Picosecond–nanosecond laser photolysis studies of a photoacid generator in solutions: Transient absorption spectroscopy and transient grating measurements†
Mie
Saotome
,
Satoko
Takano
,
Asako
Tokushima
,
Syoji
Ito
,
Satoru
Nakashima
,
Yutaka
Nagasawa
,
Tadashi
Okada
and
Hiroshi
Miyasaka
*
Division of Frontier Materials Science, Graduate School of Engineering Science, and Research Center of Materials Science at Extreme Conditions Osaka University, Toyonaka, Osaka, 560-8531, Japan. E-mail: miyasaka@chem.es.osaka-u.ac.jp; Fax: +81-6-6850-6244
First published on 15th October 2004
Abstract
Photochemical reaction profiles of a photoacid generator, N-trifluoromethylsulfonyloxy-1,8-naphthalimide (NI-Sf), in solution phase were investigated by means of picosecond and nanosecond transient absorption spectroscopy as well as transient grating measurements. Picosecond transient absorption spectroscopy directly revealed that the lifetime of the S1 state and the intersystem-crossing yield decreased with increasing solvent polarity. On the other hand, photochemical reaction yield increased with an increase in the solvent polarity. These results indicated that the photochemical reaction started in the S1 state. Transient grating measurement directly detected the diffusion process of the proton and its diffusion coefficient was obtained to be 3.9 × 10−9 m2 s−1, which was several times larger than those of the usual solute molecules.
Introduction
Chemical amplification1,2 proposed in 1982 is one of the principle techniques to enhance the sensitivity of the photochemical reactions leading to the realization of the resist systems for use in short wavelength lithographic engineering in the semiconductor processing. The photoacid generator (PAG),3,4 which works through the photoproduction of a catalyst, takes an important role in the chemical amplification. The proton released in the initial photochemical reaction generates other protons in the subsequent reactions with polymers resulting in the increase of the reaction efficiency. A number of studies have been reported on the microlithography by using PAG systems to achieve the high resolution5 and to make clear the photochemical reaction profiles.3,5,6 In addition, the time-resolved measurements7–11 have been also applied to the direct detection of the reaction profiles of the PAGs.The elucidation of the reaction dynamics of the proton generation is of crucial importance for the rational designing of the efficient PAGs. In addition, the diffusion coefficient of the proton determines the efficiency of the reaction and the resolution in the lithography.5 Large diffusion coefficient increases the sensitivity in the chemical amplification but sometimes decreases the resolution in the lithography. In the present study, we have explored the photochemical reaction dynamics of one of the PAGs, N-trifluoromethylsulfonyloxy-1,8-naphthalimide, in solution phase by means of picosecond–nanosecond transient absorption spectroscopy, since a study of the solvent effect in the photochemical reaction provides basic information on the reaction mechanisms and predicts the reaction profiles in solid matrices. In addition, the transient grating method was applied in the present study to the measurement of the diffusion coefficient of the proton. This system directly detects the diffusion process of the proton and thus provides the diffusion coefficient in the medium. In the following, the reaction dynamics and the proton diffusion process will be discussed on the basis of the dynamic behaviors as revealed by time-resolved measurements and steady-state irradiation effects.
Experimental
A picosecond laser photolysis system with a repetitive mode-locked Nd3+:YAG laser was used for transient absorption spectral measurements.12,13 The third harmonic pulse (355 nm) with 15 ps fwhm and 0.5–1.0 mJ output power was used for excitation. The excitation pulse is focused into a spot with a diameter of ca. 0.15 cm. A picosecond white continuum, generated by focusing a fundamental pulse into a 10 cm quartz cell containing a D2O and H2O mixture (3 ∶ 1), was employed as a monitoring light. A sample cell with 1cm length was used in the measurement for transient absorption spectroscopy. The repetition rate of the laser excitation was kept low (<0.2 Hz) and the solution sample was circulated. For the nanosecond transient absorption spectroscopy, THG (355 nm) of nanosecond YAG laser (Quanta Ray, DCR3) with ca. 1 mJ output power and 5 ns pulse width was used as excitation light. The combination use of the Xe lamp as a monitoring light with the diode array detector with a gated image intensifier (Hamamatsu, PMA-50) provides the transient absorption spectra with ca. 10 ns temporal resolution. Steady-state fluorescence and absorption spectra were, respectively, recorded by Hitachi F-4500 fluorometer and Hitachi U-3500 spectrophotometer. Fluorescence decay curves were measured by a MCP photomultiplier (Hamamatsu, R1563U-01) combined with a digital oscilloscope (Lecroy 9362C). The time resolution of this detection method was typically 600 ps.N-sulfonyloxynaphthalimide (NI-Sf) (Midorikagaku) was used as received since the fluorescence and absorption spectra were identical with those in ref. 11. Chemical structures of NI-Sf and other reference samples are shown in Scheme 1. Methylcyclohexane (Wako, Special Grade), ethanol, acetonitrile (Wako, Infinity Pure) were used as received. Samples (ca. 10−5–10−4 M) were deaerated by irrigating with N2 gas.
 | ||
| Scheme 1 | ||
Transient grating signals were measured with the above nanosecond YAG laser as an excitation source (355 nm). A cw He–Ne laser at 632.8 nm with 2 mW output was used as a monitoring light. The block diagram of the transient grating measurement is shown in Fig. 1. The excitation pulse was split into two beams, which were crossed in the sample at an angle of θ to produce an interference pattern in the sample and induces the periodic absorption of the light in the solution, leading to the periodic variation in the complex refractive index. The pitch length of this interference pattern, Λ, is given by eqn.(1).
 | (1) |
 | (2) |
 | ||
| Fig. 1 Block diagram for the transient grating measurement. | ||
Typical signal due to the thermal diffusion process in pyrene in ethanol solution (q2 = 4.65 × 1012 m2) is shown in Fig. 2(a). The decay profile was observed over several µs. The dependence of the decay rate constant on q2 is exhibited in Fig. 2(b), showing that the decay rate constant increases linearly in proportion to q2 and the intercept is zero. The thermal diffusion coefficient, Dth, which corresponds to half of the value of the slope in Fig. 2(b), is in relation to the heat conductivity, κ, represented by κ = σCpDth. Here, σ and Cp are, respectively, the density of the liquid ethanol and the heat capacity at constant pressure. By using the reference values19 of σ and Cp, heat conductivity, κ, was obtained to be 0.165 W m−1 K−1, of which value was confirmed to be same with the reference data19 of 0.166 W m−1 K−1 within the experimental error. All the spectroscopic measurements were performed at 22 ± 2 °C.
 | ||
| Fig. 2 (a) Transient grating signal of pyrene in ethanol solution excited with nanosecond 355 nm laser pulse and monitored at 632.8 nm. (b) Dependence of the decay constant on q2. | ||
Results and discussion
Evolution of the steady-state absorption spectra under photo-irradiation
Fig. 3(a) shows the time evolution of the absorption spectra of NI-Sf in acetonitrile solution under the irradiation with the nanosecond 355 nm laser pulse with 1 mJ output power and 10 Hz repetition rate. Laser beam was moderately focused into a spot with a diameter of ca. 5 mm and the solution was circulated. With an increase in the number of the laser shots, the absorption spectra gradually evolve into the blue-shifted band shape with a maximum at 295 nm from the initial spectrum with a maximum at 332 nm. In order to confirm the photoacid generation, the time evolution of the absorption spectra of NI-Sf in acetonitrile solution with Crystal Violet was also measured under steady-state UV Xe lamp irradiation (Fig. 4). With an increase in the exposure time, the absorption spectrum gradually evolves; the decrease in the absorption band around 330 nm and the increase in the 295 nm band in the UV region as observed in Fig. 3, together with the decrease in the 585 nm band and the increase in the 630 nm absorption. The appearance of the band at 630 nm is attributed to the production of the protonated Crystal Violet.20 | ||
| Fig. 3 (a) Time evolution of the steady-state absorption spectrum of NI-Sf in acetonitrile solution under the irradiation with nanosecond 355 nm laser pulse with 1 mJ output power and 10 Hz repetition rate. (b) Steady-state absorption spectrum of NI-OH in acetonitrile solution. | ||
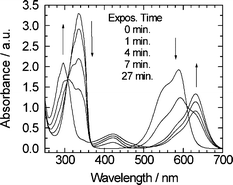 | ||
| Fig. 4 Time evolution of the absorption spectrum of NI-Sf in acetonitrile solution with Crystal Violet under the steady-state irradiation of UV Xe lamp. | ||
Since the spectral evolution keeps an isosbestic point at 308 nm, the photochemical reaction did not lead to complex decomposition of the initial NI-Sf. No remarkable spectral evolution was observed by further irradiation after 1450 shots of the nanosecond 355 nm laser in Fig. 3(a). However, the spectrum gradually evolved in several hours—a few days’ time region even, in the condition that the solution was stored in the dark place after the laser irradiation.
Prior to the discussion on the spectral evolution in longer time scale, we concentrate on the spectral change during the laser irradiation. In Fig. 3(b), we show the absorption spectrum of NI-OH (R1 in Scheme 1), indicating that the absorption maximum and spectral band shape are almost the same as those of the NI-Sf before the irradiation. In addition, it was reported that the spectrum of NI-OH was almost identical with that of NI-H (R2).21 Hence, the photoproduct observed just after the irradiation cannot be ascribed to NI-OH or NI-H. Since the spectral band shapes and absorption maximum are almost the same among NI-Sf, NI-OH, and NI-H, the ring-opening process may participate in the initial step of the photoreaction. Actually, our preliminary studies on the reaction products by NMR spectroscopy indicated that the 1,8-substituted naphthalene derivatives produced via the ring-opening reaction was confirmed.22 On the mechanisms of the acid generation process of NI-Sf, it has been proposed11 that the initial step of the reaction of the proton generation was the heterolytic cleavage, NI-Sf + hν → NI − + Sf+, and the subsequent reaction with water, Sf − + H2O → Sf-OH2+ → Sf-OH + H+. Fig. 3, 4 and 7, however, showed that the ring-opening reaction took place in the steady-state light irradiation, although the heterolytic cleavage product NI-OH might participate in the initial stage after the excitation.
In order to obtain the photochemical reaction yield, we have analyzed the time evolution of the absorbance. Fig. 5 shows the evolution of the absorbance at 335 and 295 nm as a function of the number of the laser shots, Nlaser. These figures indicate that the change of the absorbance was almost completed in the region Nlaser > ca. 1000. The solid line is the result calculated with the following equations that are derived for the steady-state light irradiation.
 | (3) |
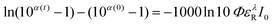 | (4) |
 | (5) |
 | ||
| Fig. 5 Time evolutions of the absorbance NI-Sf in acetonitrile solution under the irradiation with nanosecond 355 nm laser pulse with 1 mJ output power and 10 Hz repetition. (a) monitored at 335 nm and (b) at 295 nm. Solid lines are the results calculated by eqn. (4). | ||
The solid line in Fig. 5(a) is the result calculated with the excitation laser intensity, the concentration of the reactant, the amount of the sample solution, the extinction coefficients of the reactant and the product, and the parameter of the reaction quantum yield. The absorbance at 355 nm was the summation of ε355R CR(t) and ε355PCP(t). As shown in Fig. 5(a), the reaction yield of 0.22 reproduces the experimental results well. In addition, this value also reproduced the experimental result monitored at 295 nm where the rise of the product has major contribution. Since the present reaction yield is almost the same with the photo-acid generation yield of NI-Sf in acetonitrile (0.17),11 the present spectral evolution is closely related to the photo-acid generation process.
With an increasing time after the irradiation, the spectrum in acetonitrile gradually evolved as mentioned in previous sections. The spectrum after 240 hours following the irradiation is shown in Fig. 6, together with that of benzo[cd]indol-(1H)-one (R3 in Scheme 1). This figure suggests the presence of R3 in the solution after the irradiation. From the NMR spectroscopy, it was also confirmed that this species was included as one of the products in acetonitrile solution kept in dark place for several days after the photo-irradiation.
![Absorption spectrum of NI-Sf in acetonitrile observed at 240 h after the irradiation with 1500 shots of nanosecond 355 nm laser pulses (solid line) and that of benzo[cd]indol-(1H)-one (R3) (dotted line).](/image/article/2005/PP/b411283k/b411283k-f6.gif) | ||
| Fig. 6 Absorption spectrum of NI-Sf in acetonitrile observed at 240 h after the irradiation with 1500 shots of nanosecond 355 nm laser pulses (solid line) and that of benzo[cd]indol-(1H)-one (R3) (dotted line). | ||
The spectral evolution induced by the irradiation of the nanosecond laser pulse at 355 nm was also observed for NI-Sf in ethanol solution (Fig. 7(a)). With an increase in the number of the laser shots, the absorption maximum around 330 nm gradually decreases together with the increase in the absorbance in the 250–300 nm and 360–400 nm regions. As in the result in acetonitrile, the spectral evolution indicates that neither the NI-OH nor NI-H is the photoproduct as observed in the steady-state absorption spectra. Compared to the results in acetonitrile, one can find that the increase in the absorbance in the longer wavelength region is special to the ethanol solution and a much larger number of the laser shots was necessary to induce the spectral evolution. The appearance of the new absorption band in the wavelength region >360 nm suggests that photoproducts in ethanol solution are different from those in acetonitrile and/or that the long time irradiation due to small reaction yield leads to the reaction taking place in the dark place in acetonitrile.
 | ||
| Fig. 7 (a) Time evolution of the steady-state absorption spectrum of NI-Sf in ethanol solution under the irradiation with nanosecond 355 nm laser pulse with 1 mJ output power and 10 Hz repetition rate. (b) Time evolution of the absorbance of NI-Sf in ethanol solution monitored at 335 nm under the irradiation with nanosecond 355 nm laser pulse with 1 mJ output power and 10 Hz repetition. Solid lines are the results calculated by eqn. (4). | ||
Fig. 7(b) shows the time evolution of the absorbance at 335 nm of NI-Sf in ethanol solution irradiated with nanosecond 355 nm laser pulse. By assuming that the absorption spectrum obtained after several hours exposure of the steady-state Xe lamp is that of the photoproduct, the reaction yield was estimated to be 7%. Although the possibility that some other products are generated limits the reliability of this estimation, it can be safely concluded that the photochemical reaction in ethanol takes place with a much smaller reaction yield than in acetonitrile. In methylcyclohexane solution, the absorption change under the photoirradiation was very small and the quantum yield was estimated to be ≪1%.
Picosecond transient absorption spectra of NI-Sf
Fig. 8(a) shows transient absorption spectra of NI-Sf in acetonitrile solution, excited with a picosecond 355 nm laser pulse. The absorption at 450 nm and broad band in the wavelength region longer than 800 nm appeared with a time constant almost identical with the response function of the apparatus. With an increase in the delay time after the excitation, these absorption bands decreased and absorption bands at 450 and 475 nm remain in nanosecond time region. This absorption spectrum is safely ascribed to the T1 state of NI-Sf since the absorption maximum and the spectral band shape are identical with those reported previously.11 | ||
| Fig. 8 (a) Transient absorption spectra of NI-Sf in acetonitrile solution, excited with a picosecond 355 nm laser pulse. (b) Time profile of the transient absorbance at 450 nm following the excitation with a picosecond 355 nm laser pulse. | ||
Fig. 8(b) shows the time profile of the transient absorbance at 450 nm in the ps–ns time region, indicating that the transient absorbance decreases in the subnanosecond time region and a constant value region remains for several ns. This constant value is safely attributable to the contribution from the T1 state as explained above. The decay profile in subnanosecond time region was well reproduced as a single exponential process with a time constant of 520 ps. Since this decay time constant agrees with the fluorescence lifetime of NI-Sf (480 ps)11 within the experimental error, the transient absorption spectrum in the early stage after the excitation is safely assigned to the Sn ← S1 transition of NI-Sf.
Fig. 9(a) shows transient absorption spectra of NI-Sf in ethanol solution, excited with a picosecond 355 nm laser pulse. As observed in acetonitrile solution, the absorption at 450 nm and broad spectrum in the wavelength region longer than 800 nm appeared with the time constant almost identical with the response function of the apparatus and the absorption bands at 450 and 475 nm ascribable to the T1 state remain in nanosecond time region. Actually, the nanosecond transient absorption spectra observed at 500 ns following the excitation was identical with those observed at several ns following the excitation. (Fig. 9(b)).
 | ||
| Fig. 9 (a) Transient absorption spectra of NI-Sf in ethanol solution, excited with a picosecond 355 nm laser pulse. (b) Transient absorption spectra of NI-Sf in ethanol solution, observed at 500 ns after the excitation with a nanosecond 355 nm laser pulse. (c) Time profile of the transient absorbance at 450 nm following the excitation with a picosecond 355 nm laser pulse. | ||
The time profile of the transient absorbance at 450 nm shown in Fig. 9(c) indicates that the transient absorbance decreases in subnanosecond time region. The time constant of the decay was obtained to be 720 ps, of which value agrees with the fluorescence lifetime of 740 ps obtained by using the MCP detector with the picosecond 355 nm laser excitation. Since the decay time constant of the absorption at 450 nm agrees with the fluorescence lifetime and the spectral band shape and the absorption maximum are identical with those in acetonitrile, we can safely assigned this absorption spectrum to the Sn ← S1 transition.
Although the temporal evolution of the transient absorption spectra in ethanol solution is similar to that in acetonitrile solution, one can find that the remaining absorption in nanosecond time region relative to the absorption intensity of S1 state is larger in ethanol solution. The ratio A475(5 ns)/A450(40 ps) in acetonitrile solution was 0.13, while this ratio was 0.26 in ethanol solution. Here, A475(5 ns) and A450(40 ps) are, respectively, the transient absorbance at 475 nm observed at 5 ns and that at 450 nm observed at 40 ps after the excitation. This ratio provides the relative quantum yield of the intersystem crossing. Since the absorption band shapes of S1 and T1 states are similar in these two solvents, the small value of A475(5 ns)/A450(40 ps) in acetonitrile solution indicates that the intersystem crossing quantum yield is smaller than that in ethanol. The small quantum yield of the intersystem crossing with the shorter fluorescence lifetime indicates that the contribution from the some competitive processes in the decay of S1 state is larger in acetonitrile. By integrating the dynamic behaviors with the solvent effect of the reaction yields as shown in Fig. 5 and 6, it is concluded that the photochemical reaction occurs in the S1 state. That is, the increase in the reaction channel in acetonitrile solution reduced the fluorescence lifetime and the quantum yield of the intersystem crossing. Actually, longer fluorescence lifetime of 1.1 ns and larger relative quantum yield of the intersystem crossing, more than twice larger than that in ethanol solution, were confirmed in methylcyclohexane solution where the photochemical reaction yield was more than ten times smaller than that in ethanol. These results were consistent with the report that the photochemical reaction leading to the acid generation originates from the S1 state.11
Transient grating measurements
Although the S1 state was responsible for the initial reaction, the time region of the photoacid generation was not clear in the transient absorption measurements. To determine the time domain of the proton generation in the photochemical reactions, we have measured the transient grating signals in acetonitrile solution.Fig. 10 shows the transient grating signal observed in the NI-Sf in acetonitrile solution excited with a nanosecond 355 nm with q2 = 2.0 × 1012 m−2. The large grating signal appearing around the time origin decays rapidly, followed by the rise and the decay in sub-ms time region. As was mentioned in the experimental section, the transient grating signals in solutions in µs–ms time regions are usually interpreted as due to the thermal diffusion and the translational diffusion of the photoproducts. In the case where the sign of the δns in eqn. (2) are different from each other, the rise component as observed in Fig. 10 appears.14,15 The decay time constant in the hundreds of microseconds time region in Fig. 10 was obtained to be 70.0 µs.
 | ||
| Fig. 10 Transient grating signal of NI-Sf in acetonitrile solution excited with nanosecond 355 nm laser pulse and monitored at 632.8 nm. | ||
Fig. 11 exhibits the relation between the decay constant and the q2 value, showing that the decay rate constant is linearly in proportion with q2 and the intercept is zero. This result indicates that the decay signal is due to the diffusion process. From the slope of Fig. 11, the diffusion coefficient was obtained to be 3.9 × 10−9 m2 s−1. This diffusion coefficient may be attributed to the translational diffusion of the parent reactant, the photoproducts, the triplet state, and the proton in the present case. Since the triplet state in the present condition decreased within 10 µs, the present rate constant in Fig. 10 and 11 cannot be ascribed to the triplet state. Translational diffusion coefficients of many kinds of chemical species were studied by means of the transient grating methods. The present diffusion coefficient is three–four times larger than those of the solute molecules with similar size in acetonitrile solution. In actual fact, it has been reported that the diffusion coefficient of protons in methanol solution was a few times larger than that of the solute molecules.16
 | ||
| Fig. 11 Relation between the q2 value and the decay rate constant of NI-Sf in acetonitrile solution, excited with nanosecond 355 nm laser pulse and monitored at 632.8 nm. | ||
In order to clearly elucidate the origin of the signal, the effect of pyridine for the transient grating signal was investigated. In Fig. 12(a), the time profile of the transient grating signal with q2 = 2.0 × 10−12 m−2 in acetonitrile solution was shown. Compared to the time profile in Fig. 10, much slower decay was observed in Fig. 12(a). The dependence of the decay constant on q2 is shown in Fig. 12(b), indicating that the decay rate constant is linearly in proportion with q2 and the intercept is zero. This result indicates that the decay signal is due to the diffusion process. From the slope of Fig. 12(b), the diffusion coefficient was obtained to be 1.8 × 10−9 m2 s−1. Also in the solution with 10−4 M pyridine, similar result was obtained and the diffusion coefficient was 1.5 × 10−9 m2 s−1. Since the pKa value of pyridine in acetonitrile is 12.3, the proton is captured by pyridine. The present results that the large diffusion coefficient is replaced with the small coefficient in the presence of pyridine supports that the large diffusion coefficient observed in Fig. 10 is due to the diffusion process of the proton produced in the photo-acid generation process.
 | ||
| Fig. 12 (a) Transient grating signal of NI-Sf in acetonitrile solution with 10−6 M pyridine, excited with nanosecond 355 nm laser pulse and monitored at 632.8 nm. (b) Dependence of the decay constants on the q2 value. | ||
The above result show that the proton is generated in the time region ≤µs since the transient grating signal ascribable to the translational diffusion of the proton was observed in several tens–several hundreds of µs region. It is worth noting here that the diffusion coefficient of the proton in acetonitrile is close to that in methanol,16 3.1 × 10−9 m2 s−1, suggesting that the proton in acetonitrile solution is attached to some molecules and the association and dissociation process may be involved in the diffusion process. On this point, we are now going on to a more detailed study, the results of which will be reported soon.
Acknowledgements
The authors express sincere gratitude to Dr T. Itani, Dr T. Yamasaki, and Dr M. Toriumi for their advice. This work was partly supported by a Grant-in-Aid for Scientific Research on Priority Areas (417) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of the Japanese Government.References
- H. Ito and C. G. Willson, in Polymers in Electronics, ACS symposium series 242, ed. T. Davidson, American Chemical Society, Washington, DC, 1984, ch. 1 and references therein Search PubMed.
- H. Ito, Advances in chemical amplification resist systems, Jpn. J. Appl. Phys., 1992, 31, 4273–4282 CAS.
- S. P. Pappas, Photogeneration of Acid: Part 6 Review of Basic Principles for Resist Imaging Applications, J. Imaging Technol., 1985, 11, 146–157 Search PubMed and references therein.
- M. Shirai and M. Tsunooka, Photoacid and photobase generators: Chemistry and applications to polymeric materials, Prog. Polym. Sci., 1996, 21, 1–45 CrossRef CAS.
- T. Itani, H. Yoshino, S. Hashimoto, M. Yamana, N. Samoto, and K. Kasama, in Micro- and Nanopatterning Polymers, ACS symposium series 706, ed. H. Ito, E. Reichmanis, O. Nalamasu and T. Ueno, American Chemical Society, Washington, DC, 1998, ch. 9 and references therein Search PubMed.
- S. Nagahara, Y. Sakurai, M. Wakita, Y. Yamamoto, S. Tagawa, M. Komuro, E. Yano and S. Okazaki, Methods to improve radiation sensitivity of chemically amplified resists by using chain reactions of acid generation, Proc. SPIE, 2000, 3999, 386–394 Search PubMed.
- T. Tilley, B. Pappas, S. P. Pappas, Y. Yagci and J. K. Thomas, Laser flash photolysis studies on iodonium and sulfonium salts, J. Imag. Sci., 1989, 33, 62–64 Search PubMed.
- T. Iwamoto, S. Nagahara and S. Tagawa, Laser flash photolysis studies on chemically amplified resists 1. Femtosecond laser flash photolysis of diphenyliodonium salts, J. Photopolym. Sci. Technol., 1998, 11, 455–458 CAS.
- S. Tagawa, S. Nagahara, T. Iwamoto, M. Wakita, T. Kozawa, Y. Yamamoto, D. Werst and A. D. Trifunac, Radiation and photochemistry of onium salt acid generators in chemically amplified resists, Proc. SPIE, 2000, 3999, 204–213 Search PubMed.
- K.K. Iu, J. Kuczynski, S. J. Fuerniss and J. Kerry, Thomas, Laser flash photolysis of arylsulfonium salts: studies of photoproduced proton kinetics and mechanism in polar solvents by a pH-jump method, J. Am. Chem. Soc., 1992, 114, 4871–4878 CrossRef CAS.
- F. Ortica, J. C. Scaiano, G. Pohlers, J. F. Cameron and A. Zampini, Laser flash photolysis study of two aromatic n-oxyimidosulfonate photoacid generators, Chem. Mater., 2000, 12, 414–420 CrossRef CAS.
- H. Miyasaka, T. Moriyama, S. Kotani, R. Muneyasu and A. Itaya, Picosecond dynamics of photoinduced electron transfer processes in poly(N-vinylcarbazole) solid film doped with electron acceptors as revealed by transient absorption spectroscopy and dichroism measurements, Chem. Phys. Lett., 1994, 225, 315–321 CrossRef CAS.
- H. Miyasaka, T. Moriyama and A. Itaya, Direct detection of hole migration along the polymer chain: poly(N-vinylcarbazole) in 1,2-dichloroethane solution as revealed by picosecond transient absorption and dichroism measurements, J. Phys. Chem., 1996, 100, 12609–12615 CrossRef CAS.
- M. Terazima, K. Okamoto and N. Hirota, Diffusion process of methyl red in organic solvents studied by the transient grating method, J. Phys. Chem., 1993, 97, 5188–5192 CrossRef CAS.
- T. Hara, N. Hirota and M. Terazima, New Application of the Transient Grating Method to a Photochemical Reaction: The Enthalpy, Reaction Volume Change, and Partial Molar Volume Measurements, J. Phys. Chem., 1996, 100, 10194–10200 CrossRef CAS.
- K. Takeshita, N. Hirota and M. Terazima, Enthalpy changes and reaction volumes of photoisomerization reactions in solution: azobenzene and p-coumaric acid, J. Photochem. Photobiol. A: Chem., 2000, 134, 103–109 CrossRef CAS.
- A. Ukai, N. Hirota and M. Terazima, Diffusion of Organic Molecules in the Excited Triplet States Detected by the Transient Grating with a High Wavenumber, J. Phys. Chem. A, 2000, 104, 6681–6688 CrossRef CAS.
- J. Choi, N. Hirota and M. Terazima, A pH-Jump Reaction Studied by the Transient Grating Method: Photodissociation of o-Nitrobenzaldehyde, J. Phys. Chem. A, 2001, 105, 12–18 CrossRef CAS.
- Kagaku Binran II, Chemical Society of Japan, Maruzen, Tokyo, 1992, p. 68 Search PubMed.
- J. L. P. Jessop, S. N. Goldie, A. B. Scranton and G. J. Blanchard, Spectroscopic characterization of acid generation and concentration and free volume evolution in chemically amplified resists, J. Vac. Sci. Technol., B, 2002, 20, 219–225 CrossRef CAS.
- J. Gawronski, K. Gawronska, P. Skowronek and A. Holmen, 1,8-Naphthalimides as stereochemical probes for chiral amines: a study of electronic transitions and exciton coupling, J. Org. Chem., 1999, 64, 234–241 CrossRef CAS.
- M. Saotome, S. Takano, H. Miyasaka, H. Takaya, Y. Imada and T. Naota, in preparation.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |