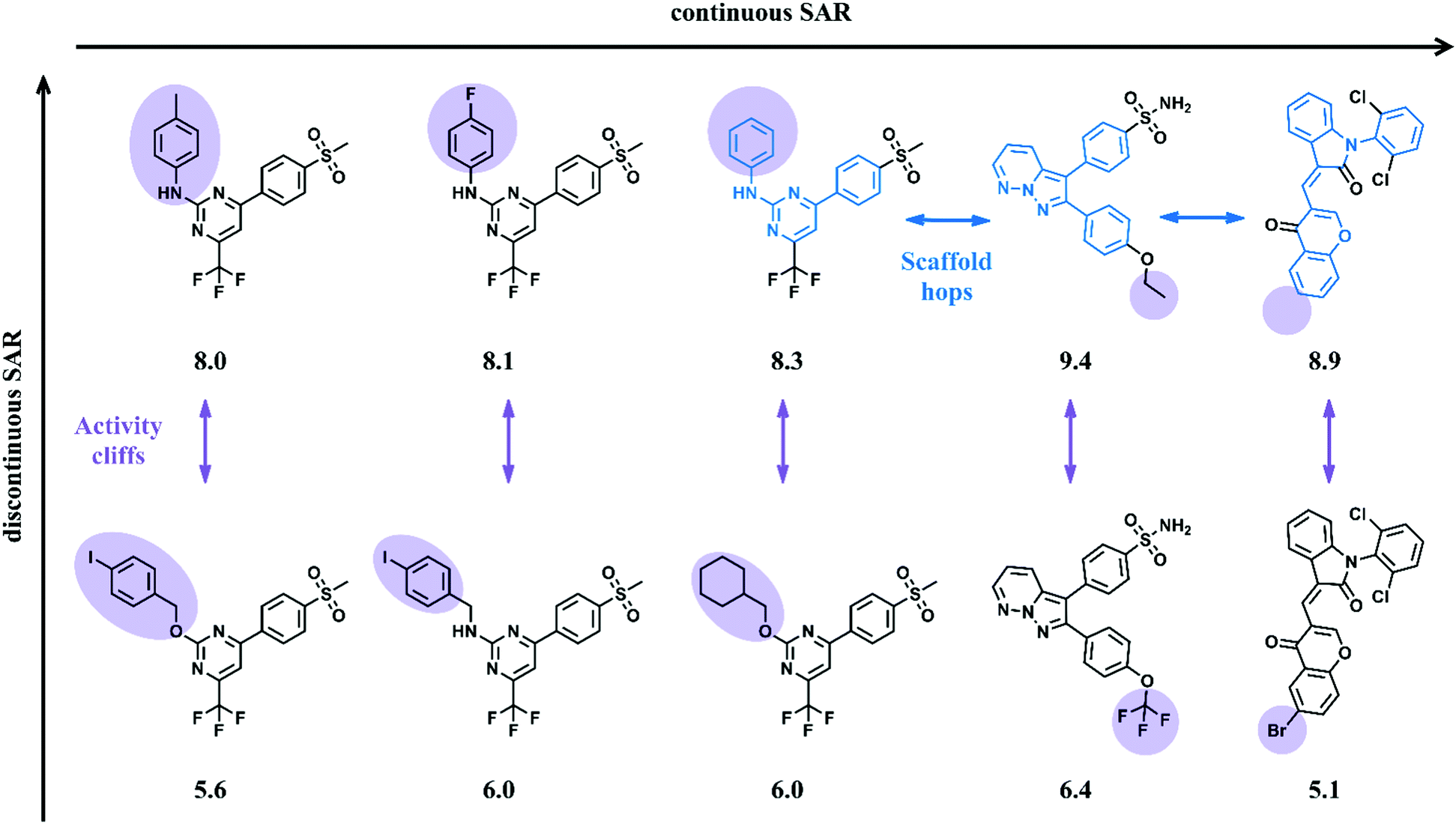
Chemical similarity is often evaluated in relation to bioactivity. Multi-dimensional structure–activity relationship (SAR) landscapes derived from models, describe similarity relationships between active molecules and their biological potency differences. These can be used to understand the effects of various structural features on biology, especially SAR continuities versus discontinuities in compound responses.22 SAR continuity is directly associated with the SPP, implicating a smooth continuous relationship between conservative structural modifications of active compounds and accompanying moderate potency alterations. In contrast, SAR discontinuities21 occur when small structural modifications lead to very large biological potency changes, not consistent with the SPP and falling outside the applicability domain of linear QSAR models. Fig. 3 shows small sets of active compounds that are characterized by SAR continuity and discontinuity, respectively. “Activity cliffs” are formed by analogs displaying the largest potency differences in a compound series for the smallest change in structure.23 The existence of activity cliffs in compound data sets is a major factor limiting QSAR predictions, often much greater than intrinsic limitations of modeling.23 Strikingly similar observations have also been made in bioinformatics where some pairs of proteins with high sequence similarity possess very different structures and functions.24 This analogy is one of many that methodologically bridge between QSAR and other fields that rely on data analytics. It should be noted that activity cliffs may be sensitive to both the choice of descriptors and the degree of the experimental variability. Importantly, SAR discontinuity limits QSAR modeling regardless of molecular representations and descriptors that are used when the corresponding compounds are close structural analogs. Activity landscapes of compound data sets might be “flattened” by using large numbers of features as molecular representations such that compounds become increasingly dissimilar (i.e., their distances in feature space increase). However, introducing artificial dissimilarity results in a loss of SAR information (and often leads to overfitting of regression models).
 | ||
| Fig. 3 Different SAR patterns. Shown are inhibitors of tyrosine kinase ABL forming different SARs. For each compound the logarithmic potency (pKi) value is reported. At the top, SAR continuity is observed where gradually changes in compound structure (traced by horizontal arrows) are accompanied by moderate potency alterations. By contrast, the inhibitors at the bottom display SAR discontinuity. Here, small structural modifications lead to large changes in potency. Vertical arrows indicate the formation of pairwise activity cliffs. | ||
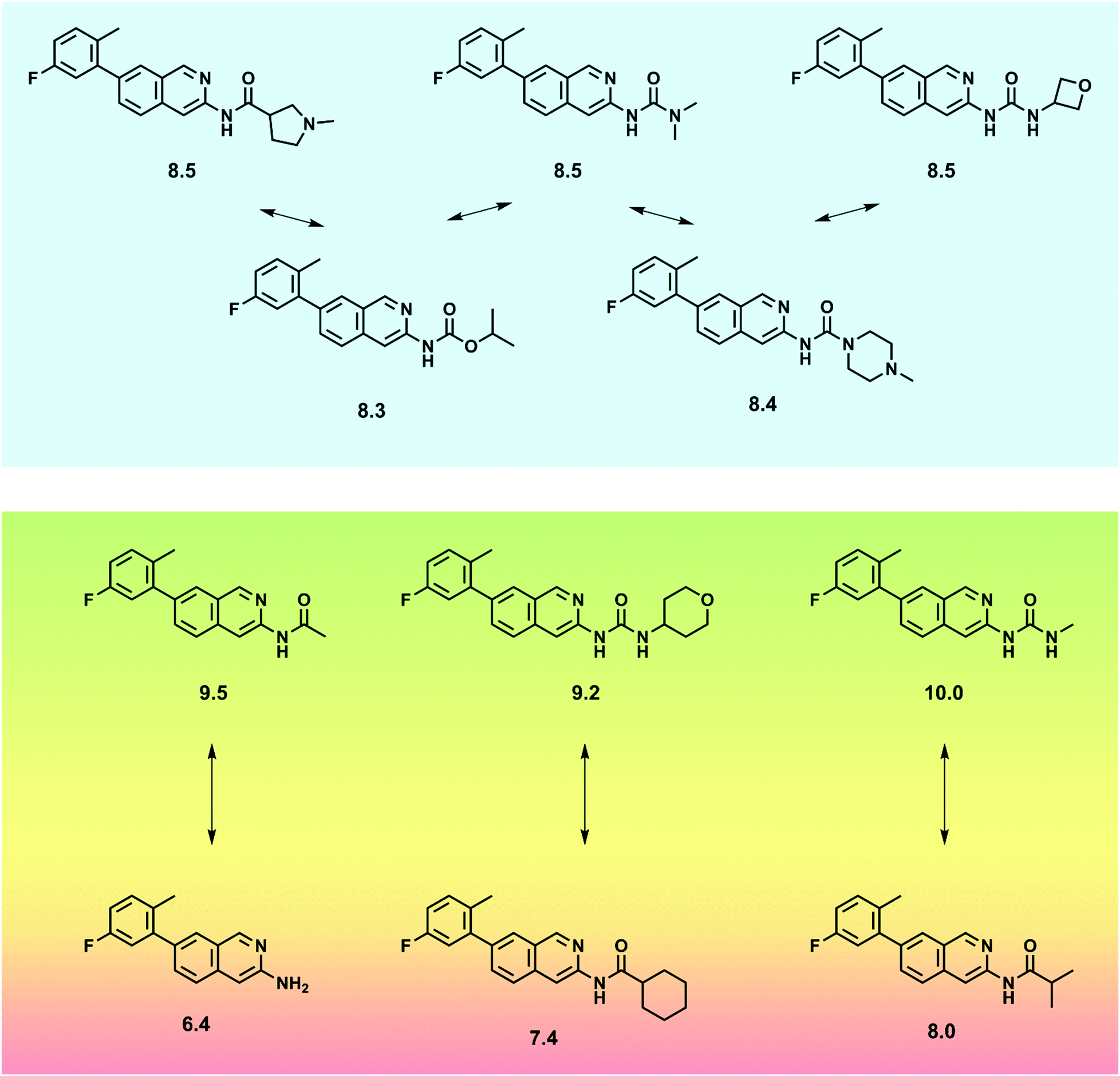
In QSAR modeling the presence of SAR continuities and discontinuities in sets of active compounds is not mutually exclusive. Rather, continuous and discontinuous SARs coexist in many data sets21 resulting in the presence of adjacent gently sloped and rugged regions in activity landscapes (Fig. 3). Focusing potency predictions around local regions of SAR continuity can often lead to QSAR models with high predictive power. To this end, numerical SAR analysis methods can be used to identify compound subsets having desirable SAR characteristics.25 Numerical similarity in QSAR is mostly quantified using the Tanimoto coefficient or related similarity metrics, which provide continuous similarity values, typically in the interval [0,1]. Numerical measures assess whole-molecule similarity on the basis of chosen descriptors. For larger compound sets, Tanimoto similarity calculations are often carried out using molecular fingerprints,26 especially in machine learning.27
Different from numerical similarity measures, substructure-based approaches yield a binary readout of similarity, i.e., either two compounds are classified as similar or not. A standard approach in substructure-based similarity assessment is clustering of compound data sets on the basis of hierarchical scaffolds extracted from them.28 Such scaffolds are conventionally used to represent core structures. In addition, substructure-based similarity can be assessed by calculating the maximum common substructure (MCS) of compounds, although MCS calculations are typically only meaningful for small compound sets.29 By contrast, similar to scaffold analysis, substructure-based similarity can be determined on large scale by applying the matched molecular pair (MMP) formalism. An MMP is defined as a pair of compounds that are only distinguished by a chemical modification at a single site.30 Accordingly, compounds forming an MMP contain a common core and the distinguishing chemical modification can be rationalized as the exchange of a pair of substructures, termed a chemical transformation. Algorithms for MMPs generation are highly efficient.31 By limiting the size of transformations, it is readily possible to restrict formation of MMPs to pairs of analogs.32 By combining MMP search with network analysis, analog series can be systematically extracted from large compound sets and subjected to SAR exploration and QSAR modeling.33
Going beyond the traditional QSAR paradigm means departing from the SPP. Modeling compounds with increasingly diverse structures with few or no common scaffolds means that structural differences between active compounds are not gradual, such as those that arise from “scaffold hopping”.34 This leads to structurally diverse active compounds that require non-linear approaches to modeling SARs satisfactorily, making bioactivity predictions more difficult. Non-linear SAR models require analysis of relationships between structure of both close and remote structural analogs and respective changes in their potency. This is beyond the capacity of classical linear regression QSAR methods and generally requires the use of machine learning (ML) as discussed in the next section.35
To summarize, the choice of molecular representations (descriptors) and assessment of molecular similarity play a critical role in QSAR.36 It should be emphasized that comparison of object representations, their similarity metrics and the interplay between object relationships and associated (latent) properties is of general relevance for data modeling irrespective of research areas. In fact, the similiar similibus curantur (“likes are cured by likes”) principle formulated by Paracelsus37 (the “father of toxicology”) could be seen as one of the most common ways of rational thinking (reflected in the SPP principle as applied in cheminformatics) and reasoning approaches in nearly any area of science. As highlighted throughout this contribution, this principle is one of key drivers of the general applicability of approaches and tools employed in cheminformatics.
Modern trends in QSAR modeling
Chemical similarity may help with qualitative assessment of compound bioactivity but its quantitative evaluation requires the use of statistical tools that can model the relationship between chemical structure and bioactivity.1 Currently, there is much talk about the use of artificial intelligence (AI) in chemistry. Here we distinguish between AI and machine learning in the following way. AI is the superset of tasks that demonstrate characteristics of human intelligence, while ML is a subset of AI which accesses data, analyses trends and generates intelligent, actionable insights. Many people use the term AI in the same context as ML in many data-rich disciplines, ranging from health care to astronomy. In this regard one can say that AI has been used in chemistry since the 1960's under the name QSAR. In general, ML represents a set of techniques for predicting a property Y based on known examples, where each example i has property Y(i) and a set of k features X(i,j), j = 1 to k. In this section we show how QSAR modeling can be applied much more broadly than has been the case previously. Theoretical organic chemistry, a highly specialized field, gave rise to the QSAR paradigm. The experience and trends in modern QSAR we summarize in this section is illustrative, and perhaps, instructional, for any data-rich area of research.Machine learning suffers from the same philosophical limitations that any type of inductive learning does: distinguishing correlation from causation and knowing when we have enough training examples to generate a model that makes accurate predictions for new cases, etc. In QSAR, the dependent variable Y is usually some biological or physical property, and the independent variable features X (called ‘descriptors’ in chemical applications) are derivable from chemical structures. In QSAR, historically the objects are drug-sized molecules, but that is not always the case. Objects can be atoms, protein sequences, pairs of proteins, etc., so long as relevant descriptors can be generated.
Chemical descriptors for drug-sized molecules fall into two main categories: substructures, which note the presence and/or frequency of certain groups, and computable properties that are representative of the entire molecule. In QSAR, the function that maps Y from X is called a model. Obviously, the same general construct is used in statistical modeling in any field, except the nature of descriptors depends on the type of the objects.
This section concentrates on trends in QSAR in the pharmaceutical industry because, arguably, that is where the opportunities and challenges for innovation and potential impact on society are greatest.38 Most pharmaceutical companies are likely to develop QSAR models for on-target (e.g., binding of ligands to targets) and off-target (secondary pharmacology) activities, as well as ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, which are discussed in the next section. Companies also develop their own best practices for building and using QSAR models. Models are used so that predictions can substitute for experiment under some circumstances. However, the current state of the art in QSAR modeling often precludes chemists from relying fully on individual quantitative predictions. Instead, the proper application of QSAR is the prediction of trends, which are accurate enough to prioritize sets of compounds for synthesis and experimental evaluation.
Researchers are always seeking ways to improve their science, and the field of QSAR is no exception. There are many recent trends but here we describe the most important ones that in our opinion, can be generalized to many other research fields:
1. Data. Data driven modeling methods are clearly highly dependent on data size, quality, and diversity.39 The size and diversity of datasets have dramatically increased in recent years due to technological advances in robotics and miniaturization (similar trends of course are observed in nearly any area of research and technology development). We can now generate very large volumes of data for a specific project, typically for 104–106 diverse molecules. Data generation is resource intensive, and data always contain experimental error. Outside of the pharmaceutical industry, the availability of large volumes of published, or otherwise public domain data in databases like ChEMBL,40 PubChem,41 or ZINC42 has transformed the field.
2. Validation methods. A common method of validating a QSAR model is by use of an external test set. Part of the data is held aside, and the remainder used to train the model. The model is used to predict the test set endpoints and a metric for the accuracy of prediction is then calculated. A better ways to simulate the natural evolution of a typical drug discovery project is to use a time-split test set,43i.e., assigning compounds tested in later phases of the project to the test set. It can be demonstrated that time-split gives a good estimate of the R2 for true prospective prediction relative to random test set selection (a standard method that can overestimate prediction accuracy) and leave-class-out validation (which is too pessimistic).43 Users of the ChEMBL database sometimes use the date of publication as a surrogate time-split threshold. Validation of QSAR models for properties of chemical mixtures is more complicated. In that regard, the points out44 approach is not different from traditional QSAR, but should be used only for predicting the same mixtures with new composition. The compounds out44 approach is suitable for predicting new mixtures of compounds from the modeling set; the mixtures out45 approach is for mixtures of one compound from the modeling set and one new compound; and the everything out46 approach (the most rigorous) is for mixtures of completely new compounds.
3. Multitask modeling. In classical QSAR only one predicted activity is modelled at a time. However, in drug development, multiple activities, both on- and off-target, are needed for prioritizing compounds. The set of techniques for prioritizing compounds based on more than one predicted activity simultaneously is called multi-parameter optimization,47 or multi-task modeling. In general, this objective can be achieved by an ensemble of single task models, or by a single model that can predict more than one activity simultaneously using either non-neural net or neural net-based techniques, including deep learning that has become popular in recent years. The multiple activities could involve related targets in one species, the same target in different species, the same target under different experimental conditions, or be completely unrelated. Multitask modeling is expected to be useful when data are sparse, i.e. not all molecules are tested on all targets, and the hope is that information will “leak” or “read across” different targets and reinforce structure–activity trends. Several methods have been proposed for multitask QSAR modeling including perturbation theory + machine learning (PTML),48 inductive learning and multi-objective optimization49 as applied in proteochemometrics modeling.50 The most common way of handling multitask modeling currently is with deep neural nets, especially convolutional neural nets. This will be discussed in more detail in the section on ML methods. Multi-task optimization represents an active area of development in QSAR modeling. However, it is still unclear whether these techniques provide a significant improvement in external predictive accuracy compared to an ensemble of single task models developed for the same end points. For example, an ensemble of individual models developed with XGBoost (gradient boosting decision trees) method exhibited the best performance in a recent 2019 IDG-DREAM Drug-Kinase Binding Prediction Challenge.51 As many compounds do have multiple biological activities, there is an obvious need to continue both methodological and application studies on multitask modeling in QSAR and other areas of statistical data analysis.
4. Applicability domain (AD). An applicability domain52 defines the space of molecular features on which the model has been trained and to which it should be applied; the AD provides a means for estimating the reliability of property predictions for new molecules from a QSAR model. It allows flagging of less reliable predictions and helps identify additional molecules that might be required to expand the model AD into more productive chemical spaces. Interestingly, AD is one area where QSAR is ahead of the general field of ML, although there is not yet a consensus on the best approach to this issue.52
5. Modelability. Whether a statistically significant model can be built from a given dataset depends on a number of issues.53,54 If the size of the experimental error in the measured dependent variable approaches the magnitude of the variation across multiple molecules in the dataset, it becomes increasingly hard to generate meaningful models. The signal to noise ratio in the data set is too low. Assuming this is not an issue, and considering activity and descriptors together, the relatively new concept of modelability55 proposes that predictivity of QSAR models is then limited by activity cliffs. As discussed above, activity cliffs exist when very similar compounds have very different activities, making the target property of compounds near the activity cliffs hard to predict.23 This difficulty is not easily overcome by changing either the QSAR method or the descriptors used. One exception is that using stereochemically-aware descriptors can reduce activity cliffs where different stereoisomers exhibit very different activities. Metrics that measure the prevalence of activity cliffs in a dataset are good predictors of the modelability of that dataset.55 Clearly, these metrics cannot distinguish activity cliffs that are intrinsic to the SAR response surface from those that are artifacts due to large experimental uncertainties in the measured activities.
6. Interpretability. Early classical QSAR methods were relatively simple and tended to deal with molecules that were close analogs. Comparative molecular field analysis (CoMFA)56 was extremely successful because of its visual appeal – it was clear where and how to modify a molecule to increase its activity. Later, projection of atom/fragment model contributions onto exemplar molecules has been suggested.57 However, as modeling methods have become more sophisticated, descriptors more arcane, and datasets more diverse, the accuracy and breadth of predictions have increased at the expense of interpretability (understanding the molecular basis for good or bad activity of molecules that guides design of improved examples). Methods that “see” into the black box of QSAR models independent of the descriptors and QSAR methods used are discussed in a recent review.58 An important process in QSAR modeling is selecting the most relevant subset of descriptors for a much larger pool in a context dependent way (sparse feature selection,59 which we also touch on in the section on biomaterials and regenerative medicine below). This improves the ability of models to generalize well and can make interpretation easier because fewer descriptors are used in the model. Subsequently, models are usually interpreted in two ways. The first is to determine which descriptors are the most important for driving improved properties of molecules. This is called “descriptor importance” for QSAR58 or “feature importance” for ML in general. The second, applicable to models trained on substructure-type descriptors, is to project the most important features from the model onto exemplar molecules to highlight structural features associated with more favorable activity.60 A molecule with atoms colored according to their contribution represents a molecular “heat map.” Another important, descriptor- and model-independent method for interpreting features is to apply small perturbations to the input descriptors one at a time, while holding the other constant, and observing the effect on the modeled property (sensitivity analysis, effectively generating partial derivatives of the response with respect to the descriptors).61 These approaches to interpretation have limitations as well.62 It is important to recall that no statistical method can distinguish correlation from causation, and interpretations cannot always be related to a mechanism. A practical approach towards mechanistic interpretability, lateral validation,63 is to observe trends across related phenomena: When the choice of variables, the sign and size of their coefficients are similar across multiple QSARs, this may help mechanistic understanding and perhaps causation.
7. ML methods. There are many standard methods of ML in QSAR.64 The current wave of enthusiasm is for deep neural nets (DNN) as the ML method. Because of their relative recency and popularity across many disciplines, comparison of DNN with other popular ML approaches is presented below.
DNN methods are attractingly widespread application across many disciplines.65 Single hidden layer neural nets were a popular ML method for developing QSAR models in the 1990's However, neural nets have undergone a renaissance in the past decade. Algorithmic improvements, advances in hardware, use of GPUs, etc., have made DNNs practical and computationally tractable. In AI applications, such as image classification or speech recognition, DNNs have been shown to be superior to any techniques that came before. DNNs began to be applied to QSAR66 after the Merck Molecular Activity Challenge in 2012.67 In less than a decade we have seen an enormous growth in publications using diverse DNN architectures for modelling chemically-related properties.
To put DNNs into context for QSAR, there are many other ML methods used in QSAR modeling including k-nearest neighbors (kNN),68 partial least squares (PLS),69 support vector machines (SVM),70 relevance vector machines, (RVM),71 random forest (RF),72 Gaussian processes (GP),73 and boosting.74 In the pharmaceutical industry (in fact, in any discipline), ML and DNN methods can be compared to older methods by the following:
1. Prediction accuracy
2. Number of sensitive and tunable hyper-parameters
3. Need for descriptor selection
4. Length of training time
5. Length of prediction time (including uploading the model into memory)
6. Domain of applicability (determined mainly by descriptors and training set characteristics)
7. Interpretability of models
RF has been a popular choice for QSAR modeling for many years as it can make very good predictions, has few adjustable parameters, and can be parallelized. Moreover, the degree of agreement of predictions of different agreement of RF trees75 can help define the AD. Boosting is also very useful because it is often one of the most accurate and fastest methods, especially with the latest implementation of extreme (XGBoost76) and light gradient boosting machine.77
The case for DNNs as a ML method would be made based on its superior predictivity. Comparison of DNNs to other ML methods like RF and XGBoost on standard industrial QSAR datasets shows a statistically significant improvement in prospective predictions as shown in studies conducted by some of the authors of this paper, and similar conclusions have been published elsewhere.78 However, in absolute terms, the improvement is less than notable. When trained on the same data sets and descriptors, DNN predictions are not different to those of other methods.79 Thus, the squared correlation coefficient (R2) of models generated with DNN was only 0.04 higher (on average) than those built with RF as shown in Fig. 4. This is consistent with the universal approximation theorem discussed below.
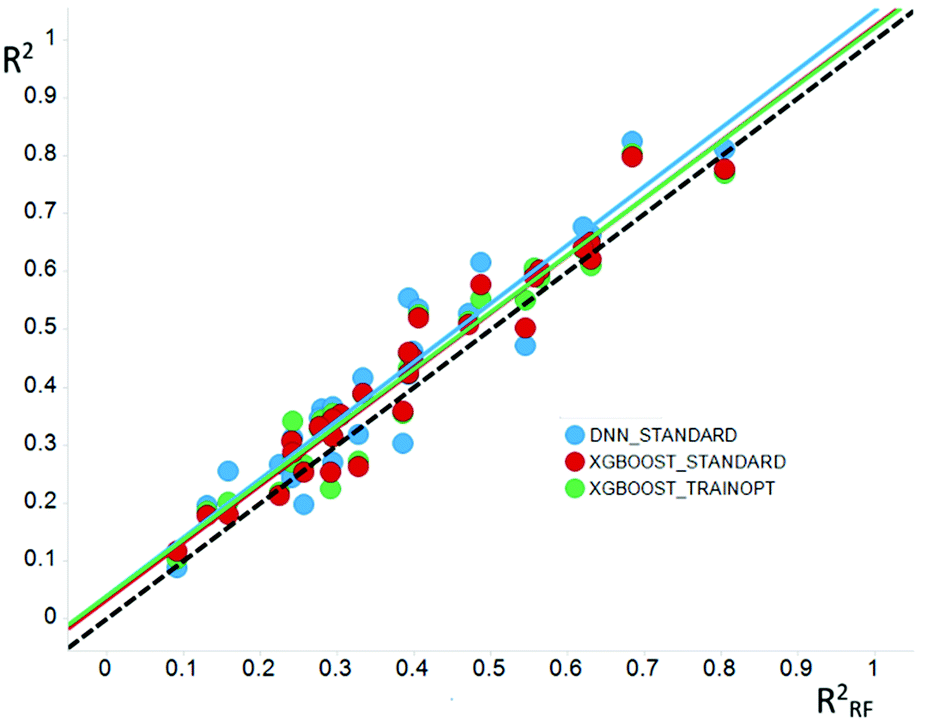 | ||
| Fig. 4 Comparison of the Pearson R2 values for models generated using DNN (blue) or XGBoost (red and green) and random forest methods. | ||
Deep neural nets also have undesirable characteristics such as requiring more tuning of training parameters for a given training set, being computationally more demanding, taking longer to predict, and being harder to interpret.
Why are DNN models not making substantially better predictions than the other ML methods? A fundamental reason is the universal approximation theorem that states that single layer neural networks (and ML methods mathematically similar) are sufficient to model any nonlinear function given sufficient data.80 Another reason may be that any pharmaceutical data set inevitably has experimental errors that will compromise very accurate model generation. Training and test sets are also not necessarily similar, and the new field of modelability suggests that all QSAR methods are limited by the presence and size of activity cliffs.81 For these reasons, more sophisticated and flexible methods will not necessary provide better predictions.
It is important to remember that in the pharmaceutical industry, unlike other areas where ML is applied, the data required to build models is limited, expensive, and resource-intensive.64 Getting marginally better predictions is not useful when the bottleneck is data paucity. However, DNNs methods do have very important advantages over most other ML methods:
1. They can straightforwardly model more than one activity at a time (multi-task models);82 the same is true for single layer NNs with multiple output nodes83 but not so for other ML methods. It has been claimed that on the average this produces better predictions than models of the individual activities. In practice, this effect can be quite modest, exhibiting both improvements and degradations in prediction for individual activities. It has been shown that improvement relies on the training set for the activities sharing similar compounds and features, and there being significant correlations between the activities.84
2. Their ability to automatically generate novel chemical features (using, e.g., graph convolutional neural networks, CNNs) is particularly important.85 This mimics how images are processed on the fly (with atoms replacing pixels), as opposed to the use of pre-generated chemical descriptors. The premise is that by generating richer molecular features, more predictive models will result. In some cases, CNN has provided more accurate predictions than descriptor based DNNs.85 For example, CNN is better at predicting quantum chemical energies.86
3. They provide the possibility of inverting the QSAR model (inverse QSAR), i.e. designing molecules directly from the model (so called generative models).87 This is in contrast to the current QSAR practice that only goes in the direction of property prediction from structures, not from properties to predicted structures. Candidate molecules must be generated by screening large virtual libraries or by assembling or swapping chemical fragments and predicting their properties by a QSAR model.
To summarize, it is still unclear from the ML literature whether DNNs are distinctly better at QSAR tasks than standard methods, because in most cases an exhaustive comparison has not been made. We would recommend that the method in question must always be compared to a good off-the-shelf ML method (such as RF or boosting) in the context of QSAR best practices.18 We would also recommend that a fairly large number of datasets (>10) should be examined in any given study. This removes the temptation to cherry-pick the results that make the method under study look better.
Another issue is the tests for DNN performance represent a low bar for success, meaning that predictivity appears better than it is in practice (an issue for the entire QSAR area). Random-split validation (which is still a literature standard) makes predictions that appear to be good because the test and training sets cover about the same chemical space, a difficult constraint as predictions outside of the model AD are likely to be poor. We recommend a time-split validation where possible, checking that the test set compounds are not too far from the model domain. Another practice in ML is to tune hyper-parameters using a validation set, where both the validation and test sets have been chosen from the same pool of compounds. In effect, this lets information about the test set to leak into the training set of the model, which makes predictions overly optimistic, and thus this practice should be avoided. The enthusiasm for DNN methods has sometimes encouraged bad practices, such as not comparing results to simpler methods (Occam's Razor) and publishing non-reproducible models, as has been reported in other areas of machine learning.88
In our opinion the current enthusiasm for DNNs in QSAR is not yet justified by its slightly increased predictive performance, given that the methods are compute-intensive and the models very hard to interpret. However, it should not be overlooked that their main advantage in in the generation of novel and useful features from relatively simple representations of molecules (or materials) and the potential for inverse QSAR. The development of new methods for DNN model interpretation such as layer-wise relevance propagation will also increase their advantage over traditional QSAR methods.89 Clearly, given how fast the field is developing, it is hard to know whether DNNs will overcome current disadvantages, although the inexorable increase in computational resources available will ease some of them. On the other hand, the enthusiasm for DL methods is driving a renaissance in the use of ML in chemistry,90 creating more opportunities.
As computational chemists, we should be actively researching other fields like data science and mathematics for advances in ML methodology. Historically, we have acquired new ML methods through serendipity, because we tend to read only the chemical literature. For example, the author of this section started applying RF to QSAR in 2003 because of a chance conversation with statisticians. We became aware of DNNs only after the Kaggle contest in 2012 and of XGBoost in 2016 because of a suggestion from a person in the IT department. However, the criteria we proposed for how DNN and ML methods should be compared, and concerns and suggestions on how best to generate dataset splits to enable robust assessment of model predictivity, have originated from our experience in QSAR modeling. These learnings will undoubtedly be valuable for other areas of statistical data modeling. The above examples suggest that exchange of best practices and methodologies between QSAR modeling and other fields will bring advances in both. Better definitions of important general concepts such as applicability domain or model interpretability are applicable to other diverse disciplines.
QSAR in chemical safety assessment
QSAR approaches have been used extensively to model important drug properties such as ADMET. Minimizing toxicity and optimizing pharmacokinetics is critical for designing new and safe medicines; incorrect estimation of these parameters can result in undesired side effects and affect in vivo efficacy, leading ultimately to a failure of a drug candidate. It should be noted that almost any chemical is toxic at a sufficiently high dose, so an important characteristic of any drug is its therapeutic index, the ratio of the effective dose causing the desired therapeutic effect in 50% of research subjects (ED50) to the drug dose causing adverse effect(s) in 50% of the subjects (TD50). Thus, it should not be surprising that even extremely toxic compounds such as snake venom toxins are useful, at proper concentrations, as diagnostic probes,91 drug leads, or even as therapeutic agents.92 Chemical toxicity is also very important for the assessment of the occupational health and environmental safety. Because toxicity is a complex multifactorial phenomenon caused by chemical effects on biological systems, it is important to understand underlying toxicity mechanisms to build mechanistically meaningful prediction models. There is a clear need to develop standardized protocols when conducting toxicity-related predictions, and the information needed for protocols to support in silico predictions for major toxicological endpoints of concern (e.g., carcinogenicity, acute, genetic, reproductive or developmental toxicity) across several industries and regulatory bodies has been discussed elsewhere.93 Below, we review several key concepts that relate to issues in chemical toxicity prediction.Adverse outcome pathways (AOP)
AOP is one of the key concepts of toxicity assessment. It assumes that toxicity is initiated by a molecular initiating event (MIE), which leads to an adverse outcome (AO).94 A single AOP describes a sequence of linked events starting from MIE, going through a cascade of linked key events (KEs), and ending at an adverse health or ecotoxicological effect. The adverse outcome pathway knowledge base is currently under active development for both health and eco-toxicology studies.95 With knowledge of AOPs, QSAR modeling can be used to identify the potential of chemical compounds to cause a MIE and/or to lead to an adverse outcome.Importantly, metabolites can also cause toxicity even when the precursor has low toxicity. Therefore, incorporation of information about metabolic activation can improve toxicity QSAR models.96 AOP facilitates mechanistic interpretation of models, provides a better understanding of toxicity, and allows the development of new in vitro tests.97 Currently, the development and validation of such tests is an emerging topic in predictive toxicology.
In vitro toxicity and Tox21
Tox2198 is a high-throughput toxicity evaluation initiative supported by several government agencies including US Environmental Protection Agency (EPA), National Institutes of Health (NIH), and Food and Drug Administration (FDA). Similar initiative exists in Europe under the REACH (Registration, Evaluation, Authorization, and Restriction of Chemicals) legislation. REACH encourages the use of so-called alternative approaches or surrogate end points to reduce animal testing. Naturally, QSAR modeling represents one of the best alternative approaches for risk assessment because it can be used both to predict in vitro activities of compounds and to combine these in vitro results with computed molecular descriptors to improve the accuracy of models in predicting in vivo effects. The requirements for using QSAR models for regulatory purposes have been reviewed elsewhere.99Tox21 data have been used actively by the cheminformatics community to test both the prediction accuracy of QSAR models and to understand current limitations of the field. The Tox21 data challenge aimed to assess the ability of QSAR models to predict important in vitro endpoints related to chemical toxicity.100 Participants predicted the outcomes of 12 cellular stress assays.100 The winning team (as determined by the AUC metric) used a DNN to build multi-task models for these outcomes.101 Model built with an associative neural network102 had similar prediction performance. The results of the Tox21 challenge indicated that recent progress in neural networks have accelerated development of robust and predictive QSAR models for in vitro toxicity. The development of new types of DNN90 has opened up new applications, allowing simpler molecular representations, such as SMILES strings or chemical graphs to be used to generate useful toxicity (and other property) models. However, these methods have generally lower prediction accuracy than ML approaches using traditional QSAR descriptors.103 DNN methods also require substantially larger datasets to fully capitalize on their advantages,90 a problem that is rapidly abating due to explosive growth in chemical data that is driven by automation.
Tox21 data also gave rise to a number of notable comprehensive studies, such as collaborative estrogen receptor (ER) activity prediction project (CERAPP)104 and collaborative modeling project for androgen receptor (AR) activity (CoMPARA), involving 17 and 25 international teams respectively. The resulting consensus QSAR models leveraged knowledge from the groups and were used to predict ER and AR potentials of 32464 new chemicals.
It should be emphasized that development of new experimental techniques such as deep-sequencing RNA-Seq,105 provides new types of data for in vitro assessment of toxicities that can also be used for QSAR modeling.106
In vivo toxicity
Given that adverse reactions could be caused by a multitude of factors, prediction of in vivo toxicity is arguably the most difficult task in QSAR modeling. The cost and ethical issues associated with direct in vivo toxicity assessment means that data to train models is scarce, so models are quite limited. This is clearly illustrated by the results of ToxCast lowest effect level prediction challenge.107 The highest prediction accuracy with the lowest RMSE of 1.08log units was achieved using a consensus prediction of associative neural network102 models developed with several sets of descriptors.107 Although the organizers of the challenge have offered a set of in vitro measurements performed within the ToxCast project, the top-ranked model was exclusively based on the calculated descriptors and was not improved by adding in vitro data as descriptors.107 The failure of this107 and QSARWorld bioavailability challenge indicates critical importance of data curation.16 Availability of more in vivo data, application of more complex methods such as those based on physiologically-based pharmacokinetic (PBPK) models,108 better data curation16 as well as new descriptors, which account for pharmacokinetics, should improve the model accuracy. Since in vitro assays in ToxCast were not predictive of such complex endpoint,109 other methods, such as those based on systems chemical biology,110 or more complex assays such as RNA-Seq used in combination with gene interaction networks, may be more successful.111 Indeed, it was reported that combination of in vitro and in silico predictions contributed better models for a number of in vivo endpoints.112
Multitask modeling: an approach that should not be overlooked
Multitask modeling leverages information from multiple correlated properties and may provide models with higher predictive power than individual QSAR endpoint models. This is attributed to read-across and the existence of mutual information in the more complex multiple end point data sets. A recent study showed that multi-task modeling consistently improved the accuracy of models for prediction of 29 in vivo endpoints using 87K chemical structures collected from the registry of toxic effects of chemical substances (RTECS) database.113 Importantly, authors suggested that the significantly improved toxicity predictions of multitask models should reduce the need for animal testing, prompting revisions to the current regulatory guidelines.Structural alerts and QSAR
Identification of molecular features associated with toxicity (structural alerts) represents a tool because it can help reduce unwanted side-effects of compounds by removal of offending moieties. However, toxicity alerts generally have lower prediction accuracies compared to QSAR models.114 It has also been suggested that a combination of alerts or any other structural rules115 and QSAR models may provide improved guidance for rationally designing new compounds with reduced toxicity.114 These combined approaches were further developed by the chemistry-wide association study (CWAS) that predicted Ames mutagenicity and an adverse drug reaction known as Stevens–Johnson syndrome.116 The identification of important chemical fragments and analysis of their co-occurrences also allows mechanistic interpretations of QSAR models without compromising their accuracy.In summary, this section provides a brief review of a special area of QSAR modeling that deals with chemical safety. However, even in this highly specialized application there are components that can be generalized to other applications. Multi-objective modelling and optimization is one such approach that will be increasingly used in other disciplines. The ability to interpret complex statistical models for any target effect is important in many fields, especially when building models of large data sets using deep neural networks.117 These examples reiterate the conceptual overlap between many elements of QSAR modeling and challenges faced by other disciplines.
Multi-target profiling and polypharmacology
Since the beginning of the 20-th century, the concept of “a magic bullet” has served as the basis for drug discovery and development.118 According to this concept, a drug should be developed with the highest selectivity toward the intended target for a particular disease. Thus, classical QSAR/QSPR studies have been performed with training sets of compounds active in a single biological assay; frequently, all compounds also belong to the same chemical series.1The advent of high-throughput screening technologies and proliferation of diverse assays have enabled screening of a larger number of molecules in more diverse assays. Consequently, it is now generally accepted that the majority of pharmaceutical agents interact with several, sometimes many, biological targets. This often generates beneficial therapeutic activities,118 due to additive or synergistic pharmacological effects.119 On the negative side, drugs can also interact with undesired molecular targets to causing adverse or toxic effects that often block further development. Clearly, there is a strong need to understand both the beneficial and adverse polypharmacology of ligands.120
Discovery of molecules with beneficial polypharmacology could be achieved by the experimental evaluation of millions of drug-like compounds against thousands of targets.121 Currently, this is an unrealistic task, particularly taking into account the variability of results obtained for the same ligand–target interaction in different assays, and relatively low hit rates of experimental screens.122 Thus, in silico prediction of biological activity profiles by (Q)SAR models is a viable alternative to these intractable experimental screens. Importantly, virtual screening approaches may be applied to millions of virtual molecules designed in silico.123 Such virtual screening greatly reduces both the number of molecules needed to be synthesized and tested, allowing pre-selection of likely hits and reduced time and cost in synthetic chemistry programs.122
Multi-target profiling of compounds has led to the concept of the biological activity spectrum,124 defined as the set of different biological activities resulting from the compound interaction with different biological systems. It therefore represents an “intrinsic” property of the compound that depends only on its chemical structure.
Several approaches for multi-target modeling have been proposed. One of the earliest developments in this area was the computer program PASS (prediction of activity spectra for substances) reported by Filimonov et al. almost 30 years ago.125 PASS employs a uniform set of multilevel neighborhoods of atoms (MNA) molecular descriptors and a Naïve Bayes classifier to model structure–activity relationships across a wide variety of biological assays. This approach allows the prediction of a wide range of biological activities at molecular, cellular, organ/tissue and organism levels. It can predict pharmacotherapeutic effects, mechanisms of action, specific toxicities, terms related to drug metabolism, gene expression, etc. The current version of PASS predicts several thousand biological activities based on the analysis of structure–activity relationships in the training set of over one million biologically active compounds.126 More recently, Gonzalez-Diaz et al.127 developed the perturbation theory machine learning (PTML) methods that search for QSAR models capable of simultaneous prediction of many target properties under several experimental conditions.
Substantial amounts of relevant chemogenomics data have recently became available from PubChem, ChEMBL, and other public sources. This has catalyzed a resurgence of freely available Web-accessible tools for bioactivity predictions and continuing development of new QSAR tools and methods.
In contrast to PASS online,124 which is an open access Web-service for predicting biological activity spectra, most other tools focus on predicting putative molecular targets for compounds of interest. They use training sets extracted from publicly available data sources, different types of chemical descriptors,128 and prediction methods based on implementations of different chemical similarity searches.129 Despite some disadvantages,130 such approaches remain an accessible way of predicting compound activity against novel pharmacological targets lacking sufficient training data for building accurate QSAR models.131 If the number of known ligands is sufficient for model building, some web portals provide an option to predict compound activities using conventional QSAR.
It is challenging to compare the performance of multi-target profiling tools. In contrast to single target models, there is a paucity of evaluation sets of compounds reproducibly tested for several types of biological activity. Thus, only a few comparative studies have been reported to date. For example, using data on affinity of drug-like compounds against several GPCRs, the performance of a collection of multiple target-specific k-nearest neighbors (kNN) QSAR models, PASS124 and similarity ensemble approach (SEA)132 was compared.133 The best results were obtained with the kNN method, while PASS demonstrated a moderate predictive accuracy and SEA shown the lowest prediction power across multiple targets.
Recently, a large evaluation set including half a million compounds tested across more than 1000 assays was constructed from ChEMBL data.134 The performance of several ML methods was evaluated, and deep feed-forward neural networks (FNN) generated the best results, while SEA showed the lowest predictivity. It is noteworthy that all ML methods showed relatively small differences in predictive accuracy and the advantage of the DNN was not readily apparent. This conclusion appears reasonable given that the principal purpose of DNN development was image feature recognition, i.e., similarity assessment but not prediction. Similar observations of the lack of advantage offered by DNN in cheminformatics compared to conventional ML was also made in the preceding section on modeling chemical toxicity.
As also noted in the preceding section of this paper, multi-task learning represents one of the major directions of QSAR development. A natural extension of multitarget QSAR is the analysis of ligand–target interactions in combined chemical–biological space, so called chemogenomics.135 Several hundred papers have been published on new methods and applications for chemogenomics (some discussed in greater detail in the following sections). For example, Gupta-Ostermann and Bajorath reported the structure–activity relationship (SAR) matrix method, which predicts activities and allows navigation in multi-target activity spaces.136 March-Vila and co-workers have summarized the promise of chemogenomics applications for drug repurposing.137
A recently proposed proteochemometrics (PCM) approach employs relevant information from target sequences and combines it with ligand descriptors to develop models predicting ligand–receptor (class of) binding affinity. This approach is more useful than ligand-based modeling in cases when the same ligands show differential binding affinity to diverse targets. Several interesting applications of the PCM approach have been reported. For instance, this approach was used to predict ligand interactions with wild-type and mutated α-adrenoceptors where it has demonstrated superior predictivity in comparison with conventional QSAR methods.138 In other study, Lapins et al.139 applied PCM method to predict inhibition of five major drug metabolizing isoforms of cytochrome P450 (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) by drug-like compounds. A recent study, has also demonstrated significant advantages of PCM approach and inductive transfer of knowledge between the targets over traditional methods.140
Careful review of the published results of PCM modeling leads to the conclusion that it may provide good estimates of ligand–target affinity in a single model by combining data from multiple assays (Fig. 5). However, to achieve this goal, substantial efforts must be applied to standardization141 and curation16 of such data.
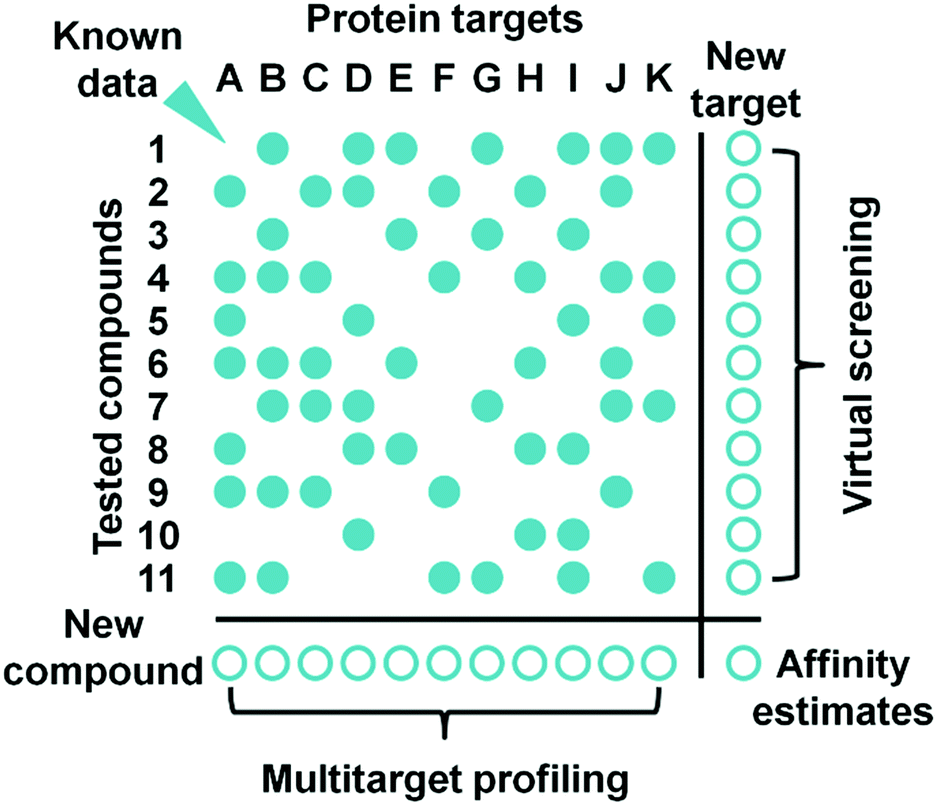 | ||
| Fig. 5 Proteochemometrics approach enables accurate affinity estimates for novel ligand–target pairs. | ||
To conclude this section, we note that training sets used to develop conventional QSAR models do not exceed millions of entries, while the estimated size of drug-like chemical space is up to 1060 molecules.142 We expect that with the growth of chemogenomic data and expansion of the studied chemical space, the multi-target QSAR modeling will become more common than single-target QSAR studies and that multi-target QSAR will lead to the discovery of novel medicines with much improved safety and potency profiles. Another important projection is that further development of multi-objective optimization methods will not only expand the field of polypharmacological QSAR but will also find use in many other predictive disciplines where multiple objectives need to be optimized.
QSAR-like approaches in genomics
Genomic and HTS (high throughput screening) data have rarely been subjected to QSAR analyses. Indeed, typical workflows require hit confirmation and validation prior to (Q)SAR modeling, and cheminformatics-based prioritization schemes based on individual compounds as well as scaffolds have been proposed.143 One of the major obstacles to date remains the absence of the gene-based descriptors suitable for ML. However, high throughput driven biomedical knowledge accumulation has created an urgent need for Big Data analytics in genomics and HTS to help with the evaluation, interpretation, and integration of data, and with development of respective models.From a life sciences perspective, the use of DNN can generate novel applications and even entirely new meaning to the field of chemical genomics by directly linking the structure of the molecule to its effect on genes, and by embedding these linkages in models that predict gene-mediated effects of chemicals in vivo. Such models require the combination of input features that characterize both small molecules (i.e., chemical descriptors) and genes (e.g., gene expression profiles) or HTS results for training. Only a few studies have been published in this area so far. For instance, it was demonstrated that gene ontology (GO) terms144 and HTS results can be translated into input features for cheminformatics models.145 In another such study, Sedykh et al.146 described and implemented a workflow for using HTS data in combination with molecular descriptors to predict in vivo toxicity. In a related work,147in vivo rat oral toxicity was predicted by combining endpoints of 499 HTS assays (biological variables) with 548 circular Morgan descriptors (chemical variables). Notably, when used separately, biological descriptors resulted in a model with lower statistical significance than the model based on chemical descriptors.
Another example of ‘hybrid’ QSAR modelling shows how QSAR descriptors and GO terms can be combined within a unified QSAR model capable of predicting the effect of a given molecule on a particular gene.148 Specifically, levels of expression of 1000 ‘hallmark genes’ in six cell lines were predicted by DNN-classifiers, where for every molecule–gene pair in the training set, circular Morgan fingerprint values (molecular descriptors) were combined with GO terms used as gene descriptors. The resulting DNN models built with back-propagated feed-forward fully connected multi-layer perceptron (MLP) with four layers yielded good prediction accuracies (cross-validated area under the curve (AUC) values were in the 0.80–0.83 range). These results suggested that ‘hybrid’ DNN models can rather accurately associate genes and small molecules to up- or down-regulation.
Seventeen different protein- and gene-centric data sources totaling over 262.3 million data points were integrated into knowledge graph representation with typed nodes and edges, which enable the conversion of the gene-based information into descriptors suitable for ML via network-based analytical algorithms.149 Specifically, a set of 103 genes having autophagy (ATG) associated annotations from GO terms, UniProt150 and KEGG,151 were used to derive ML models using the metapath approach combined with the XGBoost algorithm.152 These binary ML models were trained to distinguish ATG genes from non-autophagy genes (cross-validated AUC values were in the 0.95–0.99 range). Of the top 251 predicted novel genes, 23% were associated with ATG based on literature queries, whereas 193 were not.
These case studies offer an important example of QSAR modeling evolving towards the use of more complex datasets. Synergistic use of features representing both chemical and biological properties, including gene expression profiles, GO terms and KEGG pathway associations combined with ML methods, are generating promising results. This increase in complexity is typical for many areas of research where DNN and gradient boosting methods are finding growing applicability. The improvements in model accuracy achieved by ML approaches may have been modest so far, but the prediction power of these models may increase in near future due to cross-fertilization of ideas on using ML for data modeling both in chemical datasets as well as in many other areas of science and technology. It is tempting to speculate that DNN technology can directly screen virtual chemical libraries for compounds with bespoke, useful modulation of target genes and gene networks.
As the sources of data and sizes of datasets describing the biological properties of small molecules grow, there is also a concomitant demand for knowledge management (KM) systems, that integrate heterogeneous data into unified, predictive models and translate data into information.153 For example this might allow merging of experimental bioactivity data for small numbers of molecules, 3D information from experimentally resolved structures of protein targets for these molecules, statistics of respective drug adverse event reports, and high-volume (often lower quality) data such as genome-wide association studies (GWAS) or HTS. Such large scale datasets are already assembled into knowledge graph systems, for example Pharos,154 which supports in-depth exploration of the druggable genome.155 Modelling such data via ML, sparse feature selection, and other advanced algorithmic approaches may lead to a better understanding of the associations between chemical structures and proteins and genes in an unbiased, objective manner. They could further help identify novel gene–phenotype associations, either for diseases or for physiological phenomena such as autophagy.
QSAR in synthetic organic chemistry
The application of QSAR modeling to challenges faced by synthetic organic chemists is a recent and exciting development in predictive computational chemistry.156 Rapid growth in robotic platforms for drug and materials design has stimulated the development of reliable cheminformatics tools to assist with efficient synthesis of target molecules. These tools estimate synthetic accessibility of a target molecule and suggest feasible synthetic routes (Fig. 6). Two of the most widely used synthesis planning strategies are forward synthesis (starting from specified building blocks) and retrosynthesis (starting from a specified target molecule). Synthetic routes usually contain multiple reaction steps for which major products and, ideally, kinetic parameters must be predicted by models. Once a given elementary reaction is selected, reaction conditions (solvent, catalyst, temperature, etc.) leading to a reasonable yield should be suggested by the algorithm. The above considerations can be met by a wide range of cheminformatics tools, some of which are currently used in a computer-aided synthesis design.157 In this section we briefly describe reaction data availability, visualization, and analysis, and summarize recent studies focused on different parts of the modeling workflow described in Fig. 6.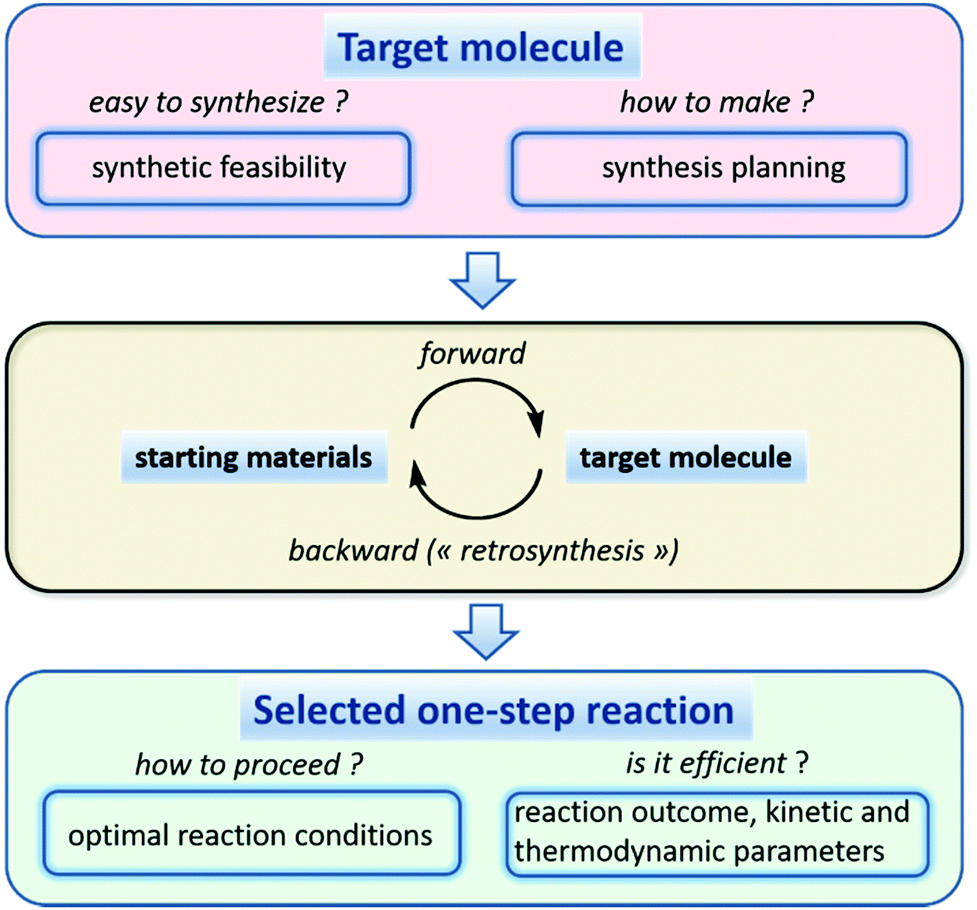 | ||
| Fig. 6 Main tasks of computer-aided synthesis design. As soon as a synthesis planning for a target molecule is established, efficiency of each one-step reaction and related optimal reaction conditions could be assessed. | ||
Reaction data availability
New modeling tools need access to large volumes of experimental reaction data stored in public and proprietary databases. In most of the recent studies, the Reaxys database (>40 M reactions including 12.5 M one-step reactions),158 the USP database extracted from US patents (>1.2 M reactions),159 and the QSRR database (∼10000 reactions) have been employed. Generally, reaction data from public databases is of mixed quality. Many of the reactions are stoichiometrically unbalanced, some important data on reaction conditions are missing, and different names are used for the same catalysts or solvents.160 However, no standards for reaction data curation have been reported so far. Ignoring the data curation step of the modeling workflow will significantly affect the quality of the training data and models derived from them.16
Reaction encoding
Chemical reactions constitute a very complex modeling problem in cheminformatics. A reaction equation involves several different types of molecular graphs (for reactants and products) and its yield depends on numerous experimental conditions. Depending on ML method used, chemical structures can be encoded by SMILES (e.g., in sequence-to-sequence models161) or by descriptor vectors, or a combined fingerprint (resulting from concatenation of descriptors of reactants and products162), or subtraction of descriptors of reactants from descriptors of products.163 The latter may require balanced reaction equations that, in turn, need a specific data curation step.163 Alternatively, a chemical reaction (balanced or unbalanced) can be encoded by the condensed graph of reaction (CGR). This merges reactant and product structures into a single molecular graph employing both conventional chemical (single, double, etc.) and “dynamic” bonds characterizing observed transformations (e.g., single and double bond breaks, single-to-double bond conversion, etc.).164 CGR can be consider a pseudomolecule to which any cheminformatics approaches can be applied. In particularly, fragment descriptors or fingerprints can easily be generated for CGR.165 Solvent can be encoded by a set of physico-chemical parameters which can be concatenated with the structural descriptors.Visualization and analysis of reaction space
Both graph-based and vector-based approaches have been used to visualize the chemical space of reactions. In graph-based approaches, chemical reactions and individual molecules (reactants and products) are represented as nodes of a large bipartite graph166 used to optimize synthetic pathways. In the vector-based case, a chemical reaction is defined as a vector in multidimensional space defined by descriptors. Dimensionality reduction is required to generate a two-dimensional map describing the data distribution. This approach was pioneered by Gasteiger et al.167,168 who generated self-organized maps (SOM) that clustered different classes of reactions effectively. Generative topographic mapping (GTM) approaches have recently been used to visualize large sets of SN2, cycloaddition, and tautomerization reactions. Unlike SOM and many other dimensionality reduction methods, GTM can be used to predict properties of new reactions projected on the map. As a predictive tool, GTM performs similarly to conventional ML methods like SVM.Planning organic synthesis using prediction of reaction products and retrosynthetic analysis
The general aim of synthesis planning is to identify a series of feasible reaction steps leading to a target compound from available starting materials. Retrosynthetic methodology, invented by Corey,169 is a real challenge because the search for precursors of a product generates a combinatorial explosion of possible reaction routes. Cheminformatics tools can help select the most feasible series of single-step reactions. The current trend in this field is to train DL models on large sets of reactions to predict probabilities of different retrosynthetic transformations. It was shown that using Monte Carlo tree searches and symbolic AI methods, it is possible to identify feasible reaction pathways.170Prediction of reaction outcomes allows one to prioritize retrosynthetic suggestions. A cheminformatics tool should predict the products of a given set of reactants under given conditions. Consideration of multistage chemical transformations and competitive reactions will significantly complicate this problem. Current trends in the modeling of reaction outcomes focus on processing large reaction databases with DL models to predict the probabilities of competitive chemical processes.171 The latter can be used directly for reaction outcome predictions. The ReactionPredictor tool172 is of particular interest because it forecasts the output of complex chemical reaction by combining mechanistic considerations with ML. This approach enumerates possible interactions and then ranks them using a pseudomolecular orbital approach.
Two orthogonal methodologies, template-based and template-free, can be applied to retrosynthesis and outcome prediction. Template-based methods rely on user-established sets of transformation rules, either suggested by expert-chemists or extracted automatically from reaction databases, the feasibility of which is assessed by the model. This concept is employed in most retrosynthetic tools, including the popular CHEMATICA program,173 which integrates more than 10000 empirical transformation rules.
Alternatively, in template-free approaches transformations between the reactants and the products of chemical reactions are deduced directly from their structures. This allows one to automatically enlarge the list of transformation rules as soon as new data are available. This methodology has become more popular in recent years. For instance, Coley et al.174 suggested using a graph-convolutional neural network and a global attention mechanism, followed by the application of rules to reaction product predictions and retrosynthetic analysis. Another template-free approach employs natural language processing methods, namely ‘sequence-to-sequence’ models. These use recurrent neural networks (RNN), commonly applied to translation of texts between languages. When applied to chemical reactions, SMILES strings of reactants and products constitute the language. This methodology was applied to model reaction products and for retrosynthetic reaction route prediction, which provided similar performance (ca. 37% for top-1) to rule-based systems (35%).161 A use of an advanced transformer architecture, which was initially used for English-to-German translation, boosted the accuracy of predictions to about 43%.175 This result indicates that retrosynthesis predictions can be significantly improved by algorithms originally developed for very different purposes.
Forward synthesis planning
One of the most impressive approaches to forward synthesis planning has been implemented in the DOGS program.176 This algorithm applies 58 well-established chemical transformation rules to a set of 25144 readily available synthetic blocks from the Sigma-Aldrich catalog. New molecules are grown in a stepwise procedure, each step consisting of complete enumeration of all possible solutions followed by selection of top scoring intermediate products to subsequent growing steps. The quality of designed products is assessed using pairwise similarity to a target molecule. Thus, DOGS can usefully suggest a synthetic plan not only for the target molecule but also for its close analogs.
Assessment of synthetic accessibility
Synthetic accessibility (or the opposite, synthetic complexity) is a scoring metric used to prioritize virtual compounds for synthesis. It is often used as an important filter for screening virtual libraries and in de novo design studies. Among scores developed so far54 the most popular is SA score.177 It is calculated using contributions from fragment occurrences in PubChem compounds and a complexity penalty based on the number of chiral centers, rings, macrocyclic fragments, and the total number of atoms. Recently, Coley et al.178 suggested the synthetic complexity score (SCS) which relies on a neural network trained on 22 million reaction pairs from the Reaxys database.Prediction of kinetic and thermodynamic characteristics
The logarithm of the reaction rate constant (logk) is a common endpoint in QSAR modeling, first used more than 70 years ago.179 Currently, quantitative structure–reactivity relationship (QSRR) modeling is performed on large and diverse datasets that account for solvent effects and temperature for many types of chemical reactions using NN approaches.180 In these models, descriptors computed for the reactants are concatenated with solvent and temperature descriptors. This technology must know the order of reactants in the reaction equation, making the development of an automatized QSRR workflow problematic. This problem can be solved using condensed graphs of reaction (CGRs) that combine the reactant and product information. Fragment descriptors generated for CGRs were concatenated with solvent and temperature descriptors and used to train logk models for bimolecular nucleophilic substitution,181 bimolecular elimination, and different types of cycloaddition.182 Similar approaches were used to develop predictive models for the equilibrium constants of tautomerization reactions.183
Prediction of optimal reaction conditions
Since the reactivity of chemicals is largely determined by the reaction conditions, their theoretical assessment is of particular importance (especially for automated robotic synthesis). Several approaches to reaction conditions modeling have been reported. For example, Marcou et al.184 used CGR-derived fragment descriptors to train SVM, RF, and Naive Bayes classification models to predict optimal solvents and catalysts for the Michael reaction. Gao et al.185 reported NN-based models trained on ∼10 million reactions from Reaxys that identify appropriate catalysts, solvents, reagents, and temperatures for a specified reactions. A 70% match with experimental conditions was found within the top-10 predictions. Lin et al.160 used the heuristic that similar reactions proceed under similar conditions to predict optimal reaction conditions. They used a simple similarity search of reaction databases with recorded conditions,160 especially effective with the CGR technology.186 The value of this approach has been demonstrated by protective group deprotection reactions. Models trained on 142111 catalytic hydrogenation reactions demonstrated high accuracy (ca. 90%) for predicting optimal experimental conditions.
In summary, the CGR technology can efficiently model optimal reaction conditions. One employs similarity searching of reaction databases to construct QSRR models, with reaction conditions as endpoints. Studies summarized in this section provide compelling examples of the impact of QSAR modeling on one of the historically most empirical areas of natural science, synthetic organic chemistry. The development of both retrosynthetic and forward synthesis prediction models, based on the analysis of an immense amount of accumulated data, represents one of the most important frontiers in modern science. It is essential for chemists to understand and begin applying these emerging approaches. When coupled with robotic synthesis methods, these synthesis prediction models are poised to transform organic chemistry as we know it and open the door to autonomous chemical synthesis systems in the future.
Closed-loop discovery and automation
Traditional serial molecular and materials discovery processes in laboratory have arguably reached a plateau. The costs of discovering materials and drug candidates remain high and the discovery and translation time is still long. Three decades ago, combinatorial chemistry (also known as high-throughput experimentation, HTE) promised to reinvigorate the discovery pipeline by carrying out synthesis and experimentation rapidly, in parallel using automation.187 HTE led to important discoveries (such as novel polymers) and, indeed, has accelerated the discovery pipeline. However, the avalanche of new drug leads that was anticipated did not occur. More recently, DNA-econded chemical libraries have made possible synthesis and testing of millions of compounds188 and many big pharma companies have embraced this approach.Furthermore, there is a growing realization that experimentation can be analyzed in terms of information theory. Questions like what is the amount of information that an experiment contains? What is the next best experiment to carry out? can be answered by modern Bayesian methods. This thinking has led to the revival of methods for developing closed-loop or autonomous approaches. By closed-loop we mean that the experimental system is designed using an information-theoretical approach, and the experimentation and assays are carried out in an automated way. By using AI or evolutionary algorithms to make decisions on what compounds to synthesize in the next cycle, in principle, an autonomous system can be developed. The term “self-driving laboratory” has been also coined to describe this type of experimental setting.189 Clearly, a self-driving closed loop laboratory is fundamentally different from existing HTE. The closed-loop approach, designed to provide rapid iterations using autonomous decision making, seeks to minimize the number of experiments required to reach a specified goal (e.g., target molecule(s)). It does not need to create large libraries, but rather employs agile experimental infrastructure, and statistics and ML to build QSAR-like models to predict the target properties for every element of the self-driving laboratory.190
Bayesian methods show promise for making closed-loop decisions. Based on prior assumptions about the nature of the experimental observations, they can propose the optimal next experiment to conduct. PHOENICS,191 for example, employs Bayesian neural networks and a kernel density estimate approximation to balance exploration vs. exploitation. Human interpretability is also an important factor in these systems. The algorithm chooses a set of experimental conditions to be generated by robot synthesizers. It is not sufficient to understand what the system generates; we must also know why certain recommendations are made. Interpretability is clearly very important for modern ML research. To aid interpretability, researchers have used hierarchical optimization approaches that operate on one or more variables. In multifactorial systems it is often necessary to understand the pareto-optimal regions of the problem space. A mathematical function called CHIMERA was recently introduced to address these problems;192 it can be used with any optimizer, such as PHOENICS.
Such systems require an operating system that is open-source and capable of controlling experimental equipment, storing data in databases, coupling with optimization approaches, and interacting with researchers. A “Cortana-” or “Alexa-like” digital assistant for scientists that is connected to the closed-loop system could accelerate adoption and innovation. Efforts such as ChemOS can help rally developers to achieve this vision.193
One of the promising applications of closed-loop discovery is in the materials space. A recent review summarized the state-of-the-art and challenges in this field.194 Examples of the application of AI to materials discovery are described in this review, as well as in following sections of this paper. One such example is the design of blue emitters for organic light-emitting diode devices accomplished by virtual screening of half a million molecules.195 This approach led the successful discovery of three lead candidate compounds with state-of-the-art performance,195 exemplifying the promise of closed loop discovery. The three good candidates required the synthesis of only ∼40 materials. In autonomous systems, experimentation becomes the bottle neck in the accelerated discovery process. This can be overcome by technological developments – creation of self-driving, closed loop robotic laboratories controlled by AI, as discussed in a recent perspective.196
Evolutionary algorithms can also be used to generate closed loop, autonomous molecule and materials discovery system. Their application to drug discovery and optimization, and materials discovery have been reviewed recently.197 ML-based QSAR can be used to model the fitness landscape of materials experiments, which can substitute for downstream experiments, improving efficiency and speed.
In summary, AI methods and models that optimally instruct every step of robotic synthesis (including the choice of both reagents and reaction conditions) represents a landmark in the extension of QSAR methods toward dramatically more efficient chemical synthesis.
Machine learning approaches in quantum chemistry
Computational chemists, physicists, and biologists commonly employ molecular potentials to evaluate energies and forces. These are used to search for novel drug compounds and materials. Hence, a faster but still accurate computational method for evaluating molecular potentials is a very important development. Potential applications include calculating the free energy of protein–ligand binding via molecular dynamics simulations, and the simulation of deformation dynamics in materials.The potential energies and forces provided by molecular potentials are obtained traditionally by quantum mechanical (QM) calculations or classical physics-based force fields (FF). QM methods solve the Schrödinger equation and are the most accurate methods for describing atomistic systems. The high computation cost of QM and long-time scales relative to experiment has limited studies of larger, realistic atomistic systems. Hence, novel robust approaches approximating QM methods without any loss in accuracy are required for continued scientific progress. Force fields are computationally efficient, allowing the simulation of up to millions of atoms, but they require explicit parametrization of classical bonding, angle, torsion, and possibly higher-order terms. The correct parametrization of force fields can be tedious and cumbersome. Further, parametrization for one atomistic system may not be transferable to new systems.
Recent breakthroughs in the development of ML methods in chemistry198 have produced general purpose models that predict potential energies and other molecular properties accurately for a broad class of chemical systems. General purpose models promise to make ML a viable alternative to classical empirical potentials (EP) and force fields since EPs are known to have many weaknesses, such as poor description of the underlying physics, lack of transferability, and are hard to systematically improve their accuracy.
Molecular representations
To develop a useful and efficient ML-based property predictor, the most critical issue is how to represent the system in question to a ML method. These representations (descriptors) consist of some numerical representation of a molecule or a system of atoms. There are a wide range of published descriptors such as the Coulomb matrix,199 or its recent Bag of Bonds (BoB)200 extension. Other popular choices include descriptors that represent molecular graphs,201 bonds and angles,202 many body expansions,203 the atomistic local chemical environment,204 and end to end models that learn the best description of the system given minimal neighborhood information.205 Many of these techniques have been successfully applied to either molecules or materials.Some recent descriptors like MBTR (many-body tensor representation) and SOAP (smooth overlap of atomic positions)206 can describe both finite- and periodic systems. MBTR is derived from the Coulomb matrix, BoB, and many-body expansion. SOAP kernel represents the local density of atoms within the environment as a sum of Gaussian functions centered on each of the neighbors of the central atom. It essentially defines the similarity between two neighboring environments and uses it as a descriptor for ML models.207
Local atomic environment vectors (AEV) are another widely used molecular representation. AEV explicitly incudes all pairwise combination of elements, which means that the size of the input layer of a ML model grows as O(N2) with the number of included chemical elements. Therefore, models can only be trained for a relatively small number of chemical elements. Adding new elements requires retraining the ML model again from scratch.
Recently, alternative weighting functions (wACSFs),208 circumventing the above issue, have been proposed. Though this is a simple re-parametrization, the number of required symmetry functions becomes independent of the actual number of elements present in the system, leading to more compact descriptors. This alternative solution to the growth problem was introduced with the deep tensor neural network (DTNN)209 and atom-in-molecule neural network (AIMNet). These constitute learnable vectors of atomic features that are used to embed atomic symmetry functions to make a unified representation of each atom's chemical environment. DTNN was subsequently refined to create the SchNet architecture205 specifically designed to model atomistic systems using continuous-filter convolutional layers.
Neural network potentials
A ML approach applicable to chemical systems containing large numbers of atoms, originally proposed by Behler and Parrinello (BP method) in 2007, used high-dimensional neural network potentials (NNP, Fig. 7).210 As in many conventional empirical potentials, the potential energy E is the sum of local atomic energies of all atoms in the system. Since this seminal publication, a substantial number of articles and reviews have been published on the use of NNPs for bulk chemical systems (e.g., bulk silicon or water) or for describing single molecule potential energy surfaces and reaction coordinates.211 | ||
Fig. 7 Depiction of a standard feed-forward neural network potential for predicting a single property Ei from input ![[G with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0047_20d1.gif) i, computing the cost C with a cost function, then back-propagated the gradient of the cost (with respect to the optimizable parameters) into the network for training. i, computing the cost C with a cost function, then back-propagated the gradient of the cost (with respect to the optimizable parameters) into the network for training. | ||
Recently, Smith et al. introduced the first NNP designed for organic molecules, ANI-1.212 It is applicable to molecular systems well outside its training set. The ANI-1 potential was trained on a dataset of small organic molecules of up to 8-heavy atoms (while sampling both conformational and configurational space). Furthermore, ANI-1 demonstrated its applicability to much larger systems, up to 70 atoms, including known drugs and molecules randomly selected from the GDB-11213 database and containing up to 10 heavy atoms. It predicted DFT energies of the test set molecules with up to 10 heavy atoms very well, with the resulting RMSE values below 0.57 kcal mol−1.
Many techniques for improving the accuracy and transferability of general-purpose ML potentials have been employed. Among these, active learning methods, already proven successful in conventional QSAR modeling, have been especially popular.214 Active learning methods provide a consistent and automated improvement in accuracy and transferability and have contributed greatly to the success of general-purpose models. An active learning algorithm decides what new QM calculations should be performed then adds the new data to the training set. Allowing the ML algorithm to drive sampling improves the transferability of an ML potential greatly. Further, transfer learning methods allow the training of accurate ML potentials by combining multiple QM approximations.
One fundamental limitation of BP-type models is the inability to pass information between atoms at larger distances. Several neural network architectures have been proposed to address this limitation. The HIP-NN (hierarchically interacting particle neural network) approach breaks molecules down into feature representations and uses a number for each atom and the pairwise distances between atoms. On-site layers encode information specific to each atom and interaction layers allow sharing of information between nearby atoms. The total energy is built hierarchically from those interactions.
Another architecture, SchNet, encompasses atom embeddings, interaction refinements, and atom-wise energy contributions. At each layer, the atomistic system is represented on atom-wise basis and is refined by continuous filter convolutions with filter-generating networks.215
In the AIMNet implementation, the solution to the short-range problem is inspired by mean field theory (MFT). The main idea of MFT is to replace all interactions of any one atom with an average or effective interaction, sometimes called a molecular field. This reduces any multi-body problem into an effective one-body problem.
Datasets
As previously stated, one of the most important aspects of building a model in chemistry is the choice of the training dataset. Various datasets of organic and materials systems for training ML models have been developed over the last decades. Two of the most popular organic molecule benchmark sets are the QM7199 and QM9216 collections. The QM7 benchmark was developed by subsampling the GDB-13217 database of small molecules. QM7 contains 7165 energy-minimized molecules consisting of up to 7 heavy atoms and several properties computed with density functional theory (DFT). This benchmark is difficult to model by ML because of its relatively small size. Initial mean absolute errors (MAE), using the coulomb matrix representation,199 were around 10 kcal mol−1.The ANI-1 dataset includes organic molecules with a large number of non-equilibrium DFT total energy calculations. It includes ∼24 M conformations for 57462 molecules from the GDB database, with the total energy values computed for each conformation. This dataset samples both chemical and conformational degrees of freedom at the same time and thus provides 100× more data. Therefore, we expect that this dataset will become a new standard for comparing the ability of current and future ML methods to improve on the best model accuracy (1 kcal mol−1) achieved for the QM9 benchmark. More importantly, this data source is a foundation for development of future general-purpose machine-learned approaches.
The COMP6 benchmark dataset214 was developed to validate the transferability of ML potentials. COMP6 is a benchmark suite containing five rigorous benchmarks that cover broad regions of organic and bio-chemical space of isolated molecules and a sixth built from the existing S66x8218 noncovalent and intermolecular interactions data.214 Properties are calculated using the ωB97x/6-31G(d) basis set, however, it could be recomputed using any desired quantum level of theory.
Advanced approaches
In addition to active learning, there are other ML techniques that aim to reduce training data requirements. Some ML-based methods (such as NN) can take advantage of information from multiple sources. The key concept is to train a model using a large dataset of medium accuracy, then retrain the model with a smaller, more accurate and difficult to obtain data set. This process called transfer learning (TL) relies on the assumption that less accurate data sets contains some information that makes it easier to learn models for the smaller datasets of higher accuracy data.For example, TL could be performed by taking a DL model that was pretrained to medium-fidelity DFT, holding some number of parameters in the model constant, then retraining the remaining parameters using a much smaller, higher accuracy CCSD(T)/CBS dataset. Such methodology resulted in the development of the ANI-1ccx potential, which represents an attractive alternative to DFT and standard force fields for conformational searches, molecular dynamics, and the calculation of reaction energies. The computed reaction energy values demonstrated that the transfer learning-based ANI-1ccx method outperforms DFT on test cases, especially those where DFT fails to capture reaction thermochemistry.
In many systems, multiple data modalities can be used to describe the same process. One such physical system is the human brain, which provides more reliable information processing based on multimodal information.219 Many ML related fields of research have successfully applied multimodal ML model training.
In chemistry, molecules, often represented by structural descriptors, can also be described by accompanying properties (dipole moments, partial atomic charges) and even electron densities. Using multimodal information as inputs has been an actively developing field in recent years.220 This boost is caused by the use of additional information that captures the implicit mapping between the learnable endpoints. We discussed the advantages of multi-objective models over traditional single task approaches in the sections on chemical safety prediction and multi-target profiling above. Here we show that the same approaches are equally useful for developing ML models of QM results.
In the previous sections we have commented on the ongoing revolution in organic chemistry brought about by advances in computational (retro)synthetic approaches and robotic chemistry. Similarly, the use of ML approaches in quantum chemistry constitutes another recent paradigm shift. These rapidly emerging approaches dramatically change current limits of the size and complexity of molecular systems accessible to QM-level structure and property calculations.
Materials informatics
Machine learning methods dependent on large experimental and computational databases, are becoming ubiquitous tools for materials development,221 extending their traditional use for organic molecules. Materials science is a very large field and space constraints permit discussion of only a small set of important questions and answers described below.Which materials are missing?
This has been a perennial question,222 but several recent studies have attempted to address this. For instance, Hautier et al.223 used experimental data to create a probabilistic framework for ionic substitution capable of dealing with sparse spaces (quaternary configurations). ML has also been used to tackle amorphous systems. For example, Perim et al.224 identified an energy spectral descriptor for de novo prediction of metallic glasses and used it to quantify the classification probability of mixtures. ML and atomic features (descriptors) were also used to identify regions of compositions prone to glass formation and demonstrated surprising accuracy.225Descriptors, the Holy Grail of optimization: where can we find them?
While the great importance of descriptors has been established,221 these parameters are often defined deus-ex-machina out of intuition. Attempts have been made to develop interpretable parameterizations with ML. Thus, Ghiringhelli et al.226 proposed compressive sensing to discover functional forms and tested stability rules for binary semiconductors. Isayev et al.227 introduced universal fragment descriptors for predicting properties of inorganic crystal and developed electronic density of states and band structure fingerprints that cluster many high temperature superconductors (materials cartography). Recently, Stanev et al.228 identified 30+ non-cuprate and non-iron-based oxides, potential new superconductors, using RF.Can enthalpies (and other properties) be predicted?
The correct calculation of enthalpies and other properties is important for ab initio computational materials design.229 Much progress has been made since the original principal components analysis of alloy thermodynamics reported by Curtarolo et al.230 Rupp et al.231 used kernel ridge regression for modeling molecular atomization energies with mean absolute error of ∼10 kcal mol−1. In a related study, De et al.207 used the smooth overlap of atomic positions (SOAPs) to introduce a very useful descriptor for comparing structures: the “alchemical similarity” for molecular and periodic structures. Gaussian process regression (GPR) was used to generate very accurate Gaussian atomic potentials (GAP) and then to train a SOAP-GAP model within a ML framework (GPR) that achieved a 99% accurate atomic-scale properties for Si surface reconstruction, stability of molecules, and protein ligands.232 Pilania et al.233 tackled melting temperatures of the octet subset of AB solids and band gaps of double perovskites. De Jong et al.234 used statistical learning to study elastic moduli of inorganic crystals, and with many other relevant studies.What material properties can we predict?
Welcoming new challenges!
Materials science properties, based on fundamental principles, are intrinsically suitable for modeling by machine learning. Success in ML approaches is a driver for the discovery and/or optimization of new materials and/or phenomena. This section has given a short—unavoidably incomplete—snapshot of the current state of the art.To conclude this section, we highlight the clear similarity between materials informatics with the traditional workflow of QSAR modeling (see Fig. 1 and 8). As with cheminformatics, the starting point of materials informatics is the accumulation of large datasets of materials with experimental or computational properties. The need for developing novel materials descriptors and their use in building property prediction models using ML techniques follows. Finally, current challenges outlined in the concluding part of this section parallel many of those facing traditional QSAR modeling of bioactive compounds. Thus, materials informatics (and a closely related field of nanomaterials informatics described in the next section) represents a prime example of a new discipline, whose development was enabled and immensely catalyzed by the experience and approaches developed in QSAR.
 | ||
| Fig. 8 Machine learning materials flow is a combination of feature extraction, descriptor analysis, structure fingerprinting (representations) of databases, and materials synthesizability. Figure reproduced with permission from the following sources: (i) ref. 240 Copyright (2020) American Chemical Society; (ii) ref. 353 under the terms of the Creative Commons CC BY license; (iii) ref. 354 under the terms of the Creative Commons CC BY license; and (iv) ref. 355 Copyright (2020) by the American Physical Society. | ||
Nanomaterials informatics
Nanotechnology is another field for which cheminformatics is becoming a key tool, especially for the quantification of diverse properties of nanomaterials and nanostructure–property modeling. Development of modern AI algorithms has stimulated an increased interest in quantitative nanostructure–activity relationships (QNAR)252 also known as nano-QSAR. Like traditional QSAR, QNAR models are based on the assumption that similar nanomaterials will induce similar biological effects. However, unlike QSAR, nanomaterials (and materials in general) are more complex than single drug molecules, as they are less well defined and feature distributions of sizes, shapes, etc.QNAR models rely on an ensemble of molecular descriptors that encode constitutional, topological, or geometrical characteristics of a given set of nanomaterials. These descriptors are derived directly from the structures of the nanomaterials using bespoke software. Moreover, experimentally determined properties (e.g., elemental composition, zeta potential, size distribution, shape) can also be appended to the computed descriptors to boost the prediction performances of QNAR models. This is analogous to the use of experimental HTS results as descriptors to model biological endpoints for drug candidates described in prior sections. QNAR models establish quantitative relationships between those experimental and computed descriptors and specified biological endpoints using ML techniques.
Importantly, QNAR models are developed using the same workflow (see Fig. 9), validation procedures, statistical criteria, and key steps as those of classical QSAR models for small molecules (see Fig. 1). However, the high structural diversity and complexity of nanomaterials typically lead to specific challenges,253 especially when it comes to the choice of molecular descriptors. Two types of representations are clearly emerging from the literature – studies in which the whole nanoparticle is characterized computationally, experimentally, or both or when such characterization is applied to the surface chemistry of the nanoparticle (especially, organic decorators) only. Naturally, the choice of descriptors and the associated software is different for these two types of QNAR modeling. For the second type of study the QNAR model is similar to a traditional QSAR model, trained using descriptors for surface chemistry, to predict biological activity of the nanomaterials. Another challenge of QNAR modeling, similar to materials informatics is the relatively small size of the datasets currently available in the public domain. This leads to lower prediction accuracy and smaller applicability domains for QNAR models compared to those of QSAR models trained on large organic molecule data sets. To mitigate this limitation, read-across techniques are increasingly used to estimate the properties of nanomaterials.254
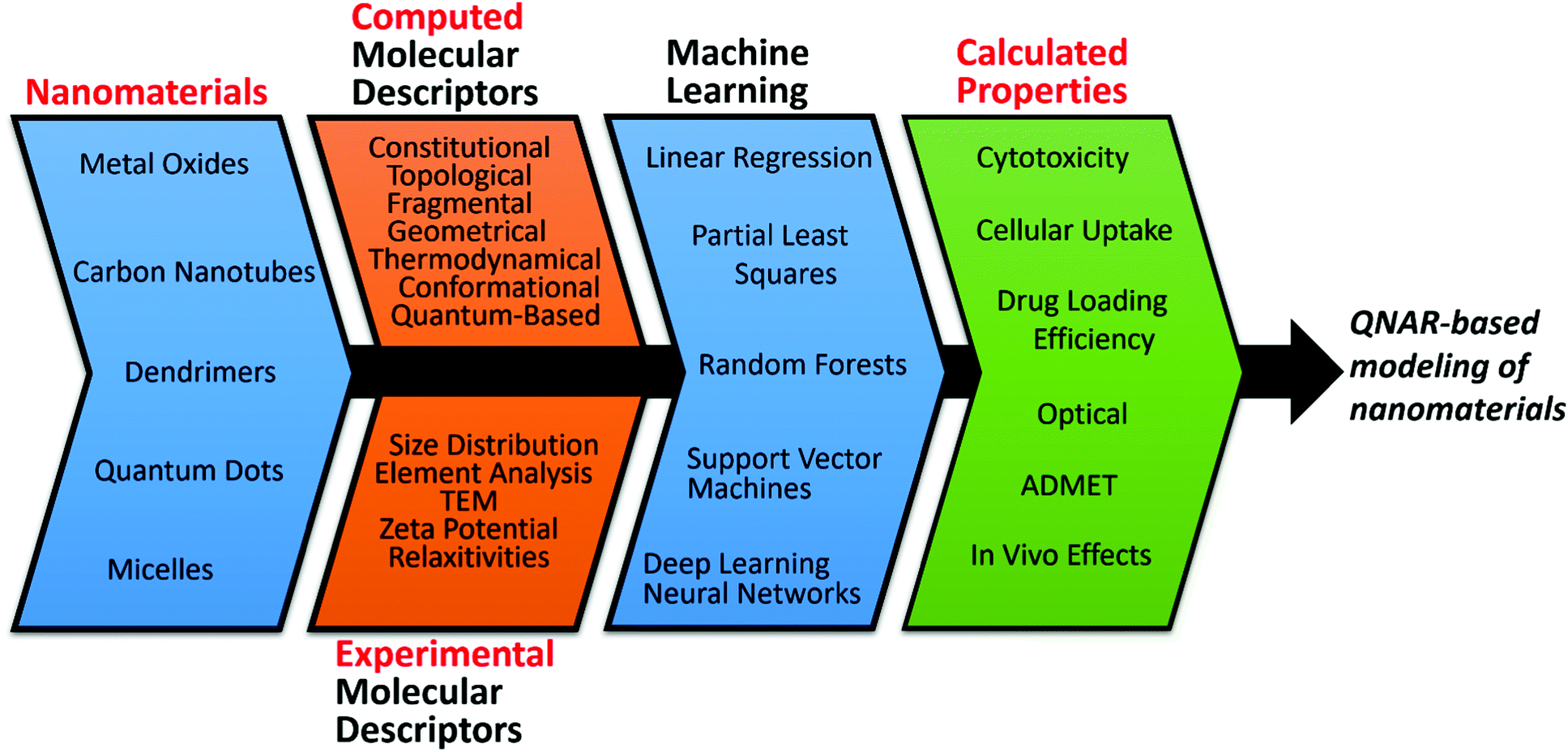 | ||
| Fig. 9 General scheme representing the development of quantitative nanostructure–activity relationships for the calculation of properties of nanomaterials using both computed and experimentally determined molecular descriptors. | ||
Assessing the environmental impact of engineered nanomaterials (ENMs, see Fig. 10) requires data on their physicochemical and bioactivity properties, as well as bioaccumulation. After data collection and validation, ML approaches can be used to generate models correlating values of ENMs descriptors (e.g., structural, physicochemical, and bioaccumulation-related) and specific toxicity outcomes associated with biological mechanisms of action under various exposure scenarios.
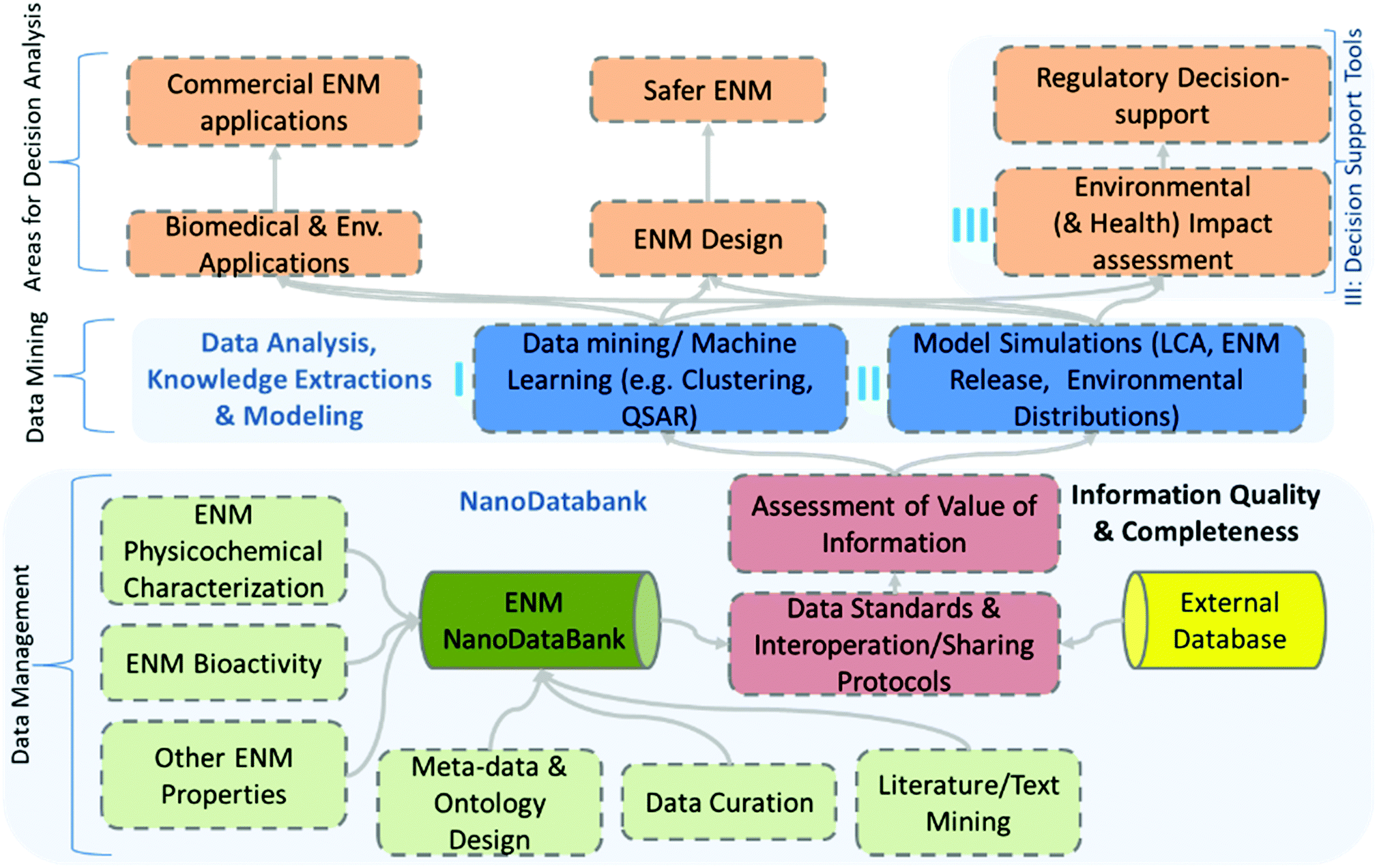 | ||
| Fig. 10 Nanoinformatics elements of environmental and health impact assessment for nanomaterials. | ||
The importance of data on ENMs structure and properties
Like other area of materials science, nanotechnology has generated various datasets of physicochemical properties, environmental fate and transport parameters, and bioactivity of nanomaterials.255 They contain both literature curated and raw data from various experimental investigations, useful for QNAR modeling. For example, the OCHEM database255 contains experimental data on ENMs and provision for generating descriptors for model building, NanoMiner256 contains data (including omics data) on 634 types of ENMs. The NM-biological interactions knowledge base contains over 200 toxicological evaluations for embryonic zebrafish exposed to metal and metal-oxide ENMs. NanoDatabank257 has raw data for over 1000 different nanomaterials and associated characterization and toxicity data.Early nanoinformatics efforts were focused on organizing data into structured datasets (i.e., with fixed fields or records).39 However, there is growing recognition that significant data are available as unstructured datasets (i.e., with no predefined fixed fields or records), often are scattered across multiple literature and online sources. Thus, significant recent efforts have been devoted to the development of public databases, meta data, and data management systems for nanomaterials. These efforts included incorporation and integration of information from multiple sources, addressing data security, effective data sharing, intelligent data queries, and data integration.258 The joint EU-US Nanoinformatics Roadmap 2030255 has stressed the need for guidelines concerning the development of nanoinformatics datasets that are structured, have controlled ontology for ENMs properties and bioactivity, and interoperability with other databases and modeling tools. Raw data (free from pre-processing by data curators) that can be curated and analyzed in a context-dependent way are most useful for QNAR development.
Substantial amounts of experimental data on the toxicity of ENMs have been generated, primarily in various cell lines such as, macrophages, pancreatic and other human cells and bacteria. There are still limited studies with simple organisms like zebrafish and even fewer on higher animals. Toxicity data include experimental results across multiple assays and cell lines/types with ENMs having different surface modifications and core compositions. There are different levels of confidence and consistency across the toxicological studies. Currently, efforts to derive generalized toxicity models based on ENMs characteristics have been based on datasets from single studies rather than integrated from the collective body of published data.259 Clearly, to develop predictive nano-SAR models of ENMs toxicity, it is useful to identify critical biological pathways that can lead to adverse outcomes.260 Understanding relationships between the structural and physicochemical properties of ENMs and the biological responses and correlation between such responses can be very useful for deriving causal relationships. Although QNAR models provide valuable insight on ENMs toxicity, they generally cannot provide direct mechanistic interpretation that can be validated and tracked back directly to experimental data. However, as with most other QSAR models, ENMs toxicity models can be very useful in the absence of mechanistic information or interpretation.
Clearly, to generate the most robust and predictive ENMs toxicity models, the quality of data is paramount. These models can then elucidate the relevance and significance of ENMs properties such as structure, surface chemistry, shape and other physicochemical parameters with respect to their biological properties. Experimental conditions can also be employed as independent variables when modeling toxicity. Several literature studies have identified causal relationships between the biological outcomes and important ENMs properties.261
QNAR modeling
Several seminal publications pioneered the field of QNAR modeling. Puzyn et al.262 built the first nano-QSAR model based on ensemble learning regression methods and CDK descriptors to predict the cytotoxicity of 17 unique metal oxide nanoparticles. Fourches et al.263 introduced the concept of QNAR modeling with a set of 109 functionalized CLIO nanoparticles and their Paca2 cell uptake. This study has been repeated and successfully reproduced several times by other research groups.264 For instance, different series of metal oxides were also modeled using the OCHEM webserver to generate reliable QNAR models.265 Drug delivery properties of nanocarriers could be successfully predicted by QNAR models as well.266Important nanomaterials, carbon nanotubes, have had their biological effects extensively modelled by QNAR. For instance, Trinh et al.267 used a combination of computed and experimental descriptors, encoded as quasi-SMILES, to build QNAR models that could accurately estimate the cytotoxicity of carbon nanotubes in human lung cells. Fourches et al.268 developed a series of QNAR models for 83 functionalized CNTs tested in vitro for protein binding and toxicity. These models reached prediction accuracies up to 74% for external test set toxicity estimates, and protein-binding classification models achieved external prediction accuracies up to 77%. A library of 240000 potential CNT surface modifiers was further screened using these models and the least toxic organic modifiers were selected for experimental validation. Subsequent synthesis and testing of these surface-modified CNTs confirmed the in silico predictions, demonstrating the utility of QNAR models for rational design of nanomaterials with enhanced properties.
In another study, a logistic regression-based QNAR model was developed269 to flag toxic outcomes; this model was trained on high-throughput toxicity screening data for BEAS2B cells exposed to nine metal oxide nanoparticles. The best-performing model had almost 100% classification accuracy and required only three nanoparticle descriptors: the period of the nanoparticle metal; the atomization energy of the metal oxide; and the nanoparticle size and volume fraction. Another study used RF classification to model cellular toxicity of metal oxide ENMs.270 The model was trained on data extracted from 216 publications, and used 14 ENMs attributes as descriptors. It demonstrated that cytotoxicity of ENMs was highly correlated with the administered dose, assay type, exposure time, and surface area of nanoparticles.270
Bayesian networks as models for predictive toxicology and for assessment of causal relationships
Models that predict toxicity of ENMs must account not only for the properties of the nanomaterials per se, but also for experimental conditions (e.g., assay types, exposure concentrations, exposure period, organism and more). It is important to quantify the relevance and significance of ENMs and experimental attributes driving toxicity while accounting for uncertainties in data, particularly that collected from multiple sources. Toxicity prediction models trained on these attribute combinations can sometimes identify causal relationships,261 which can be effectively achieved with the Bayesian network (BN, also called a Bayesian belief network, BBN) approach.271BN models construct a network where the modes are ENMs characteristics and the edges (links) represent conditional dependences of target outcomes on various attributes. This provides a visual representation of causal relationships.272 The model allows interpretation of “if/then” causal relationships where the parent (antecedent) and child (descendent) nodes are at the outgoing and incoming links in the BN structure, respectively. The set of model attributes and their conditional dependencies represents knowledge from the dataset(s) of attributes and toxicity outcomes in the form of probability distributions. BN models can identify, for example, the conditional dependence that would lead to a toxicity outcome within a specific range.
Previous studies have demonstrated the value of BNs for developing qualitative “toxicity/hazard” classification of ENMs based on using physicochemical and specialized descriptors.273 BN models identified the most relevant parameters impacting specific ENMs hazards. Thus, regression and classification models were developed274 for cause–effect relationships for hazard associated with exposure to TiO2, SiO2, Ag, CeO2, and ZnO NPs for different toxicity endpoints. A BN model predicted the hazard associated with exposure to metal and metal oxide NPs273 for eight toxicity endpoints compiled from 32 published studies. Despite the existence of significant data gaps for some NPs the resulting BN model identified the most relevant NP properties for predicting toxicity outcomes.
Data variability and curation
As is true for traditional QSAR, inter- and intra-sample variability in QNAR is a big issue that can dramatically affect the predictivity of a model. Therefore, in order to study and/or model nanomaterials, the experimental variability for both inter- and intra-sample measurements needs to be taken into account whenever possible. For instance, the size distribution of a given sample of a specified nanomaterial can vary from one instrument to another. If a series of size distribution plots is used to model a set of nanomaterials, then the experimental variability of these measured profiles needs to be considered to better understand the stability, reliability, and robustness of the model. As with small molecule drugs and/or batches of biologics, replicate measurements are necessary to understand experimental variability. All, or a subset of compounds chosen to be representative, and their associated samples are characterized in triplicate. If one endpoint (e.g., particle diameter, zeta potential) is deemed unreliable, that endpoint should not be considered as a descriptor for those nanomaterials nor should it be considered as a target property for a model. Clearly, materials characteristics measured with low accuracy and reproducibility, will limit the predictivity of the QNAR models trained using them. Nanomaterials are particularly sensitive to the protocols used for sample preparations (e.g., dilution, sonication, solvent mixtures) leading to aggregation or even degradation. Experimental variability is a general issue that the QSAR modeling field is constantly dealing with. Strict data curation prior to model development is highly recommended,16 whereas external validation ensures the stability and robustness of the models over all modeling and external prediction sets.Perspectives
Although QNAR modeling is still in its infancy, we anticipate it will grow significantly in the near future. This growth is dependent on:• development of more effective and interpretable ENMs-specific descriptors
• further development of high-throughput synthesis and screening platforms for nanomaterials, leading to the expansion of publicly available data to train QNAR models
• development of more robust and predictive, consensus models based on individual QNAR models trained on diverse ENMs descriptors using advanced ML techniques including DL
• development of nanomaterials with desired properties and pre-computed bioprofiles generated by interdisciplinary research teams. The role of QNAR modeling in the context of such multidisciplinary efforts cannot be overestimated.
Biomaterials and regenerative medicine
Previous sections have covered major underlying concepts of cheminformatics such as chemical similarity, QSAR model building and validation, and domain of applicability. These methods have been progressively extended to areas beyond their traditional applications, for instance chemical genomics and (nano)materials science as discussed above. Another emerging field is the use of QSAR methods to model control of cell phenotypes and understanding and predicting the biological response to materials. These are relatively recent, but rapidly expanding fields where the potential impact is very significant. Unlike bioinformatics,275 cell biology, and clinical medicine,276 there is a relative paucity of published examples of the application of QSAR or related ML-based methods to biomaterials, regenerative medicine, and stem cells studies. Polymers and other complex materials have been used in implantable or indwelling devices, as replacement or augmentation of natural bodily components, as scaffolds for cell culture, and as active biomaterials and drug delivery systems. Unfortunately, such materials are not as well defined as organic molecules. As discussed above in the sections on (nano)materials informatics, one of the biggest challenges in the field of biomaterials is generation of appropriate descriptors that capture relevant properties of these materials and can adequately represent their structure, often poorly understood and characterized.20 In this regard, rapid adoption of DL methods is providing useful models for this very important issue.277 The feature generation capabilities of DNN mean that simpler representations of complex materials become possible. We further anticipate that predictive material-QSAR models may be interrogated to identify the types of complex features that modulate relevant biological responses most strongly.Although the use of arcane molecular descriptors has already resulted in good predictive models of the biological effects of materials, there is increasing impatience with their inability to be related back to underlying chemical features interpretable by chemists to improve performance. The dilemma between good predictions of properties for new materials, and interpretability of models (mechanistically or in terms of molecular interactions at a surface) has been reviewed recently by Fujita and Winkler.20 This nexus has led to a rise in the popularity of signature or fragment-based descriptors for modeling of materials interaction with biological systems. For example, signature descriptors have been used to model the adhesion of bacteria to polymers.278 New ML methods such as adversarial and encoder-decoder networks have begun tackling the ‘inverse QSAR’ problem, where trained model can be used to design or suggest new molecules for synthesis with improved activity.
A second important issue that distinguishes materials modeling from small molecules modelling is that in the former case interactions are more complex. Often materials interact with mixtures of proteins, membranes, cells, and modulate the responses of a myriad signal pathways, mechanosensors, etc. Consequently, ML methods are best suited to address such complexity and uncertainty, where the mechanisms of the cell–materials interactions are largely unknown. Notably, ML methods have been successfully used already for modeling soft biological materials such as blood vessels.279
To date, QSAR methodology has been applied in regenerative medicine and biomaterials modeling in three major groups of studies. First, sparse and non-sparse feature selection methods have been used to reduce the complexity of materials-biological systems interactions. For example, sparse feature selection methods were applied to investigate stem cell behavior (see Fig. 11 for details). Similarly, an expectation maximization algorithm employing a sparse (Laplacian) prior59 was used to identify the most relevant genes in unbiased genome-wide expression studies. In one such study, mesenchymal stem cells (MSCs) were exposed to the components of a biomaterial (strontium bioglass, SrBG) with varying levels of strontium ions.280 These drive MSC differentiation down the osteogenic pathway to form bone tissue. After preliminary expression level and fold ratio filtering, the sparse feature selection method identified a handful of genes related to fatty and sterol biosynthesis – a previously unreported mechanism of bone growth modulation. Subsequent experimental validation of this mechanism by means of qPCR Raman spectroscopy and protein expression profiling led to important implications for the control osteoporosis and bone loss.
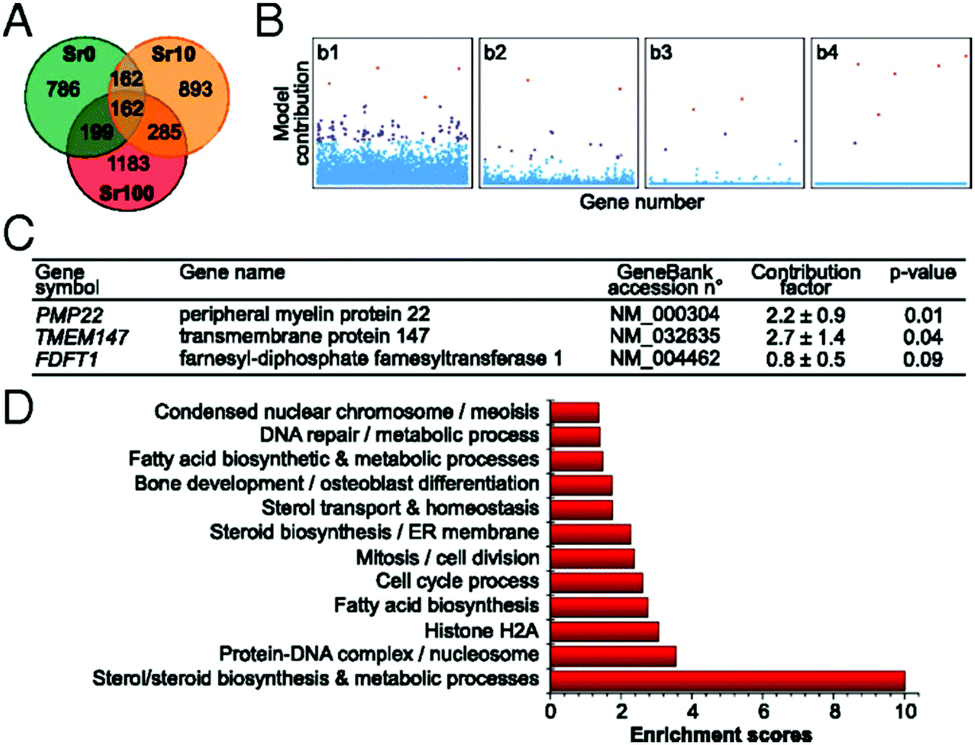 | ||
| Fig. 11 Changes in hMSC global mRNA expression mediated by treatment with BG- and SrBG-conditioned media. (A) Operation of the EM algorithm, showing progressive nulling of lower genes less relevant to the SrBG treatment. (B) The contribution (mean ± SE) of the most significant genes identified by sparse feature analysis. (C) Functional annotation clustering analysis of differentially expressed genes in response to Sr100 treatment compared with control. Figure is reproduced from ref. 280 with permission from Proceedings of the National Academy of Sciences of the United States of America. | ||
In another related investigation, unbiased sparse feature selection methods were applied to gene expression data.281 In this experiment, stem cells were forced to divide symmetrically or asymmetrically in response to several types of experimental conditions.281 Sparse feature selection methods were used to identify robust markers for symmetric cell division, which is a very important factor in stem cell proliferation and differentiation studies.281
ML methods have been increasingly applied to quantitative modeling of the responses of biological systems to interactions with materials.282 To date, most of these materials have been polymers, due to their tunable properties, ease of library generation and characterization, and generally understood biocompatibility. Early work was conducted by the Kohn group from Rutgers University who generated a library of 112 tyrosine-derived polyarylates and measured a range of their physical properties and biological responses.283 They used DRAGON descriptors284 based on the monomeric units of the polymers in combination with such parameters as glass transition temperature (Tg) and air–water contact angle to generate quantitative and predictive models of fetal rat lung fibroblast (FRLF) metabolism and fibrinogen attachment on the polymer surfaces. Subsequently, research teams at the University of Nottingham, CSIRO, Monash University, and MIT generated polymer microarrays285 and conducted high throughput screening to elucidate structure–property relationships in their interactions with cells.
The use of biomaterials as cell factories286 shows great promise, and the large generated stem cell attachment, proliferation, and differentiation datasets were modelled by ML methods. These could make robust and accurate predictions of stem cell behavior of materials not used to train the models. In one study, the attachment of embryoid bodies (a surrogate and stable cell system to mimic embryonic stem cells) to a polymer library was modelled using sparse feature selection and optimally regularized neural networks.287 These models relied on DRAGON descriptors and Bayesian regularized neural networks to quantify the attachment of embryoid bodies to the polyacrylate libraries. A more recent study modelled attachment, proliferation, and differentiation of human dental pulp stem cells to a polymer library.288 In this case study, the authors also investigated the ability of a 541 members of polyacrylate homopolymer and copolymer library to promote attachment, proliferation, and differentiation of stem cells.
Finally, advanced QSAR methods are being applied to the characterization of surfaces that interact with biological systems and to analyzes of complex high-content data such as cell imaging and phonotype recognition. Surface analysis methods such as Raman and Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) are invaluable experimental tools for characterizing the nature of surfaces interacting with biology. Surprisingly, there has been little application of statistical methods and ML to the corresponding spectroscopic data. ToF-SIMS in particular has proven to generate data that is very useful for QSPR material modeling.285 Recent work has shown how self-organizing maps (SOMS) can provide superior clustering of complex mass peak data,289 probing into the intrinsic information content (Shannon entropy) of these surface analysis methods.290
As the field of biomaterials modeling is relatively nascent, there are many issues that need resolving before the full benefit of AI/ML-based QSAR methods can be realized. The most important of these issues is how to represent a high molecular weight complex material such as a cross-linked polymer hydrogel or polymer library with distributions of chain length, block sizes, degree of cross-linking, etc. Although surprisingly effective models can be generated using descriptors based on small fragments, additional materials features may be needed where these approximations fail. More recently methods have been developed that allow many types of nanoscale topographies to be imprinted onto materials surfaces. These modulate biological properties such as macrophage polarization, so efficient ways of generating descriptors for topographical features are required. Equally important is the need to generate models that can be interrogated to guide the synthesis of subsequent generations of materials with improved characteristics.197 Biological data variability and reproducibility are also a constant struggle for high throughput materials-based experiments. Improving the reliability of these biological response data by careful statistical treatment of results and improved fabrication quality control is also important. However, as modeling of biomaterials coevolves with further development of the respective experimental research, one shall expect models to become more robust and impactful.
Clinical and health informatics
Just as advances in statistics, ML, and AI have influenced chemical research, experience accumulated in cheminformatics can be applied to clinical research. The growing linkage between QSAR modeling and clinical informatics was highlighted by the most recent 22nd EuroQSAR meeting in 2018 dedicated explicitly to “Translational and Health Informatics: Implications for Drug Discovery”.291 One example of such cross-fertilization between the fields is the development of robotic biomarkers of motor impairment of patients recovering from stroke.292One of the greatest challenges in designing clinical trials is dealing with the subjectivity and variability introduced by human assessment of clinical endpoints. This problem is particularly acute in neurology, where outcomes may be highly variable (e.g., in cognition), susceptible to the state of the patient (e.g., fatigue, pain, anxiety, depression), the lack of a gold standard definition or diagnosis (e.g., neuropathy, dementia), are high dimensional (e.g., imaging or genomic markers), or are composite in nature (e.g., clinical instruments for assessing depression or quality of life).293 These factors make it difficult to demonstrate treatment benefits, requiring larger pools of subjects in clinical trials as well as properly structured electronic health record (EMR) archiving and retrieval capabilities.
Neurological disorders such as stroke suffer from clinical assessment limitations as established methods are often subjective: scales such as the Fugl-Meyer (FM),294 motor power (MP),295 NIH stroke (NIH),296 and modified Rankin (MR),297 require properly trained personnel for evaluation, with results widely varying from rater to rater.298 While extensive training of raters and centralization of outcome assessments (whenever possible) can reduce variability, it does not completely eliminate it and comes with its own additional costs.299
One way to minimize this measurement variability issue is to replace human raters with robotic technology that can provide repeatable, reliable and speedy assessment of continuous measures of impairment and its change during recovery. Robotic devices are less sensitive to the skills and expertise of a human rater, can reduce inter- and intra-rater variability, can be used simultaneously for both assessment and rehabilitation, which can be done faster and more frequently, and can further be used in a home setting thus minimizing patient burden and inconvenience.299
The following study illustrates the use of QSAR-type approaches in clinical informatics. To test their utility in clinical trials, the four clinical scales mentioned above were used in conjunction with a robotic assay to measure arm movement in 208 patients at 7, 14, 21, 30, and 90 day time-points after acute ischemic stroke. The data were collected at two clinical sites in the US and the UK. The study had two goals. The first was to establish whether the robotic measurements could predict the scores of human raters, and the second was to develop a more sensitive robotic biomarker that could reduce the sample size of the study without compromising the predictive value. The robots were low impedance and low friction interactive devices that measured speed, position, and force.300 The robotic assessment consisted of 35 macro- and micro-metrics derived from various directed, unassisted reaching, circle drawing, resistance to external forces, and shoulder strength measurements, applied to the affected and unaffected arms.301
The relationships between these 35 robotic variables and the four clinical scales were visualized (see Fig. 12) using stochastic proximity embedding (SPE), a self-organizing nonlinear mapping algorithm that was originally invented to visualize very large combinatorial chemical libraries135 and subsequently adapted for various molecular modeling applications.302 Having established a degree of correlation, models were generated to assess whether the robotic metrics could predict the clinical scales with sufficient accuracy to serve as their surrogates. The model was trained using the data from degree of recovery from day 7 to day 90 after stroke, and all other intermediate measurements were used as test data. Specifically, 208 patients were divided into two complementary populations: those with complete data sets for days 7 and 90 (referred to as completers; N = 87) and; those with missing data on days 7 or 90 (referred to as non-completers; N = 121). The models, based on feed-forward NNs, were derived independently for each clinical scale. They were trained to predict the clinical scores of a given patient on a given day from the respective robotic metrics, using the completer population as a training set.
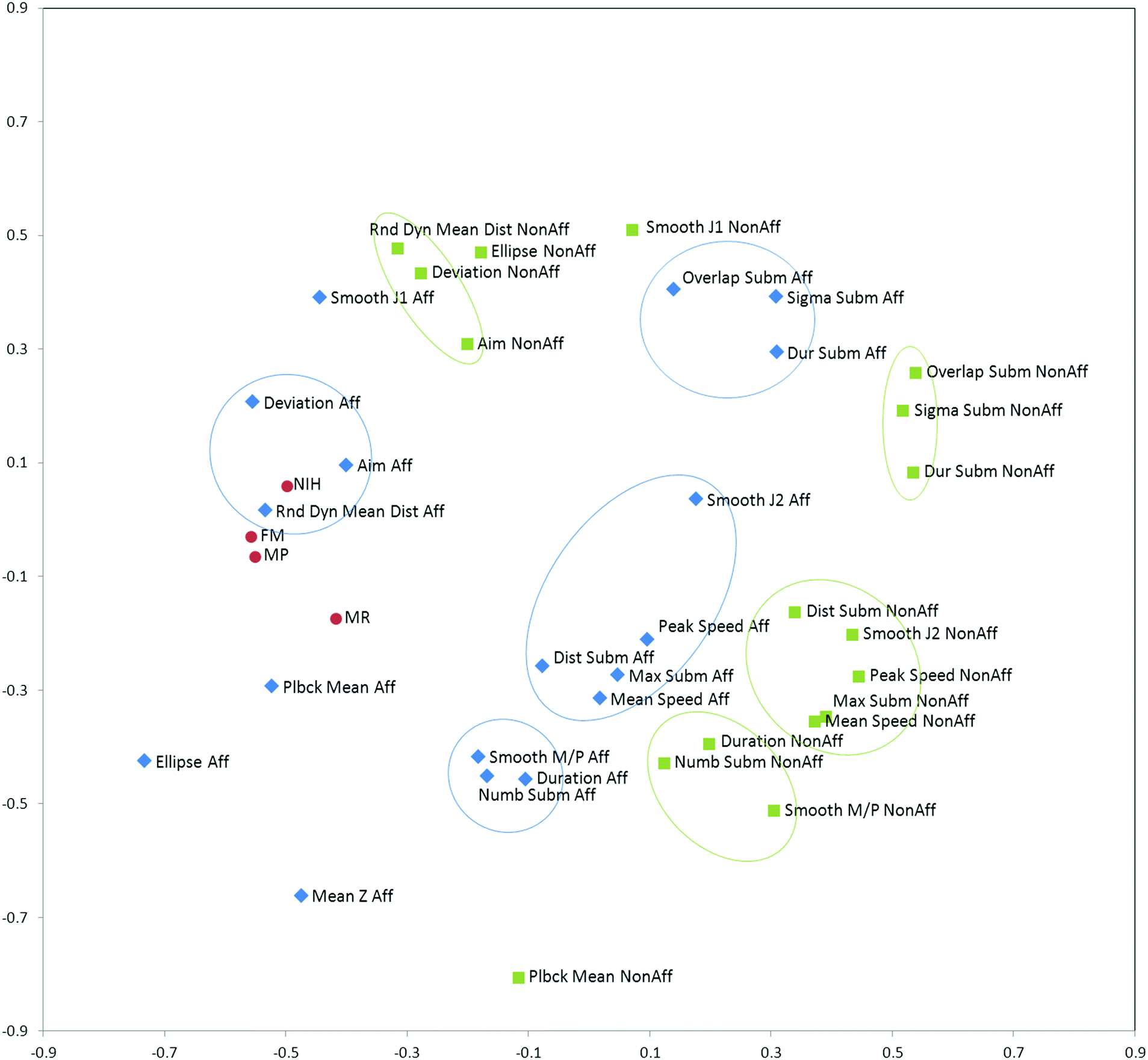 | ||
| Fig. 12 SPE map of the correlation distances of the clinical and robotic parameters for the completers cohort. The map was derived by computing the pairwise Pearson correlation coefficients (R) for all pairs of features, converting them to correlation distances (1 − abs(R)), and embedding the resulting matrix into 2 dimensions in such a way that the distances of the points on the map approximate as closely as possible the correlation distances of the respective features. The clinical parameters are highlighted in red, the robotic parameters on the affected side in blue, and the robotic parameters on the unaffected side in green. The map also shows distinct clusters of correlated variables which are preserved on both the affected and unaffected sides (outlined by green and blue ellipses, respectively). | ||
To minimize over-fitting, a feature selection algorithm based on artificial ant colonies, originally developed for QSAR applications, was used to identify the subset of robotic metrics that had the highest predictive power.303 Once the relevant features were identified, ensemble models comprising 10 neural network predictors were constructed using the same network topology and training parameters but initialized with a different random number seed. The predictions of these models were averaged to produce an ensemble prediction. All models were cross-validated using the standard jackknife approach that divided the training data into 10 disjoint subsets containing 10% of the patterns each, systematically removing each subset from the training set, building a model with the remaining patterns, and predicting the clinical scores of the removed patterns using the optimized network parameters. The resulting predictions were compared to the original clinical scores to evaluate the overall agreement with the RCV2 metrics. This process was repeated 10 times to obtain more robust cross-validation statistics. Finally, the best models identified by cross-validation were used to predict performance of the non-completers, who formed an independent test set. This protocol was virtually identical to the one used for QSAR applications.304
The resulting models recapitulated the human scored clinical scales with a cross-validated R2 of 0.73, 0.75, 0.63, and 0.60 for the FM, MP, NIH and MR scales, respectively. The models also showed lower but still useful predictive power for the external validation set (non-completers). The models had better prediction accuracy for the FM and MP scales that are more closely related to motor function than the NIH and MR metrics. Finally, the models were used to derive novel composite robotic endpoints with improved sensitivity (and effect size) compared to existing scales. To measure the effect size, Cohen's d parameter for paired observations was used, defined as the mean divided by the standard deviation of the day 7 to day 90 changes over all the completers. Since optimizing nonlinear composites is an ill-posed mathematical problem, a greedy forward-selection algorithm was employed to select up to 8 most relevant robotic features. Optimized robotic composites with as few as four features increased the effect size over a reference natural history trial305 by as much as 107% for the training and 83% for the test set. This result is highly significant as an increase of 83% in effect size would result in a 70% reduction in the number of patients required to achieve the typical 80% statistical power in a clinical trial.
While the primary purpose of EMRs is to serve patient care, the second QSAR-inspired study illustrates how structured EMR information can be processed with unsupervised learning to improve patient phenotyping in chronic obstructive pulmonary disease (COPD).306 COPD, a heterogeneous disease characterized by persistent, non-reversible airflow limitation is the fourth leading cause of death in the United States (as of 2010). While “phenotype” is a co-emergent property of the genotype–environment interaction, COPD has been classically stratified in two phenotypes,307 the “blue bloater”, which is rooted in chronic bronchitis (cyanosis due to hypoxemia), and the “pink puffer”, which is rooted in emphysema (pink skin and hyperinflation), although up to seven COPD phenotypes have been proposed, based on “clinical relevance”.308 Unsupervised learning was used to analyze EMR data from COPD patients, first to find out if common COPD patterns exist, which in turn could identify different COPD subtypes and lead to improved therapeutic management within each COPD subtype. A total of 3144 patients aged 40 or older, admitted to the University of New Mexico Hospital, a 580-bed tertiary hospital with a COPD diagnosis (ICD9 codes: 490, 491, 492 or 496) between 1 January 2011 and 1 May 2014 were processed for this study. Data processed in this analysis included demographics, comorbidities, presence of atopy, obesity, number of admissions, prescriptions for inhalers (grouped as: (i) short acting beta-agonist, (ii) long-acting beta-agonist, (iii) anticholinergics, (iv) steroids and (v) combinations), prescriptions for oral steroids, beta-blockers and statins, as well as weight loss and elevated plasma bicarbonate (used as surrogate biomarkers for disease severity). All variables, including age (40–65years and >65years) and number of admissions (one admission and ≥two admissions), were coded as binary for the study.
These data were clustered using the sphere exclusion algorithm,309 a disjoint similarity method that has been widely applied in cheminformatics. In the disjoint similarity method, a patient (object) can belong to only one cluster.310 When processing this multidimensional space that has as many dimensions as variables, dissimilarity can serve as the distance metric between patients. By definition, similarity is set to 0 if all the variables are different and is set to 1 if they are equal.310 As described elsewhere, in sphere exclusion the only user input is the similarity threshold: first, the similarity between all patients was computed. The algorithm then identified the patient with the most “neighbors” within a specified similarity cut-off, forming the first cluster. These patients were excluded from further iterations. The process was repeated until only patients without neighbors (i.e., singletons) were left. For this dataset, the optimal balance between the number of clusters and clustering overlap was found at similarity threshold 0.62. Using the sphere exclusion algorithm for clustering reduces the risk of bias since the method does not make a priori assumptions regarding numbers of clusters or similarity thresholds.
After leaving 189 patients (6%) as outliers, the following nine COPD clusters (phenotypes) were identified, with the number of patients given in brackets: 1: depression-COPD (1748); 2: malignancy-COPD (312); 3: coronary artery disease-COPD (291); 4: young age-low comorbidity-high readmission-COPD (152); 5: advanced malignancy-COPD (144); 6: cerebrovascular disease-COPD (120); 7: atopy-COPD (81); 8: diabetes mellitus-chronic kidney disease-COPD (64) and 9: advanced disease-COPD (43). The largest cluster is characterized by a large proportion of patients over age 65 and depression; two clusters (2 and 5) are associated with malignancy, although the first one has few readmissions whereas the second one has signs of advanced COPD and frequent readmissions. Cluster 3 is associated with heart disease (patients over age 65), whereas cluster 6 is associated with predominantly cerebrovascular disease and younger (under 65) patients. Cluster 4 (young patients, few comorbidities) has the highest number of prescriptions for bronchodilators; cluster 7 is also comprised of patients below age 65, but with asthma/atopy and higher numbers of readmissions; cluster 8 is associated with chronic kidney disease (CKD) and type 2 diabetes in patients aged 40–65, whereas cluster 9 has frequent readmissions, severe disease and high number of anticholinergic prescriptions. Our analysis revealed five previously unreported COPD phenotypes: two malignancy-COPD clusters (2 and 5), the COPD-CKD-diabetes cluster (8), the “advanced disease” cluster (9) and the high readmission phenotype (4). Each of these new clusters has practical implications, which may lead to better therapeutic outcomes.
To summarize, the above studies successfully adapted methods from computational chemistry and cheminformatics into in-depth analyses of health data. We anticipate that this transfer of methods and experience will continue to fuel healthcare informatics research by introducing new and improved computational methodologies.
Outlook
The field of QSAR modeling based on simple approaches used to predict chemical reactivity was initially popularized by Corwin Hansch and his colleagues more than 55 years ago.1 For many years, even decades, this field was focused on the prediction of physicochemical properties and biological activities using descriptors representing intrinsic properties of chemical structures. However, as the size and diversity of chemical datasets expanded, the QSAR modeling field has evolved to include larger and more diverse types of chemical descriptors and increasingly more complex statistical and machine learning techniques. We reflected on these trends earlier,2 and foreshadowed the impact that these developments in the QSAR modeling community would have on many other areas of research. We projected that, with the continuing strong growth of publicly accessible data, this field will become essential for extracting knowledge from, and making predictions with, these massive data sets. We forecast that the field will continue to embrace even more powerful and complex machine learning methods. Furthermore, we expect that these modeling methods will continue to find rapid acceptance not only in chemistry but also in new fields beyond chemistry, where large data sets are readily available and modeling complex relationships between a set of independent variables and given properties of interest are important. The recent expansion of QSAR studies using DL approaches (as discussed in the section on modern trends in QSAR) is an early harbinger of these expectations.We have illustrated some of non-traditional applications in this review, demonstrating how QSAR-like approaches are beginning to yield exciting results in research areas as diverse as quantum mechanics, materials and nanomaterials science, biomaterials, regenerative medicine, and health care. Impressively, many of the roadblocks and technical issues in statistical data modelling employed in different domains of knowledge had already been addressed in the QSAR modeling literature. Examples include papers on the impact of the errors on QSAR analysis311 and the importance of data curation to achieve stable and reproducible models.16 These considerations were under active discussion in the QSAR community before the reproducibility crisis brought to light by the NIH312 and biomedical scientific community at large.313 Similarly, rigorous model validation prior to prediction15 and the importance of rigor in modeling protocols314 have been articulated in several seminal publications in QSAR field315 and have already been adopted as regulatory requirements.99 Extreme examples of the application of QSAR concepts beyond its traditional domain are provided by a study into factors influencing temporal crime patterns in Chicago316 that cites a well-known work on QSAR model validation315 and a study on stock price predictions.317
We expect QSAR-like modeling techniques to continue to expand substantially even beyond the areas where it is starting to make an impact, which we discussed above. Scientists working in this field will continue to experiment with novel statistical, machine learning, and AI algorithms to accelerate the experimental discovery of novel compounds and materials with desired properties. The jury is still out on whether the newest DL approaches will improve the prediction accuracy of QSAR models. However, we expect that the answer will emerge in the next few years, given the tremendous activity in this field.
As discussed above, stunning and potentially paradigm shifting developments are occurring in the use of machine learning approaches to massively accelerate quantum mechanical calculations, without sacrificing accuracy, and the use of QSAR methods for de novo compound design. Another fascinating and emerging direction is AI-driven chemical synthesis route prediction and its synergy with robotic synthesis, also discussed above. We anticipate a multitude of new and interesting algorithmic developments in the area of retro- and forward synthesis design, with software integrated with the robotic systems. We should soon see the emergence of fully autonomous, ‘close loop’ chemical and materials synthesis and optimization systems. In addition to these methodological developments, we foresee many new and impactful experimental methods arising that lead to novel, useful, and safe chemicals when QSAR modeling is applied to these data, and the increased application of ML methodologies in drug target selection, gene–phenotype evaluation and disease modeling. Finally, besides potentially exciting developments in traditional areas of application in chemical sciences, we further expect that the experience in model development, validation, and exploitation of QSAR models for knowledge discovery in chemical sciences will lead to progressive expansion of QSAR modeling principles and approaches in many other disciplines.
Conclusions
This contribution was conceived by a group of scientists who have dedicated significant portions of their professional careers to the development and use of quantitative methods in computational chemistry and molecular modeling. Following the previous highly cited comprehensive survey of QSAR modeling that was coauthored by many contributors to this paper and published in 2014,2 we felt it was time to reflect on the new and exciting developments in QSAR modeling that have emerged in the last five years due to proliferation of large and diverse (Big Data) molecular bioactivity datasets and of burgeoning use of associated Big Data analytical methods such as DL. We also intended to share our observations and excitement concerning the prolific use of similar ML approaches in areas beyond chemical domain; the latter excitement and observations were in part influenced by the transition to other fields that some original cheminformaticians, including several co-authors of this paper, have made in their own research evolution and career development. Herein, we have summarized recent and developing trends in several areas of research where statistical data modeling has begun taking a prominent place and where experiences and generalizable approaches of QSAR modeling could catalyze new discoveries. We hope that this collective contribution will be useful for both specialists in data modeling and experimental researchers looking to expand their toolkits to include computational data analytical approaches.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This review combined a series of separately written, invited contributions from the various coauthors (some sections with multiple coauthors). Primary attributions for the various contributed sections are as follows: Introduction – E. Muratov and A. Tropsha; Chemical similarity – J. Bajorath; Modern trends in QSAR modeling – R. Sheridan; QSAR in chemical safety assessment – I. Tetko; Multi-target profiling and polypharmacology – D. Filimonov and V. Poroikov; QSAR-like approaches in chemical genomics – T. Oprea and A. Cherkasov; QSAR in synthetic organic chemistry – I. Baskin and A. Varnek; Closed-loop discovery and automation – A. Aspuru-Guzik; Machine learning approaches in quantum chemistry – O. Isayev and A. Roitberg; Materials informatics – S. Curtarolo; Nanomaterials informatics – Y. Cohen and D. Fourches; Biomaterials and regenerative medicine – D. Winkler; Clinical and health informatics – D. Agrafiotis and T. Oprea; Outlook and Conclusions – E. Muratov, A. Cherkasov, and A. Tropsha. Final editing was accomplished by A. Tropsha, who also takes primary responsibility for the final content. Mentioning of trade names or commercial products does not constitute endorsement or recommendation for use. The authors acknowledge the seminal contributions of Corwin Hansch and Toshio Fujita in the initial development of the QSAR field and of Frank Burden in the development of sparse feature selection and Bayesian regularization of neural networks for QSAR. The authors acknowledge many fruitful discussions with members of their groups. TIO acknowledge NIH funding support (U24CA224370, U24TR002278, and U01CA239108). AT and EM acknowledge NIH funding support (U01CA207160). VP and DF would like to acknowledge the support of the Russian Program for Basic Research of State Academies of Sciences for 2013–2020. AV, JB and IVT acknowledge funding by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 676434, “Big Data in Chemistry”. AER thanks NSF CHE-1802831 and OI thanks NSF CHE-1802789. DF thanks Army Research Office (ARO) W911NF1810315.Notes and references
- C. Hansch, P. Maloney, T. Fujita and R. Muir, Nature, 1962, 194, 178–180 CrossRef CAS.
- A. Cherkasov, E. N. Muratov, D. Fourches, A. Varnek, I. I. Baskin, M. Cronin, J. C. Dearden, P. Gramatica, Y. C. Martin, R. Todeschini, V. Consonni, V. E. Kuz’min, R. D. Cramer, R. Benigni, C. Yang, J. F. Rathman, L. Terfloth, J. Gasteiger, A. M. Richard and A. Tropsha, J. Med. Chem., 2014, 57, 4977–5010 CrossRef CAS PubMed.
- H. Kubinyi, Drug Discovery Today, 1997, 2, 457–467 CrossRef CAS.
- F. Ban, K. Dalal, H. Li, E. LeBlanc, P. S. Rennie and A. Cherkasov, J. Chem. Inf. Model., 2017, 57, 1018–1028 CrossRef CAS PubMed.
- V. M. Alves, E. N. Muratov, A. Zakharov, N. N. Muratov, C. H. Andrade and A. Tropsha, Food Chem. Toxicol., 2018, 112, 526–534 CrossRef CAS PubMed.
- L. Simón-Vidal, O. García-Calvo, U. Oteo, S. Arrasate, E. Lete, N. Sotomayor and H. González-Díaz, J. Chem. Inf. Model., 2018, 58, 1384–1396 CrossRef PubMed.
- R. Sheridan, W. Schafer, P. Piras, K. Zawatzky, E. C. Sherer, C. Roussel and C. J. Welch, J. Chromatogr. A, 2016, 1467, 206–213 CrossRef CAS PubMed.
- B. A. Grzybowski, S. Szymkuć, E. P. Gajewska, K. Molga, P. Dittwald, A. Wołos and T. Klucznik, Chem, 2018, 4, 390–398 CAS.
- S. J. Capuzzi, W. Sun, E. N. Muratov, C. Martínez-Romero, S. He, W. Zhu, H. Li, G. Tawa, E. G. Fisher, M. Xu, P. Shinn, X. Qiu, A. García-Sastre, W. Zheng and A. Tropsha, J. Med. Chem., 2018, 61, 3582–3594 CrossRef CAS PubMed.
- M. Hong, X. Chen, R. Zhang, D. Wang, S. Shen and V. P. Singh, Ocean Sci., 2018, 14, 301–320 CrossRef.
- D. Ghosh and R. Guha, Comput. Environ. Urban Syst., 2010, 34, 189–203 CrossRef.
- E. N. Muratov, M. Lewis, D. Fourches, A. Tropsha and W. C. Cox, Am. J. Pharm. Educ., 2017, 81, 46 Search PubMed.
- R. Hosseini, N. Newlands, C. Dean, A. Takemura, R. Hosseini, N. K. Newlands, C. B. Dean and A. Takemura, Remote Sens., 2015, 7, 2752–2780 CrossRef.
- T. Oprea, M. Olah, L. Ostopovici, R. Rad and M. Mracec, in EuroQSAR 2002—Designing Drugs and Crop Protectants: Processes Problems and Solutions, ed. M. Ford, D. Livingstone, J. Dearden and H. H. Van de Waterbeemd, Blackwell Publishing, New York, 2003, pp. 314–315 Search PubMed.
- A. Golbraikh and A. Tropsha, J. Mol. Graphics Modell., 2002, 20, 269–276 CrossRef CAS PubMed.
- D. Fourches, E. Muratov and A. Tropsha, Nat. Chem. Biol., 2015, 11, 535 CrossRef CAS PubMed.
- Editorial, Nature, 2014, 515, 7 Search PubMed.
- A. Tropsha, Mol. Inf., 2010, 29, 476–488 CrossRef CAS PubMed.
- D. Lowe, In the pipeline, https://blogs.sciencemag.org/pipeline/archives/2018/01/30/automated-chemistry-a-vision, accessed 19 August 2019.
- T. Fujita and D. A. Winkler, J. Chem. Inf. Model., 2016, 56, 269–274 CrossRef CAS.
- L. Peltason and J. Bajorath, J. Med. Chem., 2007, 50, 5571–5578 CrossRef CAS.
- L. Peltason, P. Iyer and J. Bajorath, J. Chem. Inf. Model., 2010, 50, 1021–1033 CrossRef CAS.
- G. M. Maggiora, J. Chem. Inf. Model., 2006, 46, 1535 CrossRef CAS PubMed.
- M. Kosloff and R. Kolodny, Proteins, 2008, 71, 891–902 CrossRef CAS PubMed.
- J. Bajorath, L. Peltason, M. Wawer, R. Guha, M. S. Lajiness and J. H. Van Drie, Drug Discovery Today, 2009, 14, 698–705 CrossRef CAS PubMed.
- P. Willett, Drug Discovery Today, 2006, 11, 1046–1053 CrossRef CAS PubMed.
- D. Stumpfe and J. Bajorath, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 260–282 CAS.
- Y. Hu, D. Stumpfe and J. Bajorath, J. Chem. Inf. Model., 2011, 51, 1742–1753 CrossRef CAS.
- P. Englert and P. Kovács, J. Chem. Inf. Model., 2015, 55, 941–955 CrossRef CAS PubMed.
- E. Griffen, A. G. Leach, G. R. Robb and D. J. Warner, J. Med. Chem., 2011, 54, 7739–7750 CrossRef CAS PubMed.
- J. Hussain and C. Rea, J. Chem. Inf. Model., 2010, 50, 339–348 CrossRef CAS PubMed.
- X. Hu, Y. Hu, M. Vogt, D. Stumpfe and J. Bajorath, J. Chem. Inf. Model., 2012, 52, 1138–1145 CrossRef CAS PubMed.
- D. Stumpfe, D. Dimova and J. Bajorath, J. Med. Chem., 2016, 59, 7667–7676 CrossRef CAS PubMed.
- G. Schneider, W. Neidhart, T. Giller and G. Schmid, Angew. Chem., Int. Ed., 1999, 38, 2894–2896 CrossRef CAS.
- Y.-C. Lo, S. E. Rensi, W. Torng and R. B. Altman, Drug Discovery Today, 2018, 23, 1538–1546 CrossRef CAS PubMed.
- G. Maggiora, M. Vogt, D. Stumpfe and J. Bajorath, J. Med. Chem., 2014, 57, 3186–3204 CrossRef CAS PubMed.
- T. P. A. B. Paracelsus, Opera Omnia Medico-Chemico-Chirurgica, tribus voluminibus comprehensa, Sumptibus Joan. Antonii, & Samuelis De Tournes, Geneva, Editio nov., 1658.
- A. Lavecchia, Drug Discovery Today, 2015, 20, 318–331 CrossRef PubMed.
- S.-A. Sansone, P. Rocca-Serra, D. Field, E. Maguire, C. Taylor, O. Hofmann, H. Fang, S. Neumann, W. Tong, L. Amaral-Zettler, K. Begley, T. Booth, L. Bougueleret, G. Burns, B. Chapman, T. Clark, L.-A. Coleman, J. Copeland, S. Das, A. de Daruvar, P. de Matos, I. Dix, S. Edmunds, C. T. Evelo, M. J. Forster, P. Gaudet, J. Gilbert, C. Goble, J. L. Griffin, D. Jacob, J. Kleinjans, L. Harland, K. Haug, H. Hermjakob, S. J. H. Sui, A. Laederach, S. Liang, S. Marshall, A. McGrath, E. Merrill, D. Reilly, M. Roux, C. E. Shamu, C. A. Shang, C. Steinbeck, A. Trefethen, B. Williams-Jones, K. Wolstencroft, I. Xenarios and W. Hide, Nat. Genet., 2012, 44, 121 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–D1107 Search PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2015, 44, D1202–D1213 CrossRef PubMed.
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed.
- R. P. Sheridan, J. Chem. Inf. Model., 2013, 53, 783–790 CrossRef CAS.
- E. N. Muratov, E. V. Varlamova, A. G. Artemenko, P. G. Polishchuk and V. E. Kuz’min, Mol. Inf., 2012, 31, 202–221 CrossRef CAS.
- I. Oprisiu, E. Varlamova, E. Muratov, A. Artemenko, G. Marcou, P. Polishchuk, V. Kuz’min and A. Varnek, Mol. Inf., 2012, 31, 491–502 CrossRef CAS PubMed.
- A. V. Zakharov, E. V. Varlamova, A. A. Lagunin, A. V. Dmitriev, E. N. Muratov, D. Fourches, V. E. Kuz’min, V. V. Poroikov, A. Tropsha and M. C. Nicklaus, Mol. Pharmaceutics, 2016, 13, 545–556 CrossRef CAS PubMed.
- M. D. Segall, Curr. Drug Metab., 2012, 18, 1292–1310 CrossRef CAS.
- F. J. Prado-Prado, H. González-Díaz, O. M. de la Vega, F. M. Ubeira and K.-C. Chou, Bioorg. Med. Chem., 2008, 16, 5871–5880 CrossRef CAS PubMed.
- J. B. Brown, Y. Okuno, G. Marcou, A. Varnek and D. Horvath, J. Comput.-Aided Mol. Des., 2014, 28, 597–618 CrossRef CAS PubMed.
- G. J. P. Van Westen, J. K. Wegner, A. P. Ijzerman, H. W. T. Van Vlijmen and A. Bender, MedChemComm, 2011, 2, 16–30 RSC.
- DREAM Challenges, IDG-DREAM Drug-Kinase Binding Prediction Challenge – Dream Challenges, http://dreamchallenges.org/project/idg-dream-drug-kinase-binding-prediction-challenge/, accessed 1 January 2020.
- L. Eriksson, J. Jaworska, A. P. Worth, M. T. D. Cronin, R. M. McDowell and P. Gramatica, Environ. Health Perspect., 2003, 111, 1361–1375 CrossRef CAS PubMed.
- T. Scior, J. L. Medina-Franco, Q.-T. Do, K. Martínez-Mayorga, J. a. Yunes Rojas and P. Bernard, Curr. Med. Chem., 2009, 16, 4297–4313 CrossRef CAS PubMed.
- O. Méndez-Lucio and J. L. Medina-Franco, Drug Discovery Today, 2017, 22, 120–126 CrossRef PubMed.
- A. Golbraikh, E. Muratov, D. Fourches and A. Tropsha, J. Chem. Inf. Model., 2014, 54, 1–4 CrossRef CAS PubMed.
- R. D. Cramer III, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef PubMed.
- V. E. Kuz’min, E. N. Muratov, A. G. Artemenko, L. Gorb, M. Qasim and J. Leszczynski, J. Comput.-Aided Mol. Des., 2008, 22, 747–759 CrossRef PubMed.
- P. Polishchuk, J. Chem. Inf. Model., 2017, 57, 2618–2639 CrossRef CAS PubMed.
- F. R. Burden and D. A. Winkler, QSAR Comb. Sci., 2009, 28, 645–653 CrossRef CAS.
- A. Artemenko, E. Muratov, V. Kuz’min, N. Kovdienko, A. Hromov, V. Makarov, O. Riabova, P. Wutzler and M. Schmidtke, J. Antimicrob. Chemother., 2007, 60, 68–77 CrossRef CAS PubMed.
- P. Polishchuk, V. Kuz’min, A. Artemenko and E. Muratov, Mol. Inf., 2013, 32, 843–853 CrossRef CAS PubMed.
- R. P. Sheridan, J. Chem. Inf. Model., 2019, 59, 1324–1337 CrossRef CAS PubMed.
- C. Hansch, Acc. Chem. Res., 1993, 26, 147–153 CrossRef CAS.
- S. Hochreiter, G. Klambauer and M. Rarey, J. Chem. Inf. Model., 2018, 58, 1723–1724 CrossRef PubMed.
- A. C. Mater and M. L. Coote, J. Chem. Inf. Model., 2019, 59, 2545–2559 CrossRef CAS PubMed.
- J. Ma, R. P. Sheridan, A. Liaw, G. E. Dahl and V. Svetnik, J. Chem. Inf. Model., 2015, 55, 263–274 CrossRef CAS PubMed.
- MERCK, Kaggle Merck Molecular Activity Challenge, https://www.kaggle.com/c/MerckActivity, accessed 19 August 2019.
- T. Cover and P. Hart, IEEE Trans. Inf. Theory, 1967, 13, 21–27 Search PubMed.
- S. Wold, M. Sjöström and L. Eriksson, Chemom. Intell. Lab. Syst., 2001, 58, 109–130 CrossRef CAS.
- H. Geppert, T. Horváth, T. Gärtner, S. Wrobel and J. Bajorath, J. Chem. Inf. Model., 2008, 48, 742–746 CrossRef CAS PubMed.
- F. R. Burden and D. A. Winkler, J. Chem. Inf. Model., 2015, 55, 1529–1534 CrossRef CAS PubMed.
- L. E. O. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- R. Dudley, J. Funct. Anal., 1967, 1, 290–330 CrossRef.
- V. Svetnik, T. Wang, C. Tong, A. Liaw, R. P. Sheridan and Q. Song, J. Chem. Inf. Model., 2005, 45, 786–799 CrossRef CAS PubMed.
- R. P. Sheridan, J. Chem. Inf. Model., 2013, 53, 2837–2850 CrossRef CAS PubMed.
- R. P. Sheridan, W. M. Wang, A. Liaw, J. Ma and E. M. Gifford, J. Chem. Inf. Model., 2016, 56, 2353–2360 CrossRef CAS PubMed.
- G. Ke, Q. Meng, T. Finley, T. Wang, W. Chen, W. Ma, Q. Ye and T.-Y. Liu, in Proceedings of the 31st International Conference on Neural Information Processing Systems, NIPS, Long Beach, 2017, pp. 3149–3157.
- E. B. Lenselink, N. ten Dijke, B. Bongers, G. Papadatos, H. W. T. van Vlijmen, W. Kowalczyk, A. P. IJzerman and G. J. P. van Westen, J. Cheminf., 2017, 9, 45 Search PubMed.
- D. A. Winkler and T. C. Le, Mol. Inf., 2017, 36, 1600118 CrossRef PubMed.
- G. Cybenko, Math. Control, Signals, Syst., 1989, 2, 303–314 CrossRef.
- A. Golbraikh, D. Fourches, A. Sedykh, E. Muratov, I. Liepina and A. Tropsha, in Practical Aspects of Computational Chemistry III, ed. J. Leszcynski and M. Shukla, Springer, New York, Heidelberg, Dordrecht, London, 2014, pp. 187–230 Search PubMed.
- B. Ramsundar, B. Liu, Z. Wu, A. Verras, M. Tudor, R. P. Sheridan and V. Pande, J. Chem. Inf. Model., 2017, 57, 2068–2076 CrossRef CAS PubMed.
- A. Varnek, C. Gaudin, G. Marcou, I. Baskin, A. K. Pandey and I. V. Tetko, J. Chem. Inf. Model., 2009, 49, 133–144 CrossRef CAS PubMed.
- Y. Xu, J. Ma, A. Liaw, R. P. Sheridan and V. Svetnik, J. Chem. Inf. Model., 2017, 57, 2490–2504 CrossRef CAS PubMed.
- C. W. Coley, R. Barzilay, W. H. Green, T. S. Jaakkola and K. F. Jensen, J. Chem. Inf. Model., 2017, 57, 1757–1772 CrossRef CAS PubMed.
- F. A. Faber, L. Hutchison, B. Huang, J. Gilmer, S. S. Schoenholz, G. E. Dahl, O. Vinyals, S. Kearnes, P. F. Riley and O. A. von Lilienfeld, J. Chem. Theory Comput., 2017, 13, 5255–5264 CrossRef CAS PubMed.
- D. Merk, L. Friedrich, F. Grisoni and G. Schneider, Mol. Inf., 2018, 37, 1700153 CrossRef PubMed.
- M. F. Dacrema, P. Cremonesi and D. Jannach, in Proceedings of the 13th ACM Conference on Recommender Systems – RecSys’19, ACM Press, New York, 2019, pp. 101–109.
- S. Bach, A. Binder, G. Montavon, F. Klauschen, K.-R. Müller and W. Samek, PLoS One, 2015, 10, e0130140 CrossRef PubMed.
- I. I. Baskin, D. Winkler and I. V. Tetko, Expert Opin. Drug Discovery, 2016, 11, 785–795 CrossRef CAS PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. M. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O’Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, R. Ross, B. Roth, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed.
- M. E. Garcia Denegri, S. Bustillo, C. C. Gay, A. Van De Velde, G. Gomez, S. Echeverría, M. D. C. Gauna Pereira, S. Maruñak, S. Nuñez, F. Bogado, M. Sanchez, G. P. Teibler, L. Fusco and L. C. A. Leiva, Curr. Top. Med. Chem., 2019, 19, 1962–1980 CrossRef PubMed.
- G. J. Myatt, E. Ahlberg, Y. Akahori, D. Allen, A. Amberg, L. T. Anger, A. Aptula, S. Auerbach, L. Beilke, P. Bellion, R. Benigni, J. Bercu, E. D. Booth, D. Bower, A. Brigo, N. Burden, Z. Cammerer, M. T. D. Cronin, K. P. Cross, L. Custer, M. Dettwiler, K. Dobo, K. A. Ford, M. C. Fortin, S. E. Gad-McDonald, N. Gellatly, V. Gervais, K. P. Glover, S. Glowienke, J. Van Gompel, S. Gutsell, B. Hardy, J. S. Harvey, J. Hillegass, M. Honma, J.-H. Hsieh, C.-W. Hsu, K. Hughes, C. Johnson, R. Jolly, D. Jones, R. Kemper, M. O. Kenyon, M. T. Kim, N. L. Kruhlak, S. A. Kulkarni, K. Kümmerer, P. Leavitt, B. Majer, S. Masten, S. Miller, J. Moser, M. Mumtaz, W. Muster, L. Neilson, T. I. Oprea, G. Patlewicz, A. Paulino, E. Lo Piparo, M. Powley, D. P. Quigley, M. V. Reddy, A.-N. Richarz, P. Ruiz, B. Schilter, R. Serafimova, W. Simpson, L. Stavitskaya, R. Stidl, D. Suarez-Rodriguez, D. T. Szabo, A. Teasdale, A. Trejo-Martin, J.-P. Valentin, A. Vuorinen, B. A. Wall, P. Watts, A. T. White, J. Wichard, K. L. Witt, A. Woolley, D. Woolley, C. Zwickl and C. Hasselgren, Regul. Toxicol. Pharmacol., 2018, 96, 1–17 CrossRef CAS PubMed.
- G. T. Ankley, R. S. Bennett, R. J. Erickson, D. J. Hoff, M. W. Hornung, R. D. Johnson, D. R. Mount, J. W. Nichols, C. L. Russom, P. K. Schmieder, J. a. Serrrano, J. E. Tietge and D. L. Villeneuve, Environ. Toxicol. Chem., 2010, 29, 730–741 CrossRef CAS PubMed.
- M. E. Pittman, S. W. Edwards, C. Ives and H. M. Mortensen, Toxicol. Appl. Pharmacol., 2018, 343, 71–83 CrossRef CAS PubMed.
- A. Rybacka, C. Rudén, I. V. Tetko and P. L. Andersson, Chemosphere, 2015, 139, 372–378 CrossRef CAS PubMed.
- C. Wittwehr, H. Aladjov, G. Ankley, H. J. Byrne, J. de Knecht, E. Heinzle, G. Klambauer, B. Landesmann, M. Luijten, C. MacKay, G. Maxwell, M. E. Meek, A. Paini, E. Perkins, T. Sobanski, D. Villeneuve, K. M. Waters and M. Whelan, Toxicol. Sci, 2017, 155, 326–336 CrossRef CAS PubMed.
- US EPA, Tox21, http://www.epa.gov/ncct/Tox21/, accessed 20 August 2019.
- Organisation for Economic Co-operation and Development and OECD, OECD principles for the validation, for regulatory purposes, of (Quantitative) Structure–Activity Relationship models, http://europa.eu.int/comm/environment/chemicals/reach.htm, accessed 17 February 2017.
- R. Huang, M. Xia, D.-T. Nguyen, T. Zhao, S. Sakamuru, J. Zhao, S. A. Shahane, A. Rossoshek and A. Simeonov, Front. Environ. Sci. Eng., 2016, 3, 85 Search PubMed.
- A. Mayr, G. Klambauer, T. Unterthiner and S. Hochreiter, Front. Environ. Sci. Eng., 2016, 3, 80 Search PubMed.
- I. V. Tetko, Methods in molecular biology, Humana Press, Clifton, 2008, vol. 458, pp. 180–197 Search PubMed.
- Y. Wu and G. Wang, Int. J. Mol. Sci., 2018, 19, 2358 CrossRef PubMed.
- K. Mansouri, A. Abdelaziz, A. Rybacka, A. Roncaglioni, A. Tropsha, A. Varnek, A. Zakharov, A. Worth, A. M. Richard, C. M. Grulke, D. Trisciuzzi, D. Fourches, D. Horvath, E. Benfenati, E. Muratov, E. B. Wedebye, F. Grisoni, G. F. Mangiatordi, G. M. Incisivo, H. Hong, H. W. Ng, I. V. Tetko, I. Balabin, J. Kancherla, J. Shen, J. Burton, M. Nicklaus, M. Cassotti, N. G. Nikolov, O. Nicolotti, P. L. Andersson, Q. Zang, R. Politi, R. D. Beger, R. Todeschini, R. Huang, S. Farag, S. A. Rosenberg, S. Slavov, X. Hu and R. S. Judson, Environ. Health Perspect., 2016, 124, 1023–1033 CrossRef CAS PubMed.
- Z. Wang, M. Gerstein and M. Snyder, Nat. Rev. Genet., 2009, 10, 57–63 CrossRef CAS PubMed.
- R. Liu, X. Yu and A. Wallqvist, J. Cheminf., 2015, 7, 4 Search PubMed.
- S. Novotarskyi, A. Abdelaziz, Y. Sushko, R. Körner, J. Vogt and I. V. Tetko, Chem. Res. Toxicol., 2016, 29, 768–775 Search PubMed.
- M. Jamei, Curr. Pharmacol. Rep., 2016, 2, 161–169 Search PubMed.
- B. A. Wetmore, J. F. Wambaugh, S. S. Ferguson, M. A. Sochaski, D. M. Rotroff, K. Freeman, H. J. Clewell, D. J. Dix, M. E. Andersen, K. A. Houck, B. Allen, R. S. Judson, R. Singh, R. J. Kavlock, A. M. Richard and R. S. Thomas, Toxicol. Sci, 2012, 125, 157–174 CrossRef CAS PubMed.
- T. I. Oprea, A. Tropsha, J.-L. L. Faulon and M. D. Rintoul, Nat. Chem. Biol., 2007, 3, 447–450 CrossRef CAS PubMed.
- J. Yamane, S. Aburatani, S. Imanishi, H. Akanuma, R. Nagano, T. Kato, H. Sone, S. Ohsako and W. Fujibuchi, Nucleic Acids Res., 2016, 44, 5515–5528 CrossRef PubMed.
- A. Abdelaziz, Y. Sushko, S. Novotarskyi, R. Körner, S. Brandmaier and I. V. Tetko, Comb. Chem. High Throughput Screening, 2015, 18, 420–438 CrossRef CAS PubMed.
- S. Sosnin, D. Karlov, I. V. Tetko and M. V. Fedorov, J. Chem. Inf. Model., 2019, 59, 1062–1072 CrossRef CAS PubMed.
- V. M. Alves, E. N. Muratov, S. J. Capuzzi, R. Politi, Y. Low, R. C. Braga, A. V. Zakharov, A. Sedykh, E. Mokshyna, S. Farag, C. H. Andrade, V. E. Kuz’min, D. Fourches and A. Tropsha, Green Chem., 2016, 18, 4348–4360 RSC.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- Y. S. Low, V. M. Alves, D. Fourches, A. Sedykh, C. H. Andrade, E. N. Muratov, I. Rusyn and A. Tropsha, J. Chem. Inf. Model., 2018, 58, 2203–2213 CrossRef CAS PubMed.
- G. Montavon, W. Samek and K.-R. Müller, Digit. Signal Process., 2018, 73, 1–15 CrossRef.
- B. L. Roth, D. J. Sheffler and W. K. Kroeze, Nat. Rev. Drug Discovery, 2004, 3, 353–359 CrossRef CAS PubMed.
- A. Lagunin, D. Filimonov and V. Poroikov, Curr. Pharm. Des., 2010, 16, 1703–1717 CrossRef CAS PubMed.
- S. M. Ivanov, A. A. Lagunin and V. V. Poroikov, Drug Discovery Today, 2016, 21, 58–71 CrossRef CAS PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- O. A. Tarasova, A. F. Urusova, D. A. Filimonov, M. C. Nicklaus, A. V. Zakharov and V. V. Poroikov, J. Chem. Inf. Model., 2015, 55, 1388–1399 CrossRef CAS PubMed.
- T. Scior, A. Bender, G. Tresadern, J. L. Medina-Franco, K. Martínez-Mayorga, T. Langer, K. Cuanalo-Contreras and D. K. Agrafiotis, J. Chem. Inf. Model., 2012, 52, 867–881 CrossRef CAS PubMed.
- A. Lagunin, A. Stepanchikova, D. Filimonov and V. Poroikov, Bioinformatics, 2000, 16, 747–748 CrossRef CAS PubMed.
- D. A. Filimonov, V. V. Poroĭkov, E. I. Karaicheva, R. K. Kazarian, A. P. Budunova, E. M. Mikhaĭlovskiĭ, A. V. Rudnitskikh, L. V. Goncharenko and I. V. Burov, Eksp. Klin. Farmakol., 1995, 58, 56–62 CAS.
- P. V. Pogodin, A. A. Lagunin, A. V. Rudik, D. A. Filimonov, D. S. Druzhilovskiy, M. C. Nicklaus and V. V. Poroikov, Front. Chem., 2018, 6, 133 CrossRef PubMed.
- H. González-Díaz, S. Arrasate, A. Gómez-SanJuan, N. Sotomayor, E. Lete, L. Besada-Porto and J. M. Ruso, Curr. Top. Med. Chem., 2013, 13, 1713–1741 CrossRef PubMed.
- R. C. Glen, A. Bender, C. H. Arnby, L. Carlsson, S. Boyer and J. Smith, IDrugs, 2006, 9, 199–204 CAS.
- A. Bender, H. Y. Mussa, R. C. Glen and S. Reiling, J. Chem. Inf. Comput. Sci., 2004, 44, 1708–1718 CrossRef CAS PubMed.
- Y. C. Martin, J. L. Kofron and L. M. Traphagen, J. Med. Chem., 2002, 45, 4350–4358 CrossRef CAS PubMed.
- R. P. Sheridan and S. K. Kearsley, Drug Discovery Today, 2002, 7, 903–911 CrossRef PubMed.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Nat. Biotechnol., 2007, 25, 197–206 CrossRef CAS PubMed.
- M. Luo, X. S. Wang, B. L. Roth, A. Golbraikh and A. Tropsha, J. Chem. Inf. Model., 2014, 54, 634–647 CrossRef CAS PubMed.
- H. Luo, J. Chen, L. Shi, M. Mikailov, H. Zhu, K. Wang, L. He and L. Yang, Nucleic Acids Res., 2011, 39, W492–W498 CrossRef CAS PubMed.
- D. K. Agrafiotis, V. S. Lobanov and F. R. Salemme, Nat. Rev. Drug Discovery, 2002, 1, 337–346 CrossRef CAS PubMed.
- D. Gupta-Ostermann and J. Bajorath, F1000Research, 2014, 3, 113 Search PubMed.
- E. March-Vila, L. Pinzi, N. Sturm, A. Tinivella, O. Engkvist, H. Chen and G. Rastelli, Front. Pharmacol., 2017, 8, 298 CrossRef PubMed.
- M. Lapinsh, P. Prusis, A. Gutcaits, T. Lundstedt and J. E. Wikberg, Biochim. Biophys. Acta, 2001, 1525, 180–190 CrossRef CAS.
- M. Lapins, A. Worachartcheewan, O. Spjuth, V. Georgiev, V. Prachayasittikul, C. Nantasenamat and J. E. S. Wikberg, PLoS One, 2013, 8, e66566 CrossRef CAS PubMed.
- S. Paricharak, I. Cortés-Ciriano, A. P. IJzerman, T. E. Malliavin and A. Bender, J. Cheminf., 2015, 7, 15 Search PubMed.
- S. Orchard, B. Al-Lazikani, S. Bryant, D. Clark, E. Calder, I. Dix, O. Engkvist, M. Forster, A. Gaulton, M. Gilson, R. Glen, M. Grigorov, K. Hammond-Kosack, L. Harland, A. Hopkins, C. Larminie, N. Lynch, R. K. Mann, P. Murray-Rust, E. Lo Piparo, C. Southan, C. Steinbeck, D. Wishart, H. Hermjakob, J. Overington and J. Thornton, Nat. Rev. Drug Discovery, 2011, 10, 661–669 CrossRef CAS PubMed.
- J.-L. Reymond, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- T. I. Oprea, C. G. Bologa, B. S. Edwards, E. R. Prossnitz and L. A. Sklar, J. Biomol. Screening, 2005, 10, 419–426 CrossRef CAS PubMed.
- The Gene Ontology Consortium, Nucleic Acids Res., 2017, 45, D331–D338 CrossRef PubMed.
- M. Hsing, K. Byler and A. Cherkasov, BMC Syst. Biol., 2008, 2, 80 CrossRef PubMed.
- A. Sedykh, H. Zhu, H. Tang, L. Zhang, A. Richard, I. Rusyn and A. Tropsha, Environ. Health Perspect., 2011, 119, 364–370 CrossRef CAS PubMed.
- C. G. Bologa, O. Ursu, L. Halip, R. Curpăn and T. I. Oprea, Rev. Roum. Chim., 2015, 60, 219–226 Search PubMed.
- G. Woo, M. Fernandez, M. Hsing, N. A. Lack, A. D. Cavga and A. Cherkasov, Bioinformatics, 2020, 36, 813–818 Search PubMed.
- D. S. Himmelstein and S. E. Baranzini, PLoS Comput. Biol., 2015, 11, e1004259 CrossRef PubMed.
- The UniProt Consortium, Nucleic Acids Res., 2017, 45, D158–D169 CrossRef PubMed.
- M. Kanehisa, M. Furumichi, M. Tanabe, Y. Sato and K. Morishima, Nucleic Acids Res., 2017, 45, D353–D361 CrossRef CAS PubMed.
- T. Chen and C. Guestrin, in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining – KDD’16, ACM Press, New York, New York, USA, 2016, pp. 785–794.
- P. Agarwal and D. B. Searls, Nat. Rev. Drug Discovery, 2009, 8, 865–878 CrossRef CAS PubMed.
- D.-T. Nguyen, S. Mathias, C. Bologa, S. Brunak, N. Fernandez, A. Gaulton, A. Hersey, J. Holmes, L. J. Jensen, A. Karlsson, G. Liu, A. Ma’ayan, G. Mandava, S. Mani, S. Mehta, J. Overington, J. Patel, A. D. Rouillard, S. Schürer, T. Sheils, A. Simeonov, L. A. Sklar, N. Southall, O. Ursu, D. Vidovic, A. Waller, J. Yang, A. Jadhav, T. I. Oprea and R. Guha, Nucleic Acids Res., 2017, 45, D995–D1002 CrossRef CAS PubMed.
- T. I. Oprea, C. G. Bologa, S. Brunak, A. Campbell, G. N. Gan, A. Gaulton, S. M. Gomez, R. Guha, A. Hersey, J. Holmes, A. Jadhav, L. J. Jensen, G. L. Johnson, A. Karlson, A. R. Leach, A. Ma’ayan, A. Malovannaya, S. Mani, S. L. Mathias, M. T. McManus, T. F. Meehan, C. von Mering, D. Muthas, D.-T. Nguyen, J. P. Overington, G. Papadatos, J. Qin, C. Reich, B. L. Roth, S. C. Schürer, A. Simeonov, L. A. Sklar, N. Southall, S. Tomita, I. Tudose, O. Ursu, D. Vidović, A. Waller, D. Westergaard, J. J. Yang and G. Zahoránszky-Köhalmi, Nat. Rev. Drug Discovery, 2018, 17, 317–332 CrossRef CAS PubMed.
- J. Gasteiger, J. Comput.-Aided Mol. Des., 2007, 21, 33–52 CrossRef CAS PubMed.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- Elsevier, 2018. “Reaxys Fact Sheet.”.
- D. M. Lowe, Doctoral thesis, University of Cambridge, 2012.
- A. I. Lin, T. I. Madzhidov, O. Klimchuk, R. I. Nugmanov, I. S. Antipin and A. Varnek, J. Chem. Inf. Model., 2016, 56, 2140–2148 CrossRef CAS PubMed.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS.
- P. Polishchuk, T. Madzhidov, T. Gimadiev, A. Bodrov, R. Nugmanov and A. Varnek, J. Comput.-Aided Mol. Des., 2017, 31, 829–839 CrossRef CAS.
- H. Patel, M. J. Bodkin, B. Chen and V. J. Gillet, J. Chem. Inf. Model., 2009, 49, 1163–1184 CrossRef CAS PubMed.
- F. Hoonakker, N. Lachiche, A. Varnek and A. Wagner, Int. J. Artif. Intell. Tools, 2010, 20, 253–270 CrossRef.
- A. Varnek, D. Fourches, F. Hoonakker and V. P. Solov’ev, J. Comput.-Aided Mol. Des., 2005, 19, 693–703 CrossRef CAS PubMed.
- M. Kowalik, C. M. Gothard, A. M. Drews, N. A. Gothard, A. Weckiewicz, P. E. Fuller, B. A. Grzybowski and K. J. M. Bishop, Angew. Chem., Int. Ed., 2012, 51, 7928–7932 CrossRef CAS PubMed.
- L. Chen and J. Gasteiger, J. Am. Chem. Soc., 1997, 119, 4033–4042 CrossRef CAS.
- L. Chen and J. Gasteiger, Angew. Chem., Int. Ed. Engl., 1996, 35, 763–765 CrossRef CAS.
- E. J. Corey, Chem. Soc. Rev., 1988, 17, 111–133 RSC.
- M. H. S. Segler and M. P. Waller, Chem. – Eur. J., 2017, 23, 5966–5971 CrossRef CAS PubMed.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- M. A. Kayala, C.-A. Azencott, J. H. Chen and P. Baldi, J. Chem. Inf. Model., 2011, 51, 2209–2222 CrossRef CAS PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed.
- P. Karpov, G. Godin and I. V. Tetko, Artificial Neural Networks and Machine Learning – ICANN 2019: Workshop and Special Sessions, Springer, Cham, 2019, pp. 817–830 Search PubMed.
- M. Hartenfeller, H. Zettl, M. Walter, M. Rupp, F. Reisen, E. Proschak, S. Weggen, H. Stark and G. Schneider, PLoS Comput. Biol., 2012, 8, e1002380 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2018, 58, 252–261 CrossRef CAS PubMed.
- R. W. Taft, J. Am. Chem. Soc., 1952, 74, 3120–3128 CrossRef CAS.
- O. Engkvist, P.-O. Norrby, N. Selmi, Y. Lam, Z. Peng, E. C. Sherer, W. Amberg, T. Erhard and L. A. Smyth, Drug Discovery Today, 2018, 23, 1203–1218 CrossRef CAS PubMed.
- R. I. Nugmanov, T. I. Madzhidov, G. R. Khaliullina, I. I. Baskin, I. S. Antipin and A. A. Varnek, J. Struct. Chem., 2014, 55, 1026–1032 CrossRef CAS.
- M. Glavatskikh, T. Madzhidov, D. Horvath, R. Nugmanov, T. Gimadiev, D. Malakhova, G. Marcou and A. Varnek, Mol. Inf., 2019, 38, 1800077 CrossRef CAS PubMed.
- T. R. Gimadiev, T. I. Madzhidov, R. I. Nugmanov, I. I. Baskin, I. S. Antipin and A. Varnek, J. Comput.-Aided Mol. Des., 2018, 32, 401–414 CrossRef CAS PubMed.
- G. Marcou, J. Aires de Sousa, D. A. R. S. Latino, A. de Luca, D. Horvath, V. Rietsch and A. Varnek, J. Chem. Inf. Model., 2015, 55, 239–250 CrossRef CAS PubMed.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- F. Hoonakker, N. Lachiche, A. Varnek and A. Wagner, Trends in Applied Intelligent Systems, Pt II, Proceedings, Springer, Berlin, Heidelberg, 2010, vol. 6097, pp. 318–326 Search PubMed.
- G. Schneider, Nat. Rev. Drug Discovery, 2018, 17, 97–113 CrossRef CAS PubMed.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- P. Nikolaev, D. Hooper, F. Webber, R. Rao, K. Decker, M. Krein, J. Poleski, R. Barto and B. Maruyama, npj Comput. Mater., 2016, 2, 16031 CrossRef.
- S. K. Saikin, C. Kreisbeck, D. Sheberla, J. S. Becker and A. Aspuru-Guzik, Expert Opin. Drug Discovery, 2019, 14, 1–4 CrossRef PubMed.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef PubMed.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, Chem. Sci., 2018, 9, 7642–7655 RSC.
- L. M. Roch, F. Häse, C. Kreisbeck, T. Tamayo-Mendoza, L. P. E. Yunker, J. E. Hein and A. Aspuru-Guzik, Sci. Robot., 2018, 3, eaat5559 CrossRef.
- D. P. Tabor, L. M. Roch, S. K. Saikin, C. Kreisbeck, D. Sheberla, J. H. Montoya, S. Dwaraknath, M. Aykol, C. Ortiz, H. Tribukait, C. Amador-Bedolla, C. J. Brabec, B. Maruyama, K. A. Persson and A. Aspuru-Guzik, Nat. Rev. Mater., 2018, 3, 5–20 CrossRef CAS.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, Trends Chem., 2019, 1, 282–291 CrossRef.
- T. C. Le and D. A. Winkler, Chem. Rev., 2016, 116, 6107–6132 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- M. Rupp, A. Tkatchenko, K.-R. Müller and O. A. von Lilienfeld, Phys. Rev. Lett., 2012, 108, 058301 CrossRef PubMed.
- K. Hansen, F. Biegler, R. Ramakrishnan, W. Pronobis, O. A. von Lilienfeld, K.-R. Müller and A. Tkatchenko, J. Phys. Chem. Lett., 2015, 6, 2326–2331 CrossRef CAS PubMed.
- K. Yao, J. E. Herr, S. N. Brown and J. Parkhill, J. Phys. Chem. Lett., 2017, 8, 2689–2694 CrossRef CAS PubMed.
- B. Huang and O. A. Von Lilienfeld, J. Chem. Phys., 2016, 145, 161102 CrossRef PubMed.
- K. Yao, J. E. Herr and J. Parkhill, J. Chem. Phys., 2017, 146, 014106 CrossRef PubMed.
- A. B. Keenan, S. L. Jenkins, K. M. Jagodnik, S. Koplev, E. He, D. Torre, Z. Wang, A. B. Dohlman, M. C. Silverstein, A. Lachmann, M. V. Kuleshov, A. Ma’ayan, V. Stathias, R. Terryn, D. Cooper, M. Forlin, A. Koleti, D. Vidovic, C. Chung, S. C. Schurer, J. Vasiliauskas, M. Pilarczyk, B. Shamsaei, M. Fazel, Y. Ren, W. Niu, N. A. Clark, S. White, N. Mahi, L. Zhang, M. Kouril, J. F. Reichard, S. Sivaganesan, M. Medvedovic, J. Meller, R. J. Koch, M. R. Birtwistle, R. Iyengar, E. A. Sobie, E. U. Azeloglu, J. Kaye, J. Osterloh, K. Haston, J. Kalra, S. Finkbiener, J. Li, P. Milani, M. Adam, R. Escalante-Chong, K. Sachs, A. Lenail, D. Ramamoorthy, E. Fraenkel, G. Daigle, U. Hussain, A. Coye, J. Rothstein, D. Sareen, L. Ornelas, M. Banuelos, B. Mandefro, R. Ho, C. N. Svendsen, R. G. Lim, J. Stocksdale, M. S. Casale, T. G. Thompson, J. Wu, L. M. Thompson, V. Dardov, V. Venkatraman, A. Matlock, J. E. Van Eyk, J. D. Jaffe, M. Papanastasiou, A. Subramanian, T. R. Golub, S. D. Erickson, M. Fallahi-Sichani, M. Hafner, N. S. Gray, J. R. Lin, C. E. Mills, J. L. Muhlich, M. Niepel, C. E. Shamu, E. H. Williams, D. Wrobel, P. K. Sorger, L. M. Heiser, J. W. Gray, J. E. Korkola, G. B. Mills, M. LaBarge, H. S. Feiler, M. A. Dane, E. Bucher, M. Nederlof, D. Sudar, S. Gross, D. F. Kilburn, R. Smith, K. Devlin, R. Margolis, L. Derr, A. Lee and A. Pillai, Cell Syst., 2018, 6, 13–24 CrossRef CAS PubMed.
- K. T. Schütt, H. E. Sauceda, P. J. Kindermans, A. Tkatchenko and K. R. Müller, J. Chem. Phys., 2018, 148, 241722 CrossRef PubMed.
- A. P. Bartók, R. Kondor and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 1–16 Search PubMed.
- S. De, A. P. Bartók, G. Csányi and M. Ceriotti, Phys. Chem. Chem. Phys., 2016, 18, 13754 RSC.
- M. Gastegger, L. Schwiedrzik, M. Bittermann, F. Berzsenyi and P. Marquetand, J. Chem. Phys., 2018, 148, 241709 CrossRef CAS PubMed.
- K. T. Schütt, F. Arbabzadah, S. Chmiela, K. R. Müller and A. Tkatchenko, Nat. Commun., 2017, 8, 13890 CrossRef PubMed.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- M. Gastegger, J. Behler and P. Marquetand, Chem. Sci., 2017, 8, 6924–6935 RSC.
- J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci., 2017, 8, 3192–3203 RSC.
- T. Fink, H. Bruggesser and J. L. Reymond, Angew. Chem., Int. Ed., 2005, 44, 1504–1508 CrossRef CAS PubMed.
- J. S. Smith, B. Nebgen, N. Lubbers, O. Isayev and A. E. Roitberg, J. Chem. Phys., 2018, 148, 241733 CrossRef PubMed.
- K. T. Schütt, P.-J. Kindermans, H. E. Sauceda, S. Chmiela, A. Tkatchenko and K.-R. Müller, Advances in Neural Information Processing System 30, 2017, pp. 992–1002.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
- L. C. Blum and J.-L. Reymond, J. Am. Chem. Soc., 2009, 131, 8732–8733 CrossRef CAS PubMed.
- B. Brauer, M. K. Kesharwani, S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2016, 18, 20905–20925 RSC.
- F. Pulvermüller, Nat. Rev. Neurosci., 2005, 6, 576–582 CrossRef PubMed.
- R. Caruana, Mach. Learn., 1997, 28, 41–75 CrossRef.
- S. Curtarolo, G. L. W. Hart, M. B. Nardelli, N. Mingo, S. Sanvito and O. Levy, Nat. Mater., 2013, 12, 191–201 CrossRef CAS PubMed.
- J. Maddox, Nature, 1988, 335, 201 CrossRef.
- G. Hautier, C. Fischer, V. Ehrlacher, A. Jain and G. Ceder, Inorg. Chem., 2011, 50, 656–663 CrossRef CAS PubMed.
- E. Perim, D. Lee, Y. Liu, C. Toher, P. Gong, Y. Li, W. N. Simmons, O. Levy, J. J. Vlassak, J. Schroers and S. Curtarolo, Nat. Commun., 2016, 7, 12315 CrossRef CAS PubMed.
- L. Ward, S. C. O’Keeffe, J. Stevick, G. R. Jelbert, M. Aykol and C. Wolverton, Acta Mater., 2018, 159, 102–111 CrossRef CAS.
- L. M. Ghiringhelli, J. Vybiral, S. V. Levchenko, C. Draxl and M. Scheffler, Phys. Rev. Lett., 2015, 114, 105503 CrossRef PubMed.
- O. Isayev, D. Fourches, E. N. Muratov, C. Oses, K. Rasch, A. Tropsha and S. Curtarolo, Chem. Mater., 2015, 27, 735–743 CrossRef CAS.
- V. Stanev, C. Oses, A. G. Kusne, E. Rodriguez, J. Paglione, S. Curtarolo and I. Takeuchi, npj Comput. Mater., 2018, 4, 29 CrossRef.
- A. Walsh, Nat. Chem., 2015, 7, 274–275 CrossRef CAS PubMed.
- S. Curtarolo, D. Morgan, K. Persson, J. Rodgers and G. Ceder, Phys. Rev. Lett., 2003, 91, 135503 CrossRef PubMed.
- M. Rupp, A. Tkatchenko, K.-R. Müller and O. A. von Lilienfeld, Phys. Rev. Lett., 2012, 108, 058301 CrossRef PubMed.
- A. P. Bartók, S. De, C. Poelking, N. Bernstein, J. R. Kermode, G. Csányi and M. Ceriotti, Sci. Adv., 2017, 3, e1701816 CrossRef PubMed.
- G. Pilania, A. Mannodi-Kanakkithodi, B. P. Uberuaga, R. Ramprasad, J. E. Gubernatis and T. Lookman, Sci. Rep., 2016, 6, 19375 CrossRef CAS PubMed.
- M. de Jong, W. Chen, R. Notestine, K. Persson, G. Ceder, A. Jain, M. Asta and A. Gamst, Sci. Rep., 2016, 6, 34256 CrossRef CAS PubMed.
- G. K. H. Madsen, J. Am. Chem. Soc., 2006, 128, 12140–12146 CrossRef CAS PubMed.
- F. Legrain, J. Carrete, A. van Roekeghem, S. Curtarolo and N. Mingo, Chem. Mater., 2017, 29, 6220–6227 CrossRef CAS.
- J. Carrete, N. Mingo, S. Wang and S. Curtarolo, Adv. Funct. Mater., 2014, 24, 7427–7432 CrossRef CAS.
- S. Sanvito, C. Oses, J. Xue, A. Tiwari, M. Zic, T. Archer, P. Tozman, M. Venkatesan, M. Coey and S. Curtarolo, Sci. Adv., 2017, 3, e1602241 CrossRef PubMed.
- J. Yong, Y. Jiang, D. Usanmaz, S. Curtarolo, X. Zhang, L. Li, X. Pan, J. Shin, I. Takeuchi and R. L. Greene, Appl. Phys. Lett., 2014, 105, 222403 CrossRef.
- C. Oses, E. Gossett, D. Hicks, F. Rose, M. J. Mehl, E. Perim, I. Takeuchi, S. Sanvito, M. Scheffler, Y. Lederer, O. Levy, C. Toher and S. Curtarolo, J. Chem. Inf. Model., 2018, 58, 2477–2490 CrossRef CAS PubMed.
- W. Körner, G. Krugel, D. F. Urban and C. Elsässer, Scr. Mater., 2018, 154, 295–299 CrossRef.
- J. J. Möller, W. Körner, G. Krugel, D. F. Urban and C. Elsässer, Acta Mater., 2018, 153, 53–61 CrossRef.
- D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, in Advances in Neural Information Processing Systems 28, ed. C. Cortes, Curran Associates, Inc, New York, 2015, pp. 2224–2232 Search PubMed.
- M. Widom, J. Mater. Res., 2018, 33, 2881–2898 CrossRef CAS.
- Y. Lederer, C. Toher, K. S. Vecchio and S. Curtarolo, Acta Mater., 2018, 159, 364–383 CrossRef CAS.
- A. A. Emery, J. E. Saal, S. Kirklin, V. I. Hegde and C. Wolverton, Chem. Mater., 2016, 28, 5621–5634 CrossRef CAS.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Nature, 2018, 556, 43–50 CrossRef CAS PubMed.
- S. A. Tawfik, O. Isayev, C. Stampfl, J. Shapter, D. A. Winkler and M. J. Ford, Adv. Theory Simul., 2019, 2, 1800128 CrossRef.
- A. Jain, G. Hautier, S. P. Ong and K. Persson, J. Mater. Res., 2016, 31, 977–994 CrossRef CAS.
- E. N. Muratov, A. G. Artemenko, E. V. Varlamova, P. G. Polischuk, V. P. Lozitsky, A. S. Fedchuk, R. L. Lozitska, T. L. Gridina, L. S. Koroleva, V. N. Sil’nikov, A. S. Galabov, V. A. Makarov, O. B. Riabova, P. Wutzler, M. Schmidtke and V. E. Kuz’min, Future Med. Chem., 2010, 2, 1205–1226 CrossRef CAS PubMed.
- F. Rose, C. Toher, E. Gossett, C. Oses, M. B. Nardelli, M. Fornari and S. Curtarolo, Comput. Mater. Sci., 2017, 137, 362–370 CrossRef.
- D. Fourches, D. Pu and A. Tropsha, Comb. Chem. High Throughput Screening, 2011, 14, 217–225 CrossRef CAS PubMed.
- D. Fourches, J. Barnes, N. C. Day, P. Bradley, J. Z. Reed and A. Tropsha, Chem. Res. Toxicol., 2010, 23, 171–183 Search PubMed.
- A. Gajewicz, Nanoscale, 2017, 9, 8435–8448 RSC.
- H. Haase and A. Klaessig, EU US Roadmap Nanoinformatics 2030, 2018.
- Turku Centre for Biotechnology, NanoMiner, https://bioscience.fi/, accessed 1 September 2019.
- Nanoinfo.org, NanoDatabank, https://nanoinfo.org/nanodatabank/, accessed 2 September 2019.
- R. L. Marchese Robinson, I. Lynch, W. Peijnenburg, J. Rumble, F. Klaessig, C. Marquardt, H. Rauscher, T. Puzyn, R. Purian, C. Aberg, S. Karcher, H. Vriens, P. Hoet, M. D. Hoover, C. O. Hendren and S. L. Harper, Nanoscale, 2016, 8, 9919–9943 RSC.
- M. S. Ehrenberg, A. E. Friedman, J. N. Finkelstein, G. Oberdörster and J. L. McGrath, Biomaterials, 2009, 30, 603–610 CrossRef CAS PubMed.
- S. Y. Shaw, E. C. Westly, M. J. Pittet, A. Subramanian, S. L. Schreiber and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7387–7392 CrossRef CAS PubMed.
- E. Oh, R. Liu, A. Nel, K. B. Gemill, M. Bilal, Y. Cohen and I. L. Medintz, Nat. Nanotechnol., 2016, 11, 479–486 CrossRef CAS PubMed.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H.-M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175–178 CrossRef CAS PubMed.
- D. Fourches, D. Pu, C. Tassa, R. Weissleder, S. Y. Shaw, R. J. Mumper and A. Tropsha, ACS Nano, 2010, 4, 5703–5712 CrossRef CAS PubMed.
- P. K. Ojha, S. Kar, K. Roy and J. Leszczynski, Nanotoxicology, 2018, 1–21 Search PubMed.
- V. Kovalishyn, N. Abramenko, I. Kopernyk, L. Charochkina, L. Metelytsia, I. V. Tetko, W. Peijnenburg and L. Kustov, Food Chem. Toxicol., 2018, 112, 507–517 CrossRef CAS PubMed.
- V. M. Alves, D. Hwang, E. Muratov, M. Sokolsky-Papkov, E. Varlamova, N. Vinod, C. Lim, C. H. Andrade, A. Tropsha and A. Kabanov, Sci. Adv., 2019, 5, eaav9784 CrossRef PubMed.
- T. X. Trinh, J.-S. Choi, H. Jeon, H.-G. Byun, T.-H. Yoon and J. Kim, Chem. Res. Toxicol., 2018, 31, 183–190 Search PubMed.
- D. Fourches, D. Pu, L. Li, H. Zhou, Q. Mu, G. Su, B. Yan and A. Tropsha, Nanotoxicology, 2016, 10, 374–383 CrossRef CAS PubMed.
- R. Liu, R. Rallo, S. George, Z. Ji, S. Nair, A. E. Nel and Y. Cohen, Small, 2011, 7, 1118–1126 CrossRef CAS PubMed.
- M. K. Ha, T. X. Trinh, J. S. Choi, D. Maulina, H. G. Byun and T. H. Yoon, Sci. Rep., 2018, 8, 3141 CrossRef PubMed.
- E. S. Money, L. E. Barton, J. Dawson, K. H. Reckhow and M. R. Wiesner, Sci. Total Environ, 2014, 473–474, 685–691 CrossRef CAS PubMed.
- R. E. Neapolitan, Mol. Biol., 2003, 6, 674 Search PubMed.
- H. J. P. Marvin, Y. Bouzembrak, E. M. Janssen, M. van der Zande, F. Murphy, B. Sheehan, M. Mullins and H. Bouwmeester, Nanotoxicology, 2017, 11, 123–133 CrossRef CAS PubMed.
- F. Murphy, B. Sheehan, M. Mullins, H. Bouwmeester, H. J. P. Marvin, Y. Bouzembrak, A. L. Costa, R. Das, V. Stone and S. A. M. Tofail, Nanoscale Res. Lett., 2016, 11, 503 CrossRef PubMed.
- C. Cheng and W. P. Worzel, in Genetic Programming Theory and Practice XII, ed. R. Riolo, W. P. Worzel and M. Kotanchek, 2014, pp. 1–15 Search PubMed.
- E. Molina, E. Uriarte, L. Santana, M. Matos and F. Borges, Curr. Bioinf., 2013, 8, 438–451 CrossRef CAS.
- E. Gawehn, J. A. Hiss and G. Schneider, Mol. Inf., 2016, 35, 3–14 CrossRef CAS PubMed.
- P. Mikulskis, M. R. Alexander and D. A. Winkler, Adv. Intell. Syst., 2019, 1900045 CrossRef.
- M. Cilla, I. Pérez-Rey, M. A. Martínez, E. Peña and J. Martínez, Int. J. Numer. Meth. Bio., 2018, 34, e3121 CrossRef PubMed.
- H. Autefage, E. Gentleman, E. Littmann, M. A. B. Hedegaard, T. Von Erlach, M. O’Donnell, F. R. Burden, D. A. Winkler and M. M. Stevens, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4280–4285 CrossRef CAS PubMed.
- Y. H. Huh, M. Noh, F. R. Burden, J. C. Chen, D. A. Winkler and J. L. Sherley, Stem Cell Res., 2015, 14, 144–154 CrossRef CAS PubMed.
- A. L. Hook, D. G. Anderson, R. Langer, P. Williams, M. C. Davies and M. R. Alexander, Biomaterials, 2010, 31, 187–198 CrossRef CAS PubMed.
- J. R. Smith, V. Kholodovych, D. Knight, W. J. Welsh and J. Kohn, QSAR Comb. Sci., 2005, 24, 99–113 CrossRef CAS.
- R. Todeschini and V. Consonni, Handbook of Molecular Descriptors, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2000, vol. 11 Search PubMed.
- A. L. Hook, C. Y. Chang, J. Yang, J. Luckett, A. Cockayne, S. Atkinson, Y. Mei, R. Bayston, D. J. Irvine, R. Langer, D. G. Anderson, P. Williams, M. C. Davies and M. R. Alexander, Nat. Biotechnol., 2012, 30, 868–875 CrossRef CAS PubMed.
- A. D. Celiz, J. G. W. Smith, R. Langer, D. G. Anderson, D. A. Winkler, D. A. Barrett, M. C. Davies, L. E. Young, C. Denning and M. R. Alexander, Nat. Mater., 2014, 13, 570–579 CrossRef CAS PubMed.
- V. C. Epa, J. Yang, Y. Mei, A. L. Hook, R. Langer, D. G. Anderson, M. C. Davies, M. R. Alexander and D. A. Winkler, J. Mater. Chem, 2012, 22, 20902–20906 RSC.
- S. Rasi Ghaemi, B. Delalat, S. Gronthos, M. R. Alexander, D. A. Winkler, A. L. Hook and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2018, 10, 38739–38748 CrossRef CAS PubMed.
- R. M. T. Madiona, S. E. Bamford, D. A. Winkler, B. W. Muir and P. J. Pigram, Anal. Chem., 2018, 90, 12475–12484 CrossRef CAS PubMed.
- R. M. T. Madiona, N. G. Welch, S. B. Russell, D. A. Winkler, J. A. Scoble, B. W. Muir and P. J. Pigram, Surf. Interface Anal., 2018, 50, 713–728 CrossRef CAS.
- 22nd EuroQSAR—Discngine - Enhancing Life Science Research, https://www.discngine.com/events1/2018/9/16/22nd-euroqsar, accessed 1 January 2020.
- H. I. Krebs, M. Krams, D. K. Agrafiotis, A. DiBernardo, J. C. Chavez, G. S. Littman, E. Yang, G. Byttebier, L. Dipietro, A. Rykman, K. McArthur, K. Hajjar, K. R. Lees and B. T. Volpe, Stroke, 2014, 45, 200–204 CrossRef PubMed.
- S. R. Evans, J. Exp. Stroke Transl. Med., 2010, 3, 19–27 CrossRef PubMed.
- A. R. Fugl-Meyer, L. Jääskö, I. Leyman, S. Olsson and S. Steglind, Scand. J. Rehabil. Med., 1975, 7, 13–31 CAS.
- J. Gregson, M. J. Leathley, A. P. Moore, T. L. Smith, A. K. Sharma and C. L. Watkins, Age Ageing, 2000, 29, 223–228 CrossRef CAS PubMed.
- NIH, NIH Stroke Scale, https://www.stroke.nih.gov/resources/scale.htm, accessed 29 August 2019.
- J. Rankin, Scott. Med. J., 1957, 2, 200–215 CrossRef CAS PubMed.
- H. I. Krebs, B. T. Volpe, M. Ferraro, S. Fasoli, J. Palazzolo, B. Rohrer, L. Edelstein and N. Hogan, Top. Stroke Rehabil., 2002, 8, 54–70 CrossRef CAS PubMed.
- A. C. Lo, P. D. Guarino, L. G. Richards, J. K. Haselkorn, G. F. Wittenberg, D. G. Federman, R. J. Ringer, T. H. Wagner, H. I. Krebs, B. T. Volpe, C. T. Bever, D. M. Bravata, P. W. Duncan, B. H. Corn, A. D. Maffucci, S. E. Nadeau, S. S. Conroy, J. M. Powell, G. D. Huang and P. Peduzzi, N. Engl. J. Med., 2010, 362, 1772–1783 CrossRef CAS PubMed.
- N. Hogan and H. I. Krebs, Prog. Brain Res., 2011, 192, 59–68 Search PubMed.
- T. Flash and N. Hogan, J. Neurosci., 1985, 5, 1688–1703 CrossRef CAS PubMed.
- F. Zhu and D. K. Agrafiotis, J. Comput. Chem., 2007, 28, 1234–1239 CrossRef CAS PubMed.
- S. Izrailev and D. Agrafiotis, J. Chem. Inf. Comput. Sci., 2000, 41, 176–180 CrossRef PubMed.
- D. K. Agrafiotis, W. Cedeno and V. S. Lobanov, J. Chem. Inf. Comput. Sci., 2002, 42, 903–911 CrossRef CAS PubMed.
- D. M. Kerr, R. L. Fulton, K. R. Lees and VISTA Collaborators, Stroke, 2012, 43, 1401–1403 CrossRef PubMed.
- R. Vazquez Guillamet, O. Ursu, G. Iwamoto, P. L. Moseley and T. Oprea, Health Informatics J., 2018, 24, 394–409 CrossRef PubMed.
- B. Burrows, C. M. Fletcher, B. E. Heard, N. L. Jones and J. S. Wootliff, Lancet, 1966, 1, 830–835 CrossRef CAS.
- S. Mirza and R. Benzo, Mayo Clin. Proc., 2017, 92, 1104–1112 CrossRef PubMed.
- R. Taylor, J. Chem. Inf. Model., 1995, 35, 59–67 CrossRef CAS.
- J. MacCuish, C. Nicolaou and N. E. MacCuish, J. Chem. Inf. Comput. Sci., 2001, 41, 134–146 CrossRef CAS PubMed.
- D. Young, D. Martin, R. Venkatapathy, P. Harten, T. Martin, R. Venkatapathy and P. Harten, QSAR Comb. Sci., 2008, 27, 1337–1345 CrossRef CAS.
- F. S. Collins and L. A. Tabak, Nature, 2014, 505, 612–613 CrossRef PubMed.
- M. Baker, Nature, 2016, 533, 452–454 CrossRef CAS PubMed.
- J. C. Dearden, M. T. D. Cronin and K. L. E. Kaiser, SAR QSAR Environ. Res., 2009, 20, 241–266 CrossRef CAS PubMed.
- A. Tropsha, P. Gramatica and V. K. Gombar, QSAR Comb. Sci., 2003, 22, 69–77 CrossRef CAS.
- S. Towers, S. Chen, A. Malik and D. Ebert, PLoS One, 2018, 13, e0205151 CrossRef PubMed.
- G. Sheelapriya and R. Murugesan, Spanish J. Financ. Account./Rev. Española Financ. y Contab., 2017, 46, 189–211 Search PubMed.
- N. Piclin, M. Pintore, C. M. Lanza, A. Scacco, S. Guccione, L. Giurato and J. R. Chrétien, J. Sens. Stud., 2008, 23, 558–569 CrossRef.
- A. G. T. Schut, D. J. Stephens, R. G. H. Stovold, M. Adams and R. L. Craig, Crop Pasture Sci., 2009, 60, 60–70 CrossRef.
- M. Xiao and J. P. Obbard, GCB Bioenergy, 2010, 2, 346–352 CrossRef CAS.
- A. H. Alavi, A. H. Gandomi, M. Modaresnezhad and M. Mousavi, J. Earthq. Eng., 2011, 15, 511–536 CrossRef.
- D. Fourches, E. Muratov and A. Tropsha, J. Chem. Inf. Model., 2010, 50, 1189–1204 CrossRef CAS PubMed.
- M. Antelio, M. G. P. Esteves, D. Schneider and J. M. de Souza, 2012 IEEE International Conference on Systems, Man, and Cybernetics (SMC), IEEE, 2012, pp. 931–936.
- S. M. Mousavi, E. S. Mostafavi and F. Hosseinpour, Comput. Ind. Eng., 2014, 74, 120–128 CrossRef.
- Y. Cao, Y. Jiang, H. Gao, H. Chen, X. Fang, H. Mu and F. Tao, Comput. Electron. Agric., 2014, 106, 49–55 CrossRef.
- J. El Haddad, L. Canioni and B. Bousquet, Spectrochim. Acta, Part A, 2014, 101, 171–182 CrossRef.
- J. C. Dearden, M. T. Cronin and K. L. Kaiser, SAR QSAR Environ. Res., 2009, 20, 241–266 CrossRef CAS.
- J. Ponomarenko, T. Dizhbite, M. Lauberts, A. Viksna, G. Dobele, O. Bikovens and G. Telysheva, BioResources, 2014, 9, 2051–2068 CrossRef.
- A. M. A. Sattar, J. Hydroinf., 2014, 16, 550–571 CrossRef.
- M. Elhakeem and A. M. A. Sattar, Earth Surf. Processes Landforms, 2015, 40, 1216–1226 CrossRef.
- S. Tajeri, E. Sadrossadat and J. B. Bazaz, Int. J. Rock Mech. Min. Sci., 2015, 80, 107–117 CrossRef.
- C. Mundava, A. G. T. Schut, P. Helmholz, R. Stovold, G. Donald and D. W. Lamb, Rangel. J., 2015, 37, 157 CrossRef.
- S. Heitzig, A. Weinebeck and H. Murrenhoff, SAE Int. J. Fuels Lubr., 2015, 8, 549–559 CrossRef CAS.
- T.-T. Pan, D.-W. Sun, J.-H. Cheng and H. Pu, Compr. Rev. Food Sci. Food Saf., 2016, 15, 529–541 CrossRef.
- E. Malaj, G. Guénard, R. B. Schäfer and P. C. von der Ohe, Ecol. Appl., 2016, 26, 1249–1259 CrossRef PubMed.
- A. Nikolaides, S. Miess, I. Auvera, R. Müller, J. Klosterkötter and S. Ruhrmann, Eur. Arch. Psychiatry Clin. Neurosci., 2016, 266, 649–661 CrossRef PubMed.
- J. Polanski, U. Kucia, R. Duszkiewicz, A. Kurczyk, T. Magdziarz and J. Gasteiger, Sci. Rep., 2016, 6, 28521 CrossRef PubMed.
- M. Tavana, A. Fallahpour, D. Di Caprio and F. J. Santos-Arteaga, Expert Syst. Appl., 2016, 61, 129–144 CrossRef.
- H. K. Ising, S. Ruhrmann, N. A. F. M. Burger, J. Rietdijk, S. Dragt, R. M. C. Klaassen, D. P. G. van den Berg, D. H. Nieman, N. Boonstra, D. H. Linszen, L. Wunderink, F. Smit, W. Veling and M. van der Gaag, Psychol. Med., 2016, 46, 1839–1851 CrossRef CAS PubMed.
- A. M. A. Sattar, B. Gharabaghi and E. A. McBean, Water Resour. Manag., 2016, 30, 1635–1651 CrossRef.
- A. H. Alavi, H. Hasni, I. Zaabar and N. Lajnef, Arch. Civ. Mech. Eng., 2017, 17, 326–335 CrossRef.
- S. M. Mousavi, E. S. Mostafavi and P. Jiao, Energy Convers. Manag., 2017, 153, 671–682 CrossRef.
- S. M. Hamze-Ziabari and A. Yasavoli, J. Adv. Concr. Technol., 2017, 15, 644–661 CrossRef CAS.
- N. Shahrara, T. Çelik and A. H. Gandomi, J. Civ. Eng. Manag., 2017, 23, 85–95 CrossRef.
- M. Atieh, G. Taylor, A. M. A. Sattar and B. Gharabaghi, J. Hydrol., 2017, 545, 383–394 CrossRef.
- G. B. Tesfahunegn and C. S. Wortmann, Nutr. Cycling Agroecosyst., 2017, 109, 269–289 CrossRef CAS.
- J. M. Cabrero and M. Yurrita, Eng. Struct., 2018, 171, 895–910 CrossRef.
- E. Hou, J. Wang and W. Chen, Geocarto Int., 2018, 33, 754–769 CrossRef.
- N. Kovdienko, P. Polishchuk, E. Muratov, A. Artemenko, V. Kuz’min, L. Gorb, F. Hill and J. Leszczynski, Mol. Inf., 2010, 29, 394–406 CrossRef CAS PubMed.
- X. Zhang, X. Li, L. Li, S. Zhang and Q. Qin, J. Arid Land, 2019, 11, 15–28 CrossRef.
- M. Najafzadeh, M. Rezaie-Balf and A. Tafarojnoruz, Int. J. River Basin Manag., 2018, 16, 505–512 CrossRef.
- T. Haidl, M. Rosen, F. Schultze-Lutter, D. Nieman, S. Eggers, M. Heinimaa, G. Juckel, A. Heinz, A. Morrison, D. Linszen, R. Salokangas, J. Klosterkötter, M. Birchwood, P. Patterson, S. Ruhrmann and European Prediction of Psychosis Study (EPOS) Group, Schizophr. Res., 2018, 199, 346–352 CrossRef PubMed.
- H. Glawe, A. Sanna, E. K. U. Gross and M. A. L. Marques, New J. Phys., 2016, 18, 093011 CrossRef.
- O. Isayev, C. Oses, C. Toher, E. Gossett, S. Curtarolo and A. Tropsha, Nat. Commun., 2017, 8, 15679 CrossRef CAS PubMed.
- R. Ouyang, S. Curtarolo, E. Ahmetcik, M. Scheffler and L. M. Ghiringhelli, Phys. Rev. Mater., 2018, 2, 083802 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |