Dynamics of Re(2,2′-bipyridine)(CO)3Cl MLCT formation and decay after picosecond pulsed X-ray excitation and femtosecond UV excitation†
Liyan
Zhao
,
Hideho
Odaka
,
Hiroshi
Ono
,
Shinji
Kajimoto
,
Koji
Hatanaka
,
Jonathan
Hobley
and
Hiroshi
Fukumura
*
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai, Miyagi, Japan. E-mail: fukumura@orgphys.chem.tohoku.ac.jp; Fax: ++81 22 217 6570; Tel: ++81 22 217 6567
First published on 16th September 2004
Abstract
The dynamics of Re(2,2′-bipyridine)(CO)3Cl MLCT state formation and decay were determined after femtosecond UV laser excitation and picosecond pulsed X-ray excitation, in an N,N-dimethylformamide (DMF) solution as well as in its solid form. At room temperature, after UV excitation, this MLCT excited state emits both in DMF solution and in the solid form. Transient absorption spectra were measured in solution at various delay times following excitation by a 160 fs, 390 nm laser pulse. There was a prompt absorption increase at around 460 nm occurring within the pump probe convolution (<1 ps), which was assigned to the formation of the 3MLCT state. This transient absorbance was constant over 100 ps. In contrast to the solution state, in the solid state, the emission maximum slightly red-shifts with increasing time after laser excitation. In both solid and solution the emission rises within the system response time. The solid sample exhibited a 1.4 ns emission decay that was not observed for the solution sample. The emission rise from a solid sample after 20 ps pulsed X-ray excitation was significantly slower than the system's time resolution. It is proposed that kinetically energetic electrons are ejected following X-ray induced ionisation, creating ionised tracks in which energetic cations and electrons take time to recombine yielding delayed 3MLCT states that emit.
Introduction
Radiation chemistry has been important in a wide range of applications such as dosimetry, industrial synthesis, sterilisation, and waste treatment.1 Further, an understanding of radiolysis is important in radioactive waste management.2,3 In terms of future application, fast scintillation media are required for uses such as positron emission tomography,4,5 nuclear and high energy physics, medical imaging, diffraction as well as nuclear treaty verification.6,7 For this reason it is necessary to develop faster pulsed radiation sources in order to understand the mechanisms of emission from scintillation detectors.8–11 Pulsed radiolysis has long played an important role in our understanding of radiation chemistry. Ultra-fast production of electron pulses has facilitated the study of a great number of transient states. For example scintillators,12 excited states13–15 and solvated electrons16 have all been studied. X-ray pulsed radiolysis has been less well studied, with a few experimenters employing expensive synchrotron sources.17–19 Recently bench-top X-ray sources have become available with X-ray pulse durations of around 100 ps9–11 and these have been used to investigate scintillation rise times as well as decay times. Fundamentally the rise time will depend upon the X-ray energy, the kinetic energy of electrons resulting from ionisation, the range of the electrons ejected by the X-ray20 as well as those ejected as secondary electrons and their counter ions. The recombination time of ions and electrons will depend on many such factors although the phenomenon is not well studied. For this reason we have employed a shorter pulsed X-ray source with a pulse duration of 20 ps.8 We have used this 20 ps pulsed X-ray source to induce emission from Re(bpy)(CO)3Cl, in which bpy is 2,2′-bipyridine.For many organometallic complexes, the metal to ligand charge transfer (MLCT) transition is one of the most important electronic transitions.21 MLCT transitions produce a transient change in the oxidation state of the central metal ion. The Re(bpy)(CO)3Cl complex contains a d6 metal and has a lowest excited state that is MLCT in character. The photophysics of this complex have been reported in various solvents,21–28 however the photophysical behaviour of this complex under femtosecond UV laser excitation has not been studied. There are no reports on the photophysical behaviour of Re(bpy)(CO)3Cl in its solid state. It is generally reported that emission from excited rhenium complexes such as Re(bpy)(CO)3Cl comes from the 3MLCT state.21,22,26,28 Convincing evidence for this is that the emission extends to microseconds at low temperature.26 In this paper we investigate the dynamics of Re(bpy)(CO)3Cl 3MLCT formation and decay under femtosecond UV laser excitation in N,N-dimethylformamide (DMF). We also report 3MLCT dynamics in the solid state after femtosecond UV laser excitation and picosecond pulsed X-ray excitation.
Experimental
All materials were reagent grade and used as received. The compound Re(bpy)(CO)3Cl was prepared as described previously.23 Equimolar quantities of Re(CO)5Cl (Aldrich) and 2,2′-bipyridine (Aldrich) were refluxed in toluene for 1 h. After cooling a yellow product precipitated from the solution, which was filtered, washed, air-dried and used without further purification. The expected elemental composition was: C, 33.81; H, 1.74; N, 6.06%. Using elemental analysis it was found that the composition was: C, 33.36; H, 1.96; N, 5.81%. Re(bpy)(CO)3Cl was dissolved in DMF to a concentration of 4 × 10−4 M. Time resolved studies were carried out at room temperature under aerated conditions. Solid samples were pressed pellets containing Re(bpy)(CO)3Cl and boron nitride with a mass ratio of 1 ∶ 1. UV-Vis absorption spectra were measured using a Shimadzu UV-1600 spectrophotometer. Uncorrected steady-state emission spectra were obtained using a Hitachi F-4500 spectrofluorimeter.The emission lifetime was determined after excitation by a NdYAG laser (Quantel, Brilliant) using the third harmonic (355 nm, FWHM = 6ns, <3 mJ pulse−1). Femtosecond transient absorption spectra were recorded after pulsed laser excitation by a Clark MXR, CPA2001 laser system. White light continuum (400–800 nm) probe pulses were generated by focusing a fraction of the 780 nm fundamental laser inside a 1 cm flow cell with a 1 ∶ 1 ratio of D2O ∶ H2O. The residual fundamental beam was doubled to provide a pump pulse at 390 nm (FWHM = 160 fs). Time resolution was achieved by sending the pump beam down an optical delay line and spectra were recorded from −1 to 100 ps after the pump pulse. The sample (100 cm3 volume) was pumped through a 1 cm quartz flow cell. The data was collected using an Ocean optics SD-2000 spectrograph with two fibre optic inputs. One was used as a reference to monitor the probe spectrum and the other was used to monitor light passing through the sample. The reference was used to account for spectral variance. The transient spectral change occurring in the sample when excited by the pump beam (IT/Iref) was related to the un-excited sample spectrum (Io/Iref) using the Beer–Lambert law in order to obtain a transient absorption difference spectrum. Each transient absorption spectrum was an average of 15 measurements with the pump beam on (3 s exposure at 1 kHz) and 15 with the pump beam off (3 s exposure at 1 kHz). The pump beam energy was 25μJ pulse−1. In total the 100 cm3 sample was irradiated for 15 minutes at 1 kHz. The solution's absorption spectrum was measured before and after the experiment and was unchanged, meaning that decomposition had not occurred to any significant degree.
Time-resolved emission spectral rises and early nanosecond decays were acquired after excitation with the same femtosecond laser. The 390 nm second harmonic was used to excite the sample. The pump pulses were adjusted to 2.8 μJ pulse−1 and the spectra and decays were collected for 1 h at 1 kHz using a Hamamatsu Photonics C2830 streak camera coupled to a thermoelectrically cooled charge coupled device (CCD) camera (Andor Technology). The sample again had a 100 cm3 volume and was flowed through a 1 cm quartz cell.
Picosecond pulsed X-ray was produced, as described previously,8 by irradiating a custom built Hamamatsu Photonics photoexcitable X-ray tube with fundamental light from the femtosecond laser. This X-ray source was used to excite the Re(bpy)(CO)3Cl to induce scintillation. Because of the weakness of this emission no spectral resolution was attempted and the entire emission was monitored, with all wavelengths binned, using the streak camera.
The X-ray source's spectrum was measured using a silicon detector (Rontec, XFlash 1000) and spectroscopy amplifier (Ortec 472) with a constant voltage of 15 kV applied to the X-ray tube.8
The pulse duration was estimated to be 20 ps by solving the equation of motion for electron flight inside the X-ray tube. This value was confirmed by measuring the cross luminescence rise time for a BaF2 crystal after X-ray excitation. The rise time for such crystals has been reported to be less than 2 ps,8,29 whereas the observed rise in our case was 20 ps.
Results and discussion
Solution photochemistry
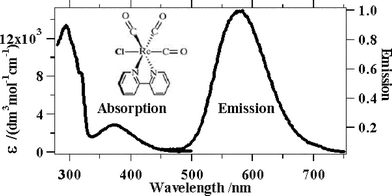 | ||
| Fig. 1 Absorption and emission spectra of Re(bpy)(CO)3Cl in DMF. | ||
The broad weaker absorption band centred at around 370 nm is assigned to the MLCT transition. The extinction coefficient of this transition is 2890 dm3 mol−1 cm−1, meaning that the transition, whilst not fully allowed compared to most charge transfer transitions, is also not fully forbidden, like singlet to triplet transitions.
The emission spectrum has a maximum at around 580 nm (uncorrected) which is moderately Stokes shifted from the absorption.
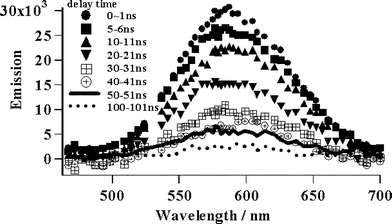 | ||
| Fig. 2 Decay of the 3MLTC state over 200 ns. | ||
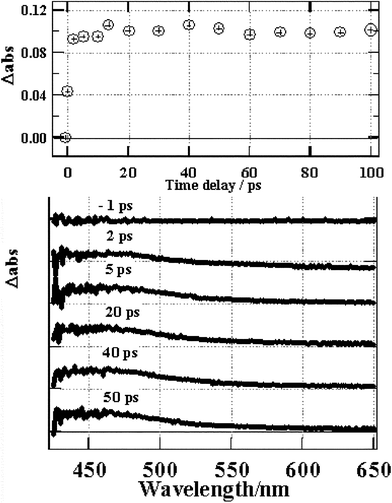 | ||
| Fig. 3 Transient absorption changes for Re(bpy)(CO)3Cl in DMF. | ||
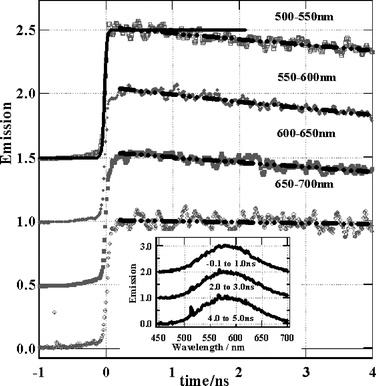 | ||
| Fig. 4 Emission spectra and rise of Re(bpy)(CO)3Cl in DMF. Solid line – instrument response function, dashed/dotted lines – fits to 26 ns lifetime decay. | ||
Solid state photochemistry
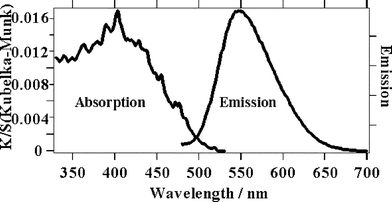 | ||
| Fig. 5 Absorption and emission spectra of solid Re(bpy)(CO)3Cl. | ||
The time resolved emission spectra of the Re(bpy)(CO)3Cl solid sample after femtosecond excitation irradiating at a fluence of 2.5 × 1013 W cm−2. (Fig. 6) exhibit a broad peak at 475–675 nm, which rises within the instrument response time. In contrast to the solution spectra, this emission peak undergoes a small red shift during 4 ns. The exact origin of the shift is unclear as the spectra are broad, however, the 390 nm excitation pulse has excess energy compared to the lowest energy of the 3MLCT transition meaning that the excited state will form vibrationally hot. In the case of solution, where the hot 3MLCT is surrounded by cold solvent, heat dissipation can be very rapid. However for solid samples the density of excited states is much higher and hot 3MLCT states will be in closer proximity to other hot 3MLCT states. In this case the migration of excess vibrational energy could be slower and it may be possible to observe hot bands for some time after excitation. Another possible reason for this red shift is that the solid surface may be inhomogeneous. It is known that the emission spectrum shifts depending on the rigidity of the medium22 and different lattice sites may have different spectra and dynamics. The kinetic traces for the emission lifetime have a 1.4 ns decay component at all wavelengths that is most pronounced in the shorter wavelength region. This 1.4 ns decay could be due to annihilation especially since the concentration of excited states at a solid surface will be extremely high even at fluences where a solution would show no second order events, but it should also contain a component of the spectral shift. It seems that two processes are occuring in tandem for us to see both a spectral shift and a 1.4 ns decay at all wavelength regions. In support of the assignment of the shift and the fast decay to a combination of annihilation and hot band, we observed that the emission spectral width was reduced at lower laser fluences (2.5 × 1013 W cm−2) and increased at higher fluence (1.6 × 1014 W cm−2). Also we found that the decay rate increased at the higher laser fluence compared to at the lower laser fluences. Such observations have been made after ps-laser excitation of solid samples of phthalocyanines.33
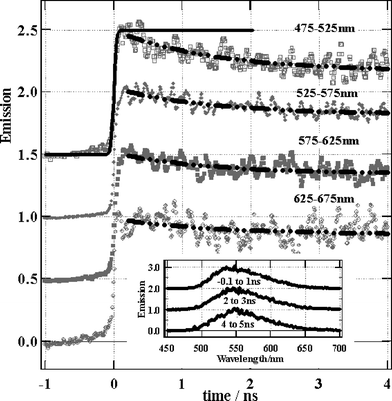 | ||
| Fig. 6 Emission spectra and rise of solid Re(bpy)(CO)3Cl. Solid line – instrument response function, dashed/dotted lines – fits to a lifetime of 1.4 ns. | ||
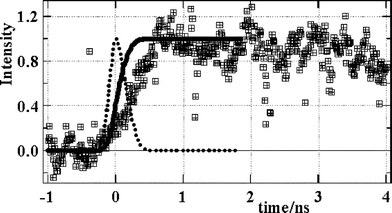 | ||
| Fig. 7 Emission rise of Re(bpy)(CO)3Cl solid pulsed X-ray excitation. Dotted line – X-ray pulse shape, solid line – integrated intensity profile, squares – emission. | ||
The ability of a medium to interact with electrons is well known to depend upon the electron energy.34,35 Therefore the excitation spectrum of the picosecond pulsed X-ray source should be considered alongside the Re(bpy)(CO)3Cl X-ray absorption spectrum36 (Fig. 8). Taking into account the excitation and absorption spectra, the X-ray excitation is stronger towards the region of the Re M-absorption edge at 1.96 keV.
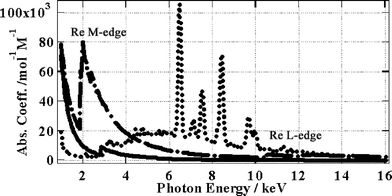 | ||
| Fig. 8 Dots – X-ray spectrum, dots and dashes – Re absorption spectrum, solid line – (bpy)(CO)3Cl absorption spectrum. | ||
From the information in Fig. 8 we can derive a photon absorption integral and then, taking into account the absorption and excitation spectra it is possible to estimate the relative numbers of electrons ejected with a given kinetic energy. In this way we can obtain the kinetic energy spectrum for ejected primary electrons (Fig. 9). Electrons with extremely high kinetic energy are, of course, harder to stop within a medium whereas those with lower kinetic energy are stopped more easily. However in our case we have a solid sample with a thickness of several mm. Since the most energetic electrons will have a maximum range of a few hundred nm and a running time of a few hundred femtoseconds, as calculated using Bethe's theory (ref. 20, Table 1), we consider that all electrons will rapidly be stopped within the sample causing large tracks of ionisation or spurs. Within these spurs electrons and excited ions will be created, which can give delayed emission from the MLCT state upon recombination. There is no reason to assume that the MLCT state initially formed by recombination is a triplet or a singlet state, however we assume that the recombined MLCT state would rapidly relax to its triplet state within a few hundred femtoseconds.30,31
| Energy/keV | Range/nm | Running time/fs |
|---|---|---|
| 1.6 | 4 | 14 |
| 2.9 | 25 | 56 |
| 4.5 | 51 | 95 |
| 6.5 | 93 | 140 |
| 13 | 290 | 320 |
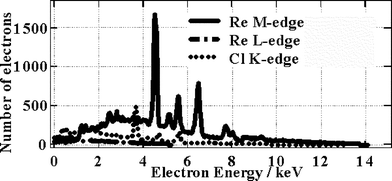 | ||
| Fig. 9 Electron energy spectrum. | ||
Note that X-ray excitation does cause inhomogeneous populations of excited states meaning that shot to shot variance can be expected, however in our case the data was accumulated over several hours meaning that we obtain a time averaged result. The amount by which the observed emission lags behind the X-ray excitation pulse is therefore not simply a direct result of the range and kinetic energy of the primary electrons that are ejected. As we have shown, these primary electrons will loose their energy within a few hundred femtoseconds. However we consider that during their lifetime these primary electrons will cause widespread radiation damage within spurs yielding secondary electrons and energetic cations. These electrons and cations can separate and survive for a longer time within the spurs, recombining later to give the evidently delayed emission that we observe experimentally. The extent of cation–electron separation should depend partly upon the original kinetic energy released during the collision with the primary electron. This in turn must depend upon the primary electron energy distribution (Fig. 9) as well as the stopping power of atoms within the medium. As can be seen, electrons with a wide range of kinetic energies are produced and these will have a variety of ranges and interactions.
Conclusion
Transient dynamics of the Re(bpy)(CO)3Cl MLCT state were determined for a solid sample after picosecond pulsed X-ray excitation. These dynamics were compared to those observed after femotosecond UV laser excitation using transient absorption and emission measurements in DMF and solid samples. In contrast to the case of femtosecond UV excitation, in the case of X-ray excitation, we observed a significant delay in the emission rise, which is explained by slower recombination of secondary electrons and cations within spurs.Acknowledgements
The authors are grateful to Hamamatsu Photonics for providing the X-ray tube and to Professor Masuhara from Osaka University for providing the high speed streak unit. H. F. is grateful to Professor Ikeda from Osaka University for helpful discussions. Z. L. is grateful to Tohoku University COE program (Unexplored Chemistry, Giant Molecules and Complex Systems) for providing a research fellowship. This work was partly financed by grant in aid 14077202.References
- A. Mozumunder, Fundamentals of Radiation Chemistry, Academic Press, San Diego, San Francisco, New York, Boston, London, Sydney, Tokyo, 1999, p. 361 Search PubMed.
- A. Barkatt and W. Sousanpour, Gamma radiolysis of aqueous media and its effects on the leaching processes of nuclear waste disposal materials, Nucl. Technol., 1983, 60(2), 218–227 CAS.
- S. Sunder and H. Christensen, Gamma radiolysis of water solutions relevant to the nuclear-fuel waste management program, Nucl. Technol., 1993, 104(3), 403–417 CAS.
- P. A. Rodnyi, Progress in fast scintillators, Radiat. Meas., 2001, 33, 605–614 CrossRef CAS.
- W. W. Moses and S. E. Derenzo, Prospects for time-of-flight PET using LSO scintillator, IEEE Trans. Nucl. Sci., 1999, 46, 474–478 CrossRef CAS.
- G. Knoll, Radiation Detection and Measurement, Wiley, New York, 3rd edn., 1999 Search PubMed.
- K. Kleinknecht, Detectors for Particle Radiation, 2nd edn., Cambridge University Press, Cambridge, UK, 1998 Search PubMed.
- (a) E. Garcia Villora, K. Hatanaka, H. Odaka, T. Sugawara, T. Miura, H. Fukumura and T. Fukuda, Luminescence of undoped β-Ga2O3 single crystals excited by picosecond X-ray and sub-picosecond UV pulses, Solid State Commun., 2003, 127, 385–388 CrossRef CAS; (b) K. Hatanaka, T. Miura, H. Odaka, H. Ono and H. Fukumura, Various methods for X-ray pulse generation using a femtosecond laser and their potential for time-resolved X-ray analyses, Bunseki Kagaku, 2003, 52(6), 373–381 CAS.
- S. E. Derenzo, W. W. Moses, S. C. Blankespoor, M. Ito and K. Oba, Design of a pulsed X-ray system for fluorescent lifetime measurements with a timing accuracy of 109 ps, IEEE Trans. Nucl. Sci., 1994, 41(3), 629–631 CrossRef CAS.
- S. C. Blankespoor, S. E. Derenzo, W. W. Moses, C. S. Rossington, M. Ito and K. Oba, Characterization of a pulsed X-ray source for fluorescent lifetime measurements, IEEE Trans. Nucl. Sci., 1994, 41(4), 698–702 CrossRef CAS.
- L. Papadopoulos, Rise time of scintillation emission in inorganic and organic scintillators, Nucl. Instrum. Methods Phys. Res., Sect. A, 1997, 401, 322–328 CrossRef CAS.
- D. L. Horrocks, High-resolution scintillation spectra obtained with nanosecond pulses of 3 MeV electrons, Photochem. Photobiol., 1972, 15(2), 239–239 CAS.
- G. Porter and J. W. Boag, Flash photolysis and pulsed radiolysis, Chem. Ind. (London), 1964,(21), 887–887.
- G. Scholes, M. Simic, G. E. Adams, J. W. Boag and B. D. Michael, Pulsed radiolysis of cyclohexane solutions - evidence for reactions of electrons, Nature, 1964, 204(496), 1187–1187 CAS.
- C. H. Marshall, Pulsed radiolysis and flash spectroscopy studies on amino acid solutions, Phys. Med. Biol., 1966, 11(1), 177–177 CrossRef.
- A. Mozumunder, Fundamentals of Radiation Chemistry, Academic Press, San Diego, San Francisco, New York, Boston, London, Sydney, Tokyo, 1999, pp. 156, 159 and 160 Search PubMed.
- V. A. Pustovarov, A. L. Krymov and E. I. Zinin, Time resolved luminescence of scintillation crystals under excitation by high-intensity synchrotron radiation, Nucl. Instrum. Methods Phys. Res., Sect. A, 1995, 359, 336–338 CrossRef CAS.
- V. A. Pustovarov, A. L. Krymov and B. V. Shulgin, Some peculiarities of the luminescence of inorganic scintillators under excitation by high-intensity synchroton radiation, Rev. Sci. Instrum., 1992, 63(6), 3521–3522 CrossRef CAS.
- N. Y. Kirikova, V. E. Klimenko, V. A. Kozlov, V. N. Makhov, N. M. Khaidukov and T. V. Uvarova, Cross luminescence of several complex fluorides excited by synchrotron-radiation, Nucl. Instrum. Methods Phys. Res., Sect. A, 1995, 359, 351–353 CrossRef CAS.
- Von H. Bethe, Zur Theorie des Durchgangs schneller Korpuskularstrahlen durch Materie, Ann. Phys., 1930, 5, 325–400 Search PubMed.
- A. J. Lees, Luminescence properties of organometalic complexes, Chem. Rev., 1987, 87, 711–743 CrossRef CAS.
- M. S. Wrighton and D. L. Morse, The nature of the lowest excited state in tricarbonyl-1,10-phenantholinerhenium(I) and related complexes, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- J. V. Casper and T. J. Meyer, Application of the energy gap law to nonradiative, excited-state decay, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- W. K. Smothers and M. S. Wrighton, Raman-spectroscopy of electronic excited organometallic complexes - a comparison of the metal to 2,2′-bipyridine charge-transfer state of FAC-(2,2′-bipyridine) tricarbonylhalorhenium and tris(2,2′-bipyridine) ruthenium(II), J. Am. Chem. Soc., 1983, 105, 1067–1069 CrossRef CAS.
- K. Kalyanasundaram, Luminescence and redox reactions of the metal-to-ligand charge-transfer excited state of tricarbonylchloro-(polypyridyl) rhenium(I) complexes, J. Chem. Soc., Faraday Trans., 1986, 2(82), 2401–2415 Search PubMed.
- L. A. Worl, R. Duesing, P. Chen, L. D. Ciana and T. J. Meyer, Photophysical properties of polypyridyl carbonyl complexes of rhenium(I), J. Chem. Soc., Dalton Trans., 1991, 849–858 RSC.
- M. W. George, F. P. A. Johnson, J. R. Westwell, P. M. Hodges and J. J. Turner, Excited state properties and reactivity of [ReCl(CO)3(2,2′-bipy)] (2,2′-bipy =2,2′-bipyridyl) studied by time-resolved infrared spectroscopy, J. Chem. Soc., Dalton Trans., 1993, 2977–2979 RSC.
- D. R. Striplin and G. A. Crosby, Nature of the emitting 3MLCT manifold of rhenium(I) (diimine) (CO)3Cl complexes, Chem. Phys. Lett., 1994, 221, 426–430 CrossRef CAS.
- C. W. E. Van Eijk, Cross-luminescence, J. Lumin., 1994, 60/61, 930–935 CrossRef.
- K-C. Tang, K. L. Liu and I-C. Chen, Rapid intersystem crossing in highly phosphorescent iridium complexes, Chem. Phys. Lett., 2004, 386, 437–441 CrossRef CAS.
- A. Vlcek, Jr., The life and times of excited states of organometallic and coordination compounds, Coord. Chem. Rev., 2000, 200–202, 933–977.
- P. Kubelka and F. Munk, Ein Beitrag zur Optik der Farbanstriche, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- M. Ichikawa, H. Fukumura and H. Masuhara, Ultrafast electron-transfer and recombination processes on copper phthalocynine solid/water interface as revealed by picosecond regular reflection spectroscopy, J. Phys. Chem., 1995, 99, 12072–12075 CrossRef CAS.
- D. Liljequist, A simple calculation of inelastic mean free-path and stopping power for 50 eV–50 keV electrons in solids, J. Phys. D: Appl. Phys., 1983, 16, 1567–1582 CrossRef CAS.
- F. Salvat and J. D. Martinez, R Mayol and J Parellada, A simple model fro electron-scattering-inelastic-collisions, J. Phys. D: Appl. Phys., 1985, 18, 299–315 CrossRef CAS.
- B. L. Henke, E. M. Gullikson and J. C. Davis, X-ray interactions: photoabsorption, scattering, transmission and reflection at E = 50–30000 eV, Z = 1–92, Atomic Data and Nuclear Data Tables, 1993, 54(2), 181–342 Search PubMed.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |