Picosecond kinetics of excited-state charge separation in 4-(dimethylamino)pyridine†
Received
29th July 2004
, Accepted 22nd September 2004
First published on 4th November 2004
Abstract
4-(Dimethylamino)pyridine (DMAP) shows solvent-dependent dual fluorescence from the initially excited state B* and a highly polar TICT state A*. Room-temperature time-resolved picosecond fluorescence investigations prove the bimodal kinetics of the excited-state electron transfer reaction B*→ A* in polar aprotic media. In medium polarity solvents (such as ethyl acetate) two emitting states of DMAP are shown to reach equilibrium within 50 ps. Both emitting states originate from the same ground state. The rate of excited-state charge separation depends on polarity and proton donating ability of the surrounding medium. The effects of temperature on the quantum yields of both fluorescences of DMAP in polar aprotic media indicate the transition from the kinetic regime (at low temperatures) to the equilibrium regime (at high temperatures). The kinetic behaviour of the dual luminescence of DMAP in protic solvents is more complex than in aprotic ones. In alcohols an efficient nonradiative channel competes with excited-state charge separation.
Introduction
One of the simplest systems which can be used to study the basic kinetic problems of photoinduced electron transfer (ET) processes are bichromophoric donor (D)–acceptor (A) molecules. A class of these shows a dual fluorescence phenomenon.1,2 Solvent-dependent dual fluorescence of 4-(dimethylamino)pyridine (DMAP)3–7 has been recently rationalized in terms of the twisted intramolecular charge transfer (TICT) state model.2,8–10 This model assumes that dielectric polarization of the solvent permits excited-state rotational isomerization of the initially excited state B* into a highly polar ICT state A* with a conformation of the D+ and A− subunits being close to perpendicular (Fig. 1). The π-electronic decoupling of the D+
(i.e., dimethylamino group) and A−
(pyridine ring) subunits of DMAP results in a nearly full charge separation and, consequently, a high dipole moment and a low electronic transition dipole moment of the CT fluorescence.6 This explanation is supported by the photophysical properties of sterically “pretwisted’
ortho-methylated derivatives of DMAP which show only a single charge transfer fluorescence band (most probably due to the reduction of the energy barrier to the excited-state reaction).5,6
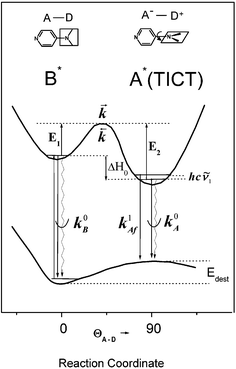 |
| | Fig. 1 Kinetic model for the TICT state formation in DMAP: schematic cross section of the ground-state and lowest excited states potential hypersurfaces along the reaction coordinate represented by a twist angle ΘA–D and solvation; k parameters denote rate constants for forward (![[k with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006b_20d1.gif) ) and back ( ) and back (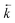 ) electron transfer and total (radiative and radiationless) deactivation of the primary excited (k0B) and TICT (k0A
+
k1Af) states where k1Af is an emission rate constant from an excited vibronic state of A*. Edest is the energy difference between the Franck–Condon ground state and the solvent equilibrated ground state. The other symbols are explained in the text. ) electron transfer and total (radiative and radiationless) deactivation of the primary excited (k0B) and TICT (k0A
+
k1Af) states where k1Af is an emission rate constant from an excited vibronic state of A*. Edest is the energy difference between the Franck–Condon ground state and the solvent equilibrated ground state. The other symbols are explained in the text. | |
The dual luminescence of DMAP, similarly to p-cyano-N,N-dimethylaniline (CDMA) and its derivatives,1,2,8–18 4-dialkylamino derivatives of benzaldehyde and acetophenone,2,9–11,19 dialkylaminobenzoic acid esters2,20 and 4-dialkylaminopyrimidines,2,4,21–23 is a manifestation of photoreaction which occurs entirely in the excited state. Therefore, both the substrate and the product can be followed in time by fluorescence spectroscopy. The investigation of their kinetics can give important clues to our understanding of how energetics and dynamics influence each other. The focus of this work is on the dependence of kinetics of the TICT state formation on solvent properties. DMAP seems to be a very good candidate for this kind of study. In aprotic solvents of medium polarity (such as ethyl acetate and 2-methyltetrahydrofuran) both emission bands of comparable intensity are spectrally well separated.6 The TICT fluorescence is dominant in highly polar media (such as butyronitrile and acetonitrile). Moreover, photophysical properties of DMAP,3,4 similarly to 4-dialkylaminopyrimidines,4,21–23 are strongly modified by hydrogen bonding. In this paper we present the temperature effects on the stationary dual fluorescence quantum yields and the results of the room-temperature picosecond fluorescence investigations of DMAP in polar aprotic and protic media.
Experimental
Materials
4-(Dimethylamino)pyridine (Aldrich) was purified by crystallization from n-hexane. Ethyl acetate (EA), acetonitrile (ACN) and the alcohols: methanol, ethanol, 1-propanol and 1-octanol were of spectroscopic or fluorescence grade (Aldrich or Merck). Butyronitrile, BN (Merck, for synthesis) was distilled successively over KMnO4
+ K2CO3, P2O5 and CaH2. 2-Methyltetrahydrofuran, MTHF (Merck, for synthesis) was dried over P2O5 and then distilled over CaH2. Water was used after four distillations over KMnO4 in a quartz apparatus. All solvents were checked for the presence of fluorescing impurities. Deuterium substituted alcohols (methanol, CH3OD, ethanol, C2H5OD, 1-butanol, C4H10OD) and water (D2O) have been kindly obtained from CHEMIPAN R&D Laboratories.
Methods
Absorption spectra were recorded on Shimadzu UV 2401 and Shimadzu UV 3100 spectrophotometers. The solutions used for isotope substitution absorption studies were prepared in a dry-box under a nitrogen atmosphere. Stationary fluorescence and fluorescence excitation spectra were measured on an Edinburgh FS 900 CDT spectrofluorimeter. The spectra were corrected using the spectral sensitivity curve of the instrument. Fluorescence spectra were recorded as a function of wavelength (λ) and subsequently multiplied by a factor of λ2 in order to convert counts per wavelength into counts per wavenumber. Temperature effects on the stationary emission spectra were determined with an OptistatDN (Oxford Instruments) cryostat. Quinine sulfate in 0.05 mol dm−3 H2SO4
(Φf
= 0.51)24 and DMAP in ACN (Φa
= 0.017)6 served for fluorescence quantum yield determination of DMAP in alcohols. Fluorescence lifetimes in a nanosecond range were obtained using an Edinburgh FL 900 CDT time-resolved fluorometer, while the fluorescence lifetimes in a picosecond range were obtained employing an experimental setup described previously.25 Briefly, the picosecond titanium-sapphire laser (Tsunami, Spectra Physics) was pumped by an argon-ion laser (BeamLok 2060, Spectra Physics). The repetition frequency of the picosecond laser was reduced to ∼ 4 MHz due to the use of a pulse selector (Model 3985, Spectra Physics). The excitation wavelength was the third harmonic of the fundamental frequency. In the emission pathway there was a polariser set at the magic angle to the vertical polarisation of the exciting beam. The electronic detection pathway was composed of an upgraded quad constant fraction discriminator CFD TC 454 (Tennelec), time to amplitude converter TAC-TC 864 (Tennelec), a multichannel analyser Norland 5000 MCA and proximity type (6 µm) microchannel plate MCP-PMT R3809U-05 (Hamamatsu). The TAC worked in the reverse mode. Picosecond fluorescence decays were analyzed by a homemade software, which allowed the use of the Levenberg–Marquardt26,27 or simplex28,29 optimization algorithms. The capabilities of the software have been described in a previous paper.30 The instrument response function (IRF) was determined by the scattering method. FWHM of IRF was ∼ 35 ps. The χ2 test and the distribution of weighted residuals were the main criteria in evaluation of the quality of the fit.
Results and discussion
Room-temperature absorption and fluorescence
Solvent effects on the room temperature absorption and fluorescence spectra of DMAP are presented in Fig. 2. The near-UV absorption spectra of DMAP show a shoulder in the low-energy part of the spectrum and a dominant band centered at ∼ 39![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 300 cm−1 in EA, 39025 cm−1 in BN and ACN and 38750 cm−1 in 1-propanol. Our previous magnetic circular dichroism (MCD) investigations indicated that these bands are related to three low-energy transitions, assigned to 1Lb, 1(n,π*) and 1La excited states.7 The absorption spectrum of DMAP (pKGa
= 9.6)31 in H2O and D2O corresponds to the cation. The room-temperature absorption spectra of the cations of DMAP and its ortho-methylated derivatives are shifted to lower energies with respect to those of the corresponding neutral forms.6 This indicates that N1 atom is the protonation centre, similarly as in aminopyridine31 and in aminopyrimidines.32,33 Contrary to the aqueous solutions, the absorption spectra of DMAP in alcohols (except methanol) do not show any evidence of the ground-state formation of the protonated forms (the corresponding spectra do not change in alkaline alcoholic solutions). Both neutral and ionic forms are observed in methanol. A similar behaviour is observed in deuterated alcohols. Ground-state complexation of DMAP with protic partners, monitored by titration experiments with mixed solvents (such as n-hexane + 1-butanol), indicates that in low concentration of alcohol (preventing the formation of alcohol oligomers) only the 1 ∶ 1 complex is formed, hydrogen bonded at the ring nitrogen atom N1.4 In bulk alcohol, however, the ground-state formation of various differently solvated complexes is expected.
300 cm−1 in EA, 39025 cm−1 in BN and ACN and 38750 cm−1 in 1-propanol. Our previous magnetic circular dichroism (MCD) investigations indicated that these bands are related to three low-energy transitions, assigned to 1Lb, 1(n,π*) and 1La excited states.7 The absorption spectrum of DMAP (pKGa
= 9.6)31 in H2O and D2O corresponds to the cation. The room-temperature absorption spectra of the cations of DMAP and its ortho-methylated derivatives are shifted to lower energies with respect to those of the corresponding neutral forms.6 This indicates that N1 atom is the protonation centre, similarly as in aminopyridine31 and in aminopyrimidines.32,33 Contrary to the aqueous solutions, the absorption spectra of DMAP in alcohols (except methanol) do not show any evidence of the ground-state formation of the protonated forms (the corresponding spectra do not change in alkaline alcoholic solutions). Both neutral and ionic forms are observed in methanol. A similar behaviour is observed in deuterated alcohols. Ground-state complexation of DMAP with protic partners, monitored by titration experiments with mixed solvents (such as n-hexane + 1-butanol), indicates that in low concentration of alcohol (preventing the formation of alcohol oligomers) only the 1 ∶ 1 complex is formed, hydrogen bonded at the ring nitrogen atom N1.4 In bulk alcohol, however, the ground-state formation of various differently solvated complexes is expected.
DMAP emits dual fluorescence a and b in polar media at room temperature,3–6 the bands being independent of the excitation wavelength or concentration (in the range of 10−6–10−3 mol dm−3). The dipole moment estimated from the solvatochromic effects on the long-wave fluorescence a is about 13 D. In aprotic solvents the relative intensity of the CT emission a increases with increasing solvent polarity (Table 1). This emission band is dominant in highly polar aprotic solvents such as BN and ACN (the quantum yield of the CT fluorescence is about 0.016). The radiative transition in emission (kAf
≅ 4 × 106 s−1) is forbidden by a factor of 10−2 with respect to the value of fully allowed transition as may be expected for a small π-orbital overlap of the nearly orthogonal D+–A− configuration.6 In protic solvents, strong quenching reduces the emission efficiency, the excitation spectra of both fluorescence bands in 1-propanol are nearly identical. The quenching is not connected with a photochemical reaction resulting in the ground-state product because the absorption spectrum does not show any change upon long-time excitation. Similarly to the case of 4-dimethylaminopyrimidines,4,23 the fluorescence quenching of DMAP increases with the acidity factor α34 and with decreasing viscosity of the alcoholic solvent. The quantum yield of the CT emission a in 1-octanol is 0.004, in 1-propanol is 0.0008 and in ethanol is as low as about 10−4. The fluorescence of DMAP in methanol or water is below the detection limits (it should be noted that the cations of DMAP do not show any detectable fluorescence at room temperature).4,6
| |
EA |
MTHF |
BN |
ACN |
1-Propanol |
1-Octanol |
|
At 293 K,6 error is about 10%.
Error is about 5%.
Eqn. (4), error is about 10%.
Eqn. (8), error is about 10%.
Eqn. (6), error is about 25%.
At 293 K, eqns. (12) and (14).
At 293 K, eqn. (13).
At 293 K, eqn. (6), kAf
=k1Af
+
k0Af
≅
Φa/τa.
At 293 K, eqns. (3) and (15), error is about 10%.
At 293 K, eqns. (14) and (15), error is about 15%.
|
|
Φ
a
a
|
0.014 |
0.014 |
0.016 |
0.017 |
0.0008 |
0.004 |
|
Φ
b
a
|
0.008 |
0.012 |
0.005 |
0.001 |
∼0.0004 |
∼0.002 |
|
T
max
b/K |
|
235 |
260 |
|
|
|
|
E
1
c/cm−1 |
|
450 |
700 |
|
|
|
|
E
3
d/cm−1 |
|
|
|
|
750 |
|
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 1
e/cm−1
1
e/cm−1 |
|
200 |
300 |
|
|
|
| ΔH0/cm−1 |
|
|
∼700 |
|
|
|
|
f/s−1 |
2.0 × 1010 |
|
|
4.2 × 1010 |
6.3 × 1010 |
|
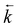 f/s−1
f/s−1 |
2.2 × 109 |
|
|
∼5.7 × 108 |
|
|
|
k
0B
+
kAg/s−1 |
6.7 × 108 |
|
|
∼6.5 × 108 |
|
|
|
k
Af
h
/s−1 |
|
|
|
∼4.6 × 106 |
5.0 × 106 |
∼7.0 × 106 |
|
k
0Bf/kAfi |
5.3 |
|
|
∼4.5 |
|
|
|
K*j |
9 |
|
|
∼75 |
|
|
Temperature effects on the position and shape of the fluorescence spectra of DMAP in BN and 1-propanol and the resulting Arrhenius-type plots for the quantum yields Φa and Φb of the both emission bands and for their ratio Φa/Φb are shown in Figs. 3 and 4. The results in aprotic solvents (such as MTHF and BN) are consistent with a two-state kinetic model (originally proposed for CDMA and its derivatives),11 which includes three thermally activated processes: forward 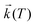 and back
and back 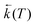 excited-state electron transfer and CT emission from thermally populated vibrational levels k1Af
(T). The kinetic scheme shown in Fig. 1 in a steady-state approximation results in the following expressions for the quantum yields:2,8,11,12
excited-state electron transfer and CT emission from thermally populated vibrational levels k1Af
(T). The kinetic scheme shown in Fig. 1 in a steady-state approximation results in the following expressions for the quantum yields:2,8,11,12| | 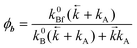 | (1) |
| | 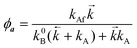 | (2) |
| | 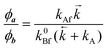 | (3) |
where:| | 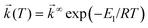 | (4) |
| | 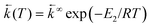 | (5) |
| | 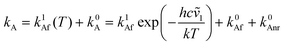 | (6) |
| | 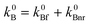 | (7) |
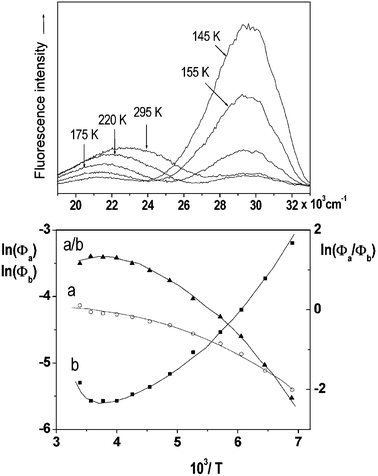 |
| | Fig. 3 Temperature effects on the position and shape of the fluorescence spectra of DMAP in BN (top) and Arrhenius-type plots of the quantum yields Φa and Φb of the both emission bands and of their ratio Φa/Φb
(bottom). | |
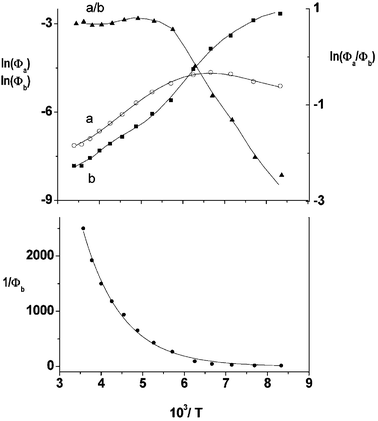 |
| | Fig. 4 Arrhenius-type plots of the quantum yields Φa and Φb and of their ratio Φa/Φb of the fluorescence spectra of DMAP in 1-propanol (top) and plot of 1/Φbvs. 1/T
(bottom). | |
The dependence of k1Af(T) on temperature for DMAP in aprotic polar solvents is consistent with the rise of the Φa values with temperature (the thermal activation is linked with the strongly forbidden character of the emission from the zero vibrational level of the TICT state (k0Af),2,9,11,12,35 the data are interpreted in terms of eqn. (6)). It should be noted, however, that the observed temperature effects can also be connected with the change of the solvent polarity. The polarity of the fluid solvents increases with lowering of temperature (due to that a considerable red shift of the CT fluorescence band of DMAP in BN, Fig. 3, and in MTHF is observed with lowering of temperature). This influences the molecular and electronic structure as well as the rates, especially of the charge recombination, contributing thus to kA(T). This indirect temperature effect has been previously observed in large conjugate D–A π-systems such as aryl derivatives of dimethylanilines,36 which show the decrease of the values of the radiative rate constants kf and of the transition dipole moments Mflu of the CT fluorescence with growing polarity of the environment (the increase of polarity induces an increase of the CT character of the fluorescent state and leads to the more decoupled conformation of these molecules).37 It seems to be not the case of relatively small compounds containing dialkylamino group as an electron donor.2 The conformation of DMAP and its ortho-methylated derivatives in the CT state is nearly orthogonal and the transition dipole moment (Mflu
≅ 0.6÷0.7 D) of the TICT fluorescence a does not depend on solvent polarity.6 Thus, the observed temperature effects are related to the thermal activation.
The maximum observed for ln(Φa/Φb) vs. 1/T is mainly due to the strong temperature dependence of Φb: at Tmax it has the minimum and increases with rising temperature corresponding to the creation of the excited state equilibrium, i.e., a significant repopulation of the initially excited state B* by back reaction. The temperature Tmax is defined by the relation 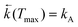 , where kA is a sum of the radiative and radiationless depopulation rate constants of the TICT state (Fig. 1, eqn. (6)). The transition from the kinetic regime, i.e., from an irreversible reaction regime (at lower temperatures), to the equilibrium (or high-temperature) regime, with the back reaction prevailing over photophysical deactivation, occurs around Tmax.2,9,11 Well below Tmax the back reaction rate constant may be neglected with respect to the other deactivation processes of A* state,
, where kA is a sum of the radiative and radiationless depopulation rate constants of the TICT state (Fig. 1, eqn. (6)). The transition from the kinetic regime, i.e., from an irreversible reaction regime (at lower temperatures), to the equilibrium (or high-temperature) regime, with the back reaction prevailing over photophysical deactivation, occurs around Tmax.2,9,11 Well below Tmax the back reaction rate constant may be neglected with respect to the other deactivation processes of A* state, 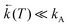 . Well above Tmax the forward and back ET reaction rate constants are larger than all other ones:
. Well above Tmax the forward and back ET reaction rate constants are larger than all other ones: 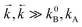 (where k0B is the rate constant of the photophysical deactivation of the primary excited state, Fig. 1, eqn. (7)).
(where k0B is the rate constant of the photophysical deactivation of the primary excited state, Fig. 1, eqn. (7)).
The temperature dependent experiments of the dual luminescence allow one to estimate the kinetic and thermodynamic data of the excited state electron transfer process.2,11,12 The activation energy E1 of the forward ET process and the enthalpy of the photoinduced charge separation ΔH0
(Fig. 1) can be obtained from the slope of the relations ln(Φb) and ln(Φa/Φb) vs. 1/T) in the low-temperature or in the high-temperature regime, respectively. The data are collected in Table 1. The activation energy E1
(eqn. (4)) of the excited-state process B*→ A* is close to, or slightly smaller than the activation energy of viscous flow in BN (780 cm−1)38 and in MTHF (620 cm−1),39 respectively. The activation energy 1
(eqn. (6)), of about 200–300 cm−1, estimated from the slope of the relation ln(Φa) vs. 1/T, can be attributed to the torsional or inversion vibrations which increase the donor–acceptor orbital overlap, and thus the radiative transition probability.2,9,11
Contrary to the finding in aprotic polar solvents, the slope of ln(Φa) vs. 1/T in 1-propanol is opposite to the expected one for the thermally activated TICT fluorescence (Fig. 4). Thus, similarly to 4-dialkylaminopyrimidines4,23 and carbonyl derivatives of N,N-dimethylaniline,40 the quenching of the fluorescence b with increasing temperature seems to have another origin than the depopulation of the B* state by transformation to the TICT state in a neutral molecule. In view of the evidence from Figs. 3 and 4 we can state that an efficient thermally activated nonradiative channel appears in protic solvents. The enhancement of the radiationless processes in hydrogen-bonded systems is usually explained in terms of the change of the hydrogen bonding strength upon excitation. This leads to different shapes and positions of the minima in the potential energy hypersurfaces of the two states which, in turn, may activate rapid internal conversion (IC) from the lowest excited singlet state.41 Another mechanism which may be responsible for the decrease of fluorescence is an excited-state formation of a nonfluorescent product, e.g., the TICT state of a protonated form of DMAP. The latter mechanism might operate for the system, which change significantly their acid–base equilibria upon excitation. INDO/S computed change of ΔpKa
= pKa*
− pKGa
≅ 44
(based on the concept of the effective valence electron potentials42) results in the pKa* value of DMAP in the lowest excited singlet state of 13.6. The large value of pKa* could be responsible for the excited-state formation of the non-emitting cations of DMAP in alcoholic solutions. The observed correlation between the CT fluorescence quenching and the proton donating ability34
(and mobility) of the solvating alcohol partner supports this mechanism. There are two possible sequences for an efficient fluorescence quenching involving such an event: (i) efficient IC occurring during formation of the cations, before reaching the CT fluorescent state, and (ii) rapid deactivation of the ionic product, faster than the rate of its creation. In order to gain more insight into the nature of the quenching process, the analysis of the temperature effects on the deactivation of the initially excited B* state has been performed. If one assumes that the nonradiative rate of deactivation of this state may be expressed as a sum of a temperature independent and a temperature dependent processes 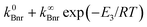 , the Arrhenius-type expression results:
, the Arrhenius-type expression results:
| | 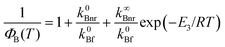 | (8) |
Fitting of the experimental data for the emission b to this equation enabled the evaluation of the Arrhenius parameters, k∞Bnr and E3. An example of the fit is shown in Fig. 4. Assuming that the rate constant of the radiative depopulation of the B* state is similar to that in polar aprotic solvents, e.g., in ACN (from 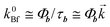 and k0Bf
≅ 4.5kAf one can obtain k0Bf
≅ 3 (±1)
× 107s−1, Table 1), the values of the preexponential factor k∞Bnr are of the order of 5 × 1012 s−1, close to those typical for internal conversion43 and about two orders of magnitude larger than the values obtained for intramolecular electron transfer in neutral forms (Table 1). The activation energy E3 of the temperature-dependent quenching process in the hydrogen bonded complexes of DMAP is about 750–900 cm−1. This value is significantly lower than the activation energy of the mobility of 1-propanol (being ∼1500 cm−1).44
and k0Bf
≅ 4.5kAf one can obtain k0Bf
≅ 3 (±1)
× 107s−1, Table 1), the values of the preexponential factor k∞Bnr are of the order of 5 × 1012 s−1, close to those typical for internal conversion43 and about two orders of magnitude larger than the values obtained for intramolecular electron transfer in neutral forms (Table 1). The activation energy E3 of the temperature-dependent quenching process in the hydrogen bonded complexes of DMAP is about 750–900 cm−1. This value is significantly lower than the activation energy of the mobility of 1-propanol (being ∼1500 cm−1).44
Bimodal kinetics in polar aprotic media
The fluorescence kinetic data obtained on the nanosecond time scale for DMAP in EA and dichloroethane showed the common decay of both fluorescence bands.6 The bimodal kinetics and the reversibility of the excited-state ET reaction in DMAP in aprotic polar media such as EA and ACN at room temperature is proved by the picosecond fluorescence decay investigations. The typical kinetic curves of both fluorescence bands in EA are presented in Fig. 5. A conventional precursor–successor kinetic model which does not involve a time-dependent rate constants results in the following equations:45| | 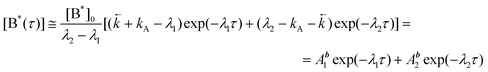 | (9) |
| | 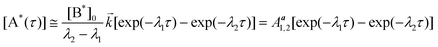 | (10) |
where: [B*(τ)] and [A*(τ)] are the concentrations of the primary excited species and the product of the excited-state ET process, respectively; [B*]0 is the initial concentration of the substrate at τ
= 0; λ1 and λ2 are expressed by the following relations:| | 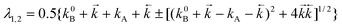 | (11) |
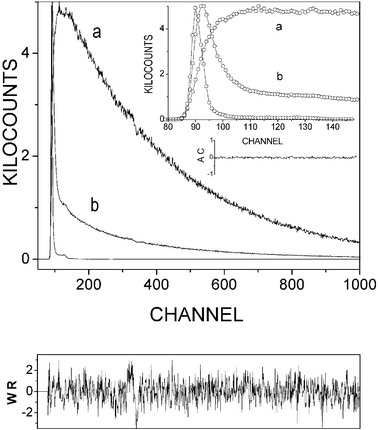 |
| | Fig. 5 Kinetic curves of room temperature dual fluorescence bands of DMAP in ethyl acetate: b monitored at 30300 cm−1, a at 21300 cm−1
(the instrument response function is also shown for comparison). One channel corresponds to 9.8 ps. The plot of weighted residuals (WR) and correlation function of weighted residuals (AC) is related to emission a. | |
In the high-temperature approximation (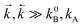 ):
):
| | 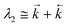 | (12) |
| | 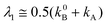 | (13) |
| | 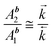 | (14) |
The picosecond kinetic data (Table 1) for DMAP in EA indicate that at room temperature the equilibrium B*⇔ A* is established within 50 ps. The fluorescence bands, b and a, were monitored at 30300 and 21300 cm−1, respectively (the spectral width of the observation was about 800 cm−1 to prevent any overlap of both emissions). The decay of the fluorescence b involves two components: τ1
≡ 1/λ1
≊ 2.8 (±0.3) ns and τ2
≡ 1/λ2
≊ 46 (±5) ps, with the ratio of the amplitudes Ab2/Ab1
≊ 9 (±1). Both lifetimes can be recovered in the decay (3.1 ± 0.1 ns) and rise (45 (±3) ps) of the TICT fluorescence a. The ratio of the amplitudes Aa2/Aa1
≊
−1.0 (±0.05) proves that the B* state is the precursor of the A* state (in other words both emitting states, B* and A*, originate from the same ground state). The slow and the fast component approximately corresponds to the sum of the rate constants of the photophysical decay processes of the both reaction partners B* and A*
(k0B
+
kA
≅ 6.7 × 108 s−1) and to the sum of the rate constants of the forward and back ET (
+
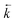 ≊ 2.2 × 1010 s−1)
≊ 2.2 × 1010 s−1)
, respectively. These values are similar to those determined previously for CDMA and its derivatives,13,15–17 4-dialkylamino derivatives of benzaldehyde19 and 4-dialkylaminopyrimidines.23
The experimental values of the decay times τ1 and τ2 and of the ratio of the amplitudes Ab2/Ab1 allows one to estimate the ratio of the rate constants of the forward and back ET (eqn. (14)), the ratio of the rate constants of the radiative depopulation of the B* and A* states (eqns. (3) and (14)), and the equilibrium constants K*:
| | 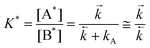 | (15) |
The bimodal kinetics and the reversibility of the excited-state ET reaction has been also observed in ACN (the short-wavelength emission b has been monitored at 30800 cm−1, the TICT band a at 19800 cm−1). The decay of both emission bands shows two-exponential behaviour, the ratio of the amplitudes Aa2/Aa1
=
−0.97 (±0.03). While the slow component τ1
≅ 3.1 (±0.2) ns has been found to be very similar to that in EA, the fast component τ2
≅ 23 (±5) ps is two times shorter. It indicates that the excited-state reaction is accelerated with increasing polarity of the environment. Moreover, the ratio of the amplitudes Ab2/Ab1
≅ 75 (±10) indicates that
≫
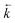 . Thus,
≅ 4.2 × 1010 s−1 and the equilibrium constant K* value in ACN is about one order of magnitude higher than that in EA (Table 1). These effects are not unexpected for the D–A systems such as DMAP possessing a double minimum potential in the excited state (Fig. 1). In strongly polar solvents the stabilization of the CT state and the resulting correlation of states leads to a smaller energy barrier to internal rotation on the excited-state relaxation path to the TICT state and to the higher barrier to the back reaction.2,4
. Thus,
≅ 4.2 × 1010 s−1 and the equilibrium constant K* value in ACN is about one order of magnitude higher than that in EA (Table 1). These effects are not unexpected for the D–A systems such as DMAP possessing a double minimum potential in the excited state (Fig. 1). In strongly polar solvents the stabilization of the CT state and the resulting correlation of states leads to a smaller energy barrier to internal rotation on the excited-state relaxation path to the TICT state and to the higher barrier to the back reaction.2,4
Complex kinetics in alcohol solutions
The dual luminescence of DMAP in 1-propanol, contrary to the finding in polar aprotic solvents, can hardly be explained in terms of the bimodal reversible kinetic model involving only single ground-state species. In spite of the experimental problems connected with the very low fluorescence intensity (especially of the short-wave fluorescence b), both emissions are not simply kinetically coupled. The decay of b
(monitored at 31250 cm−1) could not be satisfactorily fitted, the best two-exponential fit (χ2
= 1.28) for b is characterized by the decay times of ∼9 ps and ∼ 480 ps, with the ratio of the amplitudes Ab2/Ab1
≈ 250. The three-exponential approximation does not improve the fit. On the other hand, the decay of the CT emission a
(monitored at 19800 cm −1) is well described (χ2
≅ 1.1) by the rise time τ2
≅ 16 (±2) ps and decay time τ1
≅ 160 (±5) ps. The ratio of the amplitudes Aa2/Aa1
=
−1.0 (±0.02) indicates that the CT state is not formed by direct excitation from the ground-state species. The clear explanation of these results is difficult because we can hardly state if the observed difference between the fast components of the kinetics curves of both fluorescences is meaningful (the investigations with the better time resolution are planned to clarify the problem). The results suggest, however, that of several possible hydrogen-bonded complexes of DMAP with 1-propanol, only one undergoes the excited-state ET reaction in neutral form, the other sites lead to the fast quenching. Moreover, the formation of the TICT state in 1-propanol is accelerated (
≅ 6.3 × 1010 s−1) with respect to that in ACN (i.e., the aprotic solvent of similar polarity). Complexing increases the electron affinity of the acceptor ring, both thermodynamic and kinetic factors thus favour the photoinduced ET reaction (see Fig. 3 in ref. 22).
The preliminary measurements of the dual luminescence of DMAP in 1-octanol show even more complex kinetics than that observed in 1-propanol. Using the kinetic model involving only single ground-state species undergoing the excited-state charge separation, a rather poor fit (χ2
≅1.4) to a two-exponential decay function of the CT emission a
(monitored at 20000 cm−1) leads to the rise time τ2
≈ 30 ps and decay time τ1
≈ 560 ps, with the ratio of the amplitudes Aa2/Aa1
≈
−1.1. The best two-exponential fit (χ2
≅ 1.15) of the decay of b
(monitored at 29850 cm−1) results in the values of τ2
≅ 20 (±3) ps and τ1
≅ 700 (±40) ps, with the ratio of the amplitudes Ab2/Ab1
≊ 16 (±4). The corresponding value of is about 3 × 1010 s−1. The three-exponential approximation improves the fits (χ2
≅ 1.0) of the kinetic curves of the both emission bands: (i) the decay of b is described by the components of about 17 and 720 ps (with a significant yield each) and of about 90 ps (with a small contribution), (ii) the observed biexponential rise of the CT emission a of about 16 and 250 ps is followed by the decay of about 525 ps. The systematic kinetic studies of the dual luminescence of DMAP in 1-octanol and 1-decanol are in progress.
Conclusions
The effects of temperature on the dual luminescence, a and b, of DMAP and the room-temperature time-resolved picosecond fluorescence investigations prove the bimodal kinetics of the excited-state electron transfer reaction B*→ A* in polar aprotic solvents. Two emitting states of DMAP were shown to reach equilibrium at room temperature. The activation energies of the excited-state charge separation reaction B*→ A* in MTHF and BN appeared to be slightly smaller than the activation energies of the viscous flow in a given solvent. The rate of this reaction is accelerated with increasing polarity and proton donating ability of the surrounding medium. The peculiarity of the hydrogen-bonded complexes of DMAP, similarly to 4-dimethylaminopyrimidines,4,21–23 is the appearance of an efficient nonradiative channel (most probably connected with internal conversion during the formation of the TICT state of an ionic form of DMAP), which effectively competes with charge separation in neutral molecules. It should be pointed out, that the low-temperature phosphorescence investigations of DMAP do not show any evidence for the acceleration of the intersystem crossing (ISC) process in alcoholic glasses (such as 1-propanol and methanol) with respect to polar aprotic ones (e.g., butyronitrile). In order to clarify the role of the triplet states in the appearance of an effective radiationless process in protic media the transient absorption investigations are in progress.
The formation of the TICT state consists of the geometry change of the D-A molecule (against the viscosity of the medium), of the solvent reorientation (dependent on the solvent relaxation times, related to viscosity) and of electron transfer step. Our future plans include more systematic temperature-dependent kinetics studies of DMAP and its dialkylamino derivatives in solvents of varying polarity and viscosity to identify the rate-determining steps in the TICT state formation.
Acknowledgements
Technical assistance from Mrs Anna Zielińska is deeply appreciated. Dynamics investigations were performed at the Centre for Ultrafast Laser Spectroscopy at the Adam Mickiewicz University in Poznań
(we are grateful to Dr Krzysztof Dobek for his kind help). This work was partly sponsored by the grant 7 T09A 044 21 from the Polish Committee for Scientific Research.
References
-
E. Lippert, W. Lüder and H. Boss, Fluoreszenzspectrum und Franck–Condon-prinzip in lösungen aromatischer verbindungen, in Advances in Molecular Spectroscopy, ed. A. Mangini, Pergamon Press, Oxford, 1962, p. 443 Search PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge transfer states and structures, Chem. Rev., 2003, 103, 3899 CrossRef.
- C. Cazeau-Dubroca, G. Nouchi, M. Ben Brahim, M. Pesquer, D. Gorse and Ph. Cazeau, Dual luminescence of 4-N,N-dimethylaminopyridine. Role of hydrogen-bonded complex in the ground state, J. Photochem. Photobiol. Chem. A, 1994, 80, 125 CrossRef CAS.
- J. Herbich and J. Waluk, Excited charge transfer states in 4-aminopyrimidines, 4-(dimethylanilino)pyrimidine and 4-(dimethylamino)pyridine, Chem. Phys., 1994, 188, 247 CrossRef CAS.
- S. Mishina, M. Takayanagi, M. Nakata, J. Otsuki and K. Araki, Dual fluorescence of 4-dimethylaminopyridine and its derivatives. Effects of methyl substitution at the pyridine ring, J. Photochem. Photobiol. Chem. A, 2001, 141, 153 CrossRef CAS.
- I. Szydlowska, A. Kyrychenko, J. Nowacki and J. Herbich, Photoinduced intramolecular electron transfer in 4-dimethylaminopyridines, Phys. Chem. Chem. Phys., 2003, 5, 1032 RSC.
- I. Szydlowska, A. Kyrychenko, A. Gorski, J. Waluk and J. Herbich, Excited states of 4-dimethylaminopyridines: magnetic circular dichroism and computational studies, Photochem. Photobiol. Sci., 2003, 2, 187 RSC.
- K. Rotkiewicz, K. H. Grellmann and Z. R. Grabowski, Reinterpretation of the anomalous fluorescence of p-N,N-dimethylamino-benzonitrile, Chem. Phys. Lett., 1973, 19, 315 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz, A. Siemiarczuk, D. J. Cowley and W. Baumann, Twisted intramolecular charge transfer states (TICT). A new class of excited states with a full charge separation, Nouv. J. Chim., 1979, 3, 443 Search PubMed.
- Z. R. Grabowski and J. Dobkowski, Twisted intramolecular charge transfer (TICT) excited states: energy and molecular structure, Pure Appl. Chem., 1983, 55, 245 CAS.
- Z. R. Grabowski, K. Rotkiewicz, W. Rubaszewska and E. Kirkor-Kaminska, Spectroscopy and kinetics of the twisted internal charge-transfer (TICT) excited state formation in p-substituted dialkylanilines, Acta Phys. Polonica A, 1978, 54, 767 Search PubMed.
- W. Rettig, External and internal parameters affecting the dual fluorescence of p-cyano-dialkylanilines, J. Lumin., 1980, 26, 21 CrossRef CAS.
- Y. Wang and K. B. Eisenthal, Picosecond dynamics of twisted internal charge transfer phenomena, J. Chem. Phys., 1982, 77, 6076 CrossRef.
- S. R. Meech and D. Phillips, Complex kinetics of a* state formation in the DMABN-ethanol system, Chem. Phys. Lett., 1985, 116, 262 CrossRef CAS.
- U. Leinhos, W. Kühnle and K. A. Zachariasse, Intramolecular charge transfer and thermal exciplex dissociation with p-aminobenzonitriles in toluene, J. Phys. Chem., 1991, 95, 2013 CrossRef CAS.
- W. Schuddeboom, S. A. Jonker, J. M. Warman, U. Leinhos, W. Kühnle and K. A. Zachariasse, Excited-state dipole moments of dual fluorescent 4-(dialkylamino)benzonitriles. Influence of alkyl chain length and effective solvent polarity, J. Phys. Chem., 1992, 96, 10809 CrossRef CAS.
- K. A. Zachariasse, M. Grobys, T. von der Haar, A. Hebecker, Y. V. Il'ichev, Y.-B. Jiang, O. Morawski and W. Kühnle, Intramolecular charge transfer in the excited state. Kinetics and configurational changes, J. Photochem. Photobiol. Chem. A, 1996, 102, 59 CrossRef CAS.
- J. Dobkowski, J. Wójcik, W. Koźmiński, R. Kołos, J. Waluk and J. Michl, An experimental test of C–N bond twisting in the TICT state: syn-anti photoisomerization in 2-(N-methyl-N-isopropylamino)-5-cyanopyridine, J. Am. Chem. Soc., 2002, 124, 2406 CrossRef CAS.
- C. Rullière, Z. R. Grabowski and J. Dobkowski, Picosecond absorption spectra of carbonyl derivatives of dimethylaniline: the nature of the TICT excited states, Chem. Phys. Lett., 1987, 137, 408 CrossRef CAS.
- P. C. M. Weisenborn, A. H. Huizer and C. A. G. O. Varma, Excited-state dynamics of ethyl 4-(N,N-dimethylamino)benzoate and ethyl 4-(N,N-diethylamino)benzoate in apolar and polar solvents, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 1895 RSC.
-
J. Smagowicz, K. Berens and K. L. Wierzchowski, Unique properties of the lowest excited singlet state of 4-dialkylaminopyrimidines, in Excited States of Biological Molecules, ed. J. B. Birks, Wiley, London, 1976, p. 24 Search PubMed.
- J. Herbich, Z. R. Grabowski, H. Wójtowicz and K. Golankiewicz, Dual fluorescence of 4-(dialkylamino)pyrimidines. Twisted intramolecular charge transfer state formation favored by hydrogen bond or by coordination to the metal ion, J. Phys. Chem., 1989, 93, 3439 CrossRef CAS.
- J. Herbich, J. Karpiuk, Z. R. Grabowski, N. Tamai and K. Yoshihara, Modification of the intramolecular electron transfer by hydrogen bonding: 4-(dialkylamino)pyrimidines, J. Lumin., 1992, 54, 165 CrossRef CAS.
-
R. A. Velapoldi, Considerations on organic compounds in solution and inorganic ions in glasses as fluorescent standard reference materials, in National Bureau of Standards Special Publication 378, Accuracy in Spectrophotometry and Luminescence Measurements, Proc. Conf. NBS, National Bureau of Standards, Gaithersburg, MD, 1972, p. 231. Search PubMed.
- J. Karolczak, D. Komar, J. Kubicki, M. Szymański, T. Wrózowa and A. Maciejewski, Fluorescence dynamics spectrometer of single-picosecond resolution: optimisation of experimental performance, Bull. Pol. Acad. Sci. Chem., 1999, 47, 361 Search PubMed.
- D. W. Marquardt, An algorithm for least-squares estimation of nonlinear parameters, J. Soc. Ind. Appl. Math., 1963, 11, 431 Search PubMed.
-
W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes in C, Cambridge University Press, U.K. Cambridge, 1992 Search PubMed.
- J. A. Nelder and R. Mead, A simplex method for function minimization, Comput. J., 1965, 7, 308 Search PubMed.
-
F. W. Walters and L. R. Parker, Jr., Sequential Simplex Optimization, CRC Press LLC, 1992 Search PubMed.
- J. Karolczak, D. Komar, J. Kubicki, T. Wrózowa, K. Dobek, B. Ciesielska and A. Maciejewski, The measurements of picosecond fluorescence lifetimes with high accuracy and subpicosecond precision, Chem. Phys. Lett., 2001, 344, 154 CrossRef CAS.
- F. Cruége, G. Girault, S. Coustal, J. Lascombe and P. Rumpf, Bull. Soc. Chim. Fr., 1970, 3889 CAS.
- D. I. Brown, E. Hoerger and S. F. Mason, Simple pyrimidines. Part III. The methylation and structure of the aminopyrimidines, J. Chem. Soc., 1955, 4035 RSC.
- Z. Proba and K. L. Wierzchowski, Synthesis an properties of some 2-, 4- and 5-aminopyrimidines. Effect of –NH2 and ortho-C methylation on protolytic equilibria and electronic absorption spectra, Acta Biochim. Pol., 1975, 22, 131 CAS.
- M. J. Kamlet, J.-L. M. Abboud, M. H. Abraham and R. W. Taft, Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, π*, α and β and some methods for simplifying the generalized solvatochromic equation, J. Org. Chem., 1983, 48, 2877 CrossRef CAS.
- M. Van der Auweraer, Z. R. Grabowski and W. Rettig, Molecular structure and temperature-dependent radiative rates in twisted intramolecular charge transfer and exciplex system, J. Phys. Chem., 1991, 95, 2084.
- J. Herbich and A. Kapturkiewicz, Radiative electron transfer in aryl derivatives of dimethylanilines, Chem. Phys., 1993, 170, 221 CrossRef CAS.
- J. Herbich and A. Kapturkiewicz, Electronic structure an molecular conformation in the excited charge transfer singlet states of 9-acridyl and other aryl derivatives of aromatic amines, J. Am. Chem. Soc., 1998, 120, 1014 CrossRef CAS.
-
J. A. Riddick and W. B. Bunger, Organic solvents, in: Techniques of Chemistry, Wiley-Interscience, New York, 3rd edn., 1970, vol. 2 Search PubMed.
-
B. P. Engler and L. A. Harrah, Viscosity and density of 2-methyltetrahydrofuran as a function of temperature, Sandia Lab. 78-1414, Albuquerque, New Mexico, USA Search PubMed.
- Z. R. Grabowski, J. Dobkowski and W. Kühnle, Model compounds in study of the photophysical behaviour of carbonyl derivatives of N,N-dimethylaniline, J. Mol. Struct., 1984, 114, 93 CrossRef CAS.
- P. F. Barbara, P. M. Rentzepis and L. E. Brus, Photochemical kinetics of salicylidenaniline, J. Am. Chem. Soc., 1980, 102, 2786 CrossRef CAS.
- J. Spanget-Larsen, A structure-reactivity relationship for the basicity of aza-arenes, J. Chem. Soc., Perkin Trans. 2, 1985, 417 RSC.
- J. Herbich, C.-Y. Hung, R. P. Thummel and J. Waluk, Solvent-controlled excited state behaviour: 2-(2′-pyridyl)indoles in alcohols, J. Am. Chem. Soc., 1996, 118, 3508 CrossRef CAS.
- Calculated from the data given in: D. S. Viswanath, G. Natarajan, Data book on the viscosity of liquids, Hemisphere Publishing: New York, 1989 Search PubMed;
Landolt-Börnstein, Bd. II, 5 Teil, Springer Verlag, 1969, pp. 209–214 Search PubMed.
- J. B. Birks, Reversible photophysical processes, Nouv. J. Chim., 1977, 1, 453 Search PubMed.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |
Click here to see how this site uses Cookies. View our privacy policy here. 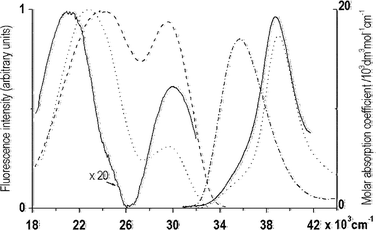
![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ), and corrected and normalized fluorescence spectra of DMAP in ethyl acetate (
), and corrected and normalized fluorescence spectra of DMAP in ethyl acetate (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), butyronitrile (⋯) and 1-propanol (—). Concentrations of the compounds were in the range from 10−5 to 10−4 mol dm−3.
), butyronitrile (⋯) and 1-propanol (—). Concentrations of the compounds were in the range from 10−5 to 10−4 mol dm−3.