Photophysical behavior and intramolecular energy transfer in Os(II) diimine complexes covalently linked to anthracene†
G. Christian Balazs, André del Guerzo and Russell H. Schmehl*
Department of Chemistry, Tulane University, New Orleans, LA 70118, USA. E-mail: russ@tulane.edu
First published on 4th November 2004
Abstract
The photophysical behavior of two Os(II) complexes having a bipyridine ligand with anthracene attached directly to the bipyridine (4-(9-anthryl)-2,2′-bipyridine, bpy-AN) is reported. The two complexes [(bpy)2Os(bpy-AN)]2+ and [(bpy-AN)2Os(CO)Br]+ have 3MLCT excited states that differ in energy by less than 800 cm−1. Despite this fact, the observed photophysical behavior of the two complexes is entirely different. The complex with the higher energy 3MCLT state, [(bpy-AN)2Os(CO)Br]+, is nonemissive at room temperature, but has a long lived excited state that is localized on the 3(π–π*) state of the anthracene substituent. The other complex, [(bpy)2Os(bpy-AN)]2+, exhibits emission at room temperature and has a transient absorption spectrum that is consistent with a localized 3MLCT state. The excited state decay behavior of the two complexes can be fit well assuming a model in which noninteracting 3MLCT and 3(π–π*) states are in equilibrium.
Introduction
The photophysical behavior of transition metal complexes having metal-to-ligand charge transfer (MLCT) excited states has been examined in great detail.1–7 Many complexes in this class are excellent sensitizers for one-electron transfer photoredox processes not only because the excited states are good oxidants and reductants, but also because they have reasonably long excited state lifetimes and are stable to photodegradation. Thus, understanding the factors that control excited state relaxation in complexes with MLCT states is directly related to the design of complexes with photophysical behavior optimized for sensitization of photoredox reactions.Most emissive complexes of this class have singlet ground states and the emitting state is believed to have a high degree of triplet character, coupled to the singlet state to some degree through spin–orbit coupling interactions.8,9 Excited state decay follows the energy gap formalism developed by Freed, Jortner and others, stating that the nonradiative relaxation rate constant increases exponentially with decreasing energy gap between the ground and excited state potential surfaces.10–13 However, it is clear that other factors can dramatically influence nonradiative relaxation rate constants of MLCT states. For instance, early studies of temperature-dependent excited state decay clearly showed that rate constants for decay were dependent on the degree of singlet character in the emitting excited state. Meyer and co-workers have presented convincing evidence that MLCT excited state distortion can also have a major impact on excited state relaxation dynamics (the rate constant for nonradiative decay decreases with decreasing distortion).4,14,15
Nonradiative relaxation is also influenced by thermally activated internal conversion to close lying states.6,16 For instance bis-terpyridyl Ru(II) ([Ru(tpy)2]2+) has an excited state lifetime of less than 1 ns in solution at room temperature while tris-bipyridyl Ru(II) ([Ru(bpy)3]2+) has a lifetime in deaerated acetonitrile of more than 800 ns.6 The large difference in lifetimes is due to very facile thermally activated internal conversion from the emitting 3MLCT state to a triplet ligand field (3LF) state in [Ru(tpy)2]2+; the stronger ligand field of the bipyridyl complex results in a larger barrier for population of the 3IL state. More recently it has been observed that ligands having triplet excited states (3IL states) that are nearly isoenergetic with the 3MLCT state can lead to large increases in the lifetime of the MLCT state via reversible energy transfer between the 3MLCT state and the triplet state localized on the ligand.17–19Fig. 1 illustrates a state diagram typical of complexes of this type. With aromatic hydrocarbon ligands (or aromatic hydrocarbon substituents on coordinated ligands), the singlet–triplet energy gap of the ligand (or substituent) is generally much larger than the same energy gap for the MLCT state of the metal complex. As a result, the MLCT state can be selectively excited and energy migration to populate the triplet state localized on the ligand (substituent) occurs via the 3MLCT state. For example, Ru(II) bipyridyl complexes having covalently tethered pyrene have been shown to have excited state lifetimes more than 50 times that of the parent [Ru(bpy)3]2+ as a result of reversible population of a triplet state localized on the pyrene.20–24
 | ||
| Fig. 1 Energy level diagram for complexes having MLCT states interacting with ligand localized states. | ||
The rate of excited state relaxation in such complexes depends on a variety of factors. Fig. 1 represents the simplest possible diagram for explaining decay in complexes having a 3MLCT state that interacts with a 3IL state. If emission occurs only from the 3MLCT state, the observed lifetime will be a function of the four rate constants shown in Fig. 1. If energy transfer, kMA, is much faster than decay to the ground state of both the 3MLCT and 3IL states, the two excited states will be in equilibrium and the relative population of each state during the excited state lifetime will be a function of the energy gap between the two states.18 If, however, one state decays at a rate fast relative to the either forward or reverse energy transfer, the two states will not be in equilibrium and the population of one of the two states will dominate for kinetic reasons.
In this paper we report the photophysical behavior of two Os(II) complexes having the anthracene substituted bipyridine derivative shown below (bpy-AN). The two complexes, [(bpy-AN)Os(bpy)2]2+ and [(bpy-AN)2Os(CO)Br]+, have 3MLCT excited states differing only slightly in energy; however, in one complex the 3MLCT state is the predominant state occupied and in the other the 3IL (anthracene 3π–π* state) is the only state observed. The behavior is discussed in terms of the kinetic scheme presented in Fig. 1. There have been a few earlier studies of energy transfer between 3MLCT states of Os(II) complexes and anthracene. Intermolecular energy transfer between a series of Os(II) diimine complexes and either anthracene or benzanthracene was discussed by Meyer and co-workers. The evaluation of vibrational overlap factors allowed estimation of electronic coupling for the outer sphere bimolecular exchange energy transfer process.25 More recent work by Ziessel26 and Campagna27 discusses intramolecular energy transfer between Os(II) diimine or triimine chromophores and covalently linked anthracene. In both cases evidence is presented for reversible energy transfer between the 3MLCT state of the Os(II) complex and the 3(π–π*) of the anthracene. This work demonstrates that slight changes in the energy of the Os(II) diimine 3MLCT state can have a dramatic impact on the photophysical behavior observed. In more practical terms, subtle changes in the chromophore energetics can lead to large changes in the excited-state lifetime of the potential sensitizer.
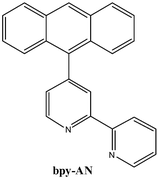
Experimental
Syntheses: 4-(9-anthryl)-2,2′-bipyridine (bpy-AN)
The synthesis of this ligand has been reported earlier;28 however, the approach employed in this work differs and is reported as follows. 9-Anthraldehyde (Lancaster Synthesis Inc., 3.59 g, 0.0174 mol) was dissolved in absolute ethanol (1.0 L). 300 ml of deionized water was slowly added to the ethanol solution. Pyruvic acid sodium salt (TCI, 5.63 g, 0512 mol) dissolved in 50 ml of water was slowly added and the solution was immersed in an ice-bath. 10.97 g KOH dissolved in 25 ml water was added dropwise over a period of approximately 3 min, and the resultant yellow–orange solution was stirred for 0.5 h. An additional 9.67 g KOH in 25 ml water was added, and the solution was allowed to stir an additional 2 h. After acidification with 50% aqueous HCl, the solution was reduced to half volume, and the precipitate was filtered and washed with water and cold ethanol. Drying in a vacuum oven at 85 °C overnight gave 3.4 g of an orange–red solid.The intermediate (2.0 g, 7.2 mmol) was dissolved 150 ml water in a round-bottom flask, and 2-acetylpyridine pyridinium hexafluorophosphate (2.74 g, 8.0 mmol) and ammonium acetate (4.28 g, 56 mmol) were added. The solution was refluxed under Ar for 9 h, during which time a light brown precipitate formed. This precipitate was filtered off, placed in a tube furnace, and heated under vacuum at 210 °C for 10 min. The resultant black gum was dissolved in chloroform and run through a short plug of alumina (Aldrich, activated, neutral, Brockmann I, ∼150 mesh). HCl gas was bubbled through the collected eluent, yielding a precipitate, which was filtered, washed multiple times with chloroform, and dried under vacuum to yield a light yellow solid ((Hbpy-AN)Cl, 0.324 g, 0.97 mmol, 13.4%).
[Os(bpy-AN)(bpy)2](PF6)2
[Os(bpy)2(Br)2] was prepared using previously described methods.29 [Os(bpy)2(Br)2] (20 mg, 3.0 × 10−5 mol) and bpy-AN (50 mg, 1.5 × 10−4 mol) were dissolved in 20 ml 2-methoxyethanol and heated at reflux under argon for 12 h. The solution was cooled, reduced to 10 ml, and 30 ml water was added. The solution was filtered to remove unreacted ligand. The addition of a small amount of ammonium hexafluorophosphate precipitated the product as a dark green solid (28 mg, 2.5 × 10−5 mol, 83%). Analysis for C48H45N6OsP2F12: C, 48.61; H, 3.82; N, 7.09. Found: C, 48.43; H, 3.65; N 6.88%.[Os(bpy-AN)2(CO)Br](PF6)
This procedure is a modification of a previously described method.11 [Os(bpy-AN)2(Br)2] (50 mg, 7.5 × 10−5 mol) was dissolved in 20 ml ethylene glycol, and heated at reflux for 1 h with CO gas bubbled through the solution continuously. The solution was cooled, and 200 ml water added. Addition of a small amount of ammonium hexafluorophosphate precipitated the product as a dark red solid (44 mg, 4.0 × 10−5 mol, 53%). νCO = 1967 cm−1. Analysis for C51H38N4BrOOsPF6: C, 53.83; H, 3.37; N, 4.92. Found: C, 53.56; H, 3.25; N, 5.01%.Instrumental methods
All absorption spectra were obtained on a Cary 100 Scan UV-Visible spectrophotometer. All spectra were baseline corrected using reagent grade solvent. Luminescence spectra for all complexes were obtained using a SPEX fluorolog spectrofluorometer equipped with a 450 W xenon arc lamp for excitation, 0.22 m excitation monochromator, 0.34 m emission monochromator, and either a Hammamatsu R928 photomultiplier tube (for excitation scans), or a liquid-nitrogen cooled CCD detector (for emission scans). Room temperature spectra were obtained in N2 bubble-degassed CH3CN unless otherwise noted. 77 K emission spectra were obtained in 4 ∶ 1 ethanol–methanol glasses. Spectra were not corrected for detector response. Quantum yields were obtained relative to [Ru(bpy)3](PF6)2 in CH3CN.Luminescence lifetimes between 77 K and room temperature were acquired using N2-pumped dye laser excitation, filtering the emitted light through a Heath-McPherson Model EU-700 monochromator and detecting the emitted light with a Hammamatsu R 928 PMT. Decays typically represented the average of 1000–10![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 pulses to maximize signal-to-noise ratios, and were collected on a HP Model 54111D digital oscilloscope.
000 pulses to maximize signal-to-noise ratios, and were collected on a HP Model 54111D digital oscilloscope.
Transient absorption spectra were obtained on a homebuilt system consisting of a Quantel Brilliant Nd:YAG laser with second harmonic generation (4 ns pulse), a 150 W Xe arc lamp for analysis, a Heath-McPherson Model EU-700 monochromator for dispersion of the analyzing light, a Hammamatsu 1P-28 PMT linked to a Lecroy 1 GHz transient digitizer for detection of transient signals.
Cyclic voltammograms and differential pulse polarograms were obtained using an EG&G Versastat for potentiostating and sweep control. Voltammograms were collected in nitrogen-purged acetonitrile solutions using either platinum or glassy carbon disk working electrodes, platinum wire auxiliary, and a silver wire pseudo-reference electrode. Potentials were measured using the ferrocene/ferrocenium couple as an internal reference and were corrected to be relative to SCE. The supporting electrolyte in all experiments was tetraethylammonium perchlorate (TEAP). In general, sweep rates varied from 50 to 100 mV s−1 for cyclic voltammograms and 20 mV s−1 for differential pulse polarograms (with a pulse height of 25 mV and pulse width of 50 ms).
Results and discussion
Syntheses
The ligand 4-(9-anthryl)-2,2′-bipyridine was prepared by a variation of the Hantsch reaction originally developed by Kronke. The complexes were prepared by well established literature methods and were purified by column chromatography on alumina. The ligands were characterized by 1H and 13C NMR spectroscopy. Characterization of the complexes included elemental analysis, Uv-vis spectrophotometry, cyclic voltammetry and infrared spectroscopy and luminescence spectroscopy. The absorption spectra and voltammetric behavior of the complexes were unique and characteristic of the particular coordination environments for the complexes studied.Electronic spectra
Absorption spectra of [(bpy)2Os(bpy-AN)]2+ and [(bpy-AN)2Os(CO)Br]+ are shown in Fig. 2. The complex having a tris-diimine coordination environment has absorption maxima at 480 and 440 nm, corresponding to spin allowed metal-to-ligand charge transfer (MLCT) transitions. The presence of the anthracenyl substituent does not seem to perturb the MLCT absorption of [(bpy)2Os(bpy-AN)]2+ in a measurable way. The anthracenyl bipyridine complex has two additional maxima in the 350–400 nm range that can clearly be assigned as anthracene localized π–π* transitions. The maxima, at 383 and 365 nm are intermediate between those of anthracene (375, 355 nm) and diphenylanthracene (390, 373 nm),30 which is reasonable considering the bpy-AN ligand is effectively a mono-aryl substituted anthracene. Many 9,10-disubstituted anthracene derivatives have three strong vibronic bands in the 300–400 nm region, but bpy-AN exhibits only two; this may be a result of symmetry lowering in bpy-AN, but we have not examined this point in any detail. The higher energy transitions of the parent complex, [(bpy)3Os]2+, has a broad band at 370 nm which has not been definitively assigned, but is too low in energy to be assigned as a bpy localized π–π* transition and may be a higher energy MLCT transition. The MLCT absorptions of [(bpy-AN)2Os(CO)Br]+ are very broad and are not as easily interpreted because of significant overlap of MLCT and ligand localized absorption bands. Based on data for related Ru(II) carbonyl complexes, it is likely that the spin-allowed MLCT absorption is at higher frequency than that of [(bpy)2Os(bpy-AN)]2+, and may significantly overlap with the intraligand transitions associated with the anthracene substituent. Such behavior would be consistent with that observed for [(bpy)2Ru(CO)Cl]+, which has an MLCT absorption significantly to the blue of that for [(bpy)3Ru]2+.31 Finally, the lower energy transition observed around 500 nm in [(bpy-AN)2Os(CO)Br]+ may correspond to a spin-forbidden MLCT transition. In [(bpy)2Os(bpy-AN)]2+ the additional weaker absorption in the 550–700 nm region corresponds to spin forbidden 1GS → 3MLCT transitions characteristic of many Os(II) diimine complexes.8,9 These spin forbidden transitions are not observed in closely related derivatives of Ru(II) and Fe(II).![Absorption spectra of [Os(bpy)2(bpy-AN)]2+ (solid line) and [Os(bpy-AN)2(CO)Br]+ (dashed line) in CH3CN at room temperature.](/image/article/2005/PP/b410472m/b410472m-f2.gif) | ||
| Fig. 2 Absorption spectra of [Os(bpy)2(bpy-AN)]2+ (solid line) and [Os(bpy-AN)2(CO)Br]+ (dashed line) in CH3CN at room temperature. | ||
Luminescence spectra and lifetimes
Room-temperature luminescence spectral data are presented in Table 1. All the complexes other than [(bpy-AN)2Os(CO)Br]+ are luminescent at room temperature. Data for [(bpy)3Os]2+ and [(bpy)2Os(CO)Cl]+ are taken from earlier work by Meyer and co-workers.11,32 The 600 cm−1 difference in emission maxima between [(bpy)3Os]2+ and [(bpy)2Os(bpy-AN)]2+ may be partially due to the fact that the spectra were collected using different detectors (PMT for [(bpy)3Os]2+ and CCD for [(bpy)2Os(bpy-AN)]2+) in a spectral region where detector response is a strong function of wavelength. More important is the difference in emission maxima between [(bpy)3Os]2+ and [(bpy)2Os(CO)Cl]2+ (both reported from the same reference) which indicates that the emission of the carbonyl chloride complex is approximately 900 cm−1 higher in energy than the tris-bipyridyl complex.Luminescence decays obtained for the three emissive complexes (Table 1) indicate that the anthracene-substituted derivative has an excited state lifetime more than three times that of the parent [(bpy)3Os]2+. Such a large difference in luminescence lifetimes for chromophores having apparently the same emissive state and nearly equivalent energy gap (MLCT) suggests a significant perturbation of the excited state by the anthracene substituent. In earlier work on Ru(II) diimine complexes, Meyer and Strouse showed that, for related complexes of nearly equivalent emission energy, differences in excited state lifetimes can be related to differences in excited state distortion using the formalism for nonradiative decay derived by Jortner and Freed and stated in an abbreviated form in eqn. (1).14,15 Factors affecting the nonradiative decay rate constant include the zero–zero emission energy, Eo, an average medium frequency vibrational mode coupled to nonradiative relaxation, hν, and the electron–vibrational coupling parameter, S, which is directly proportional to the magnitude of excited state distortion (S ∝ ΔQe2). The relationship between these terms is illustrated in eqn. (1). For two
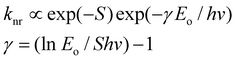 | (1) |
 | (2) |
dπ
→ bpy-ANπ* MLCT state will have a smaller nuclear distortion than the Rudπ
→ bpyπ* MLCT state. Several Ru(II) diimine complexes having aryl substituted bipyridine ligands have been examined and most have excited state lifetimes that are longer than those of the unsubstituted tris-bipyridyl complex.22,23,33–36 In addition, McCusker and co-workers have postulated that, for 4,4′-diphenyl-2,2′-bipyridine complexes of Ru(II), the phenyl substituent comes into planarity with the coordinated bipyridine on the 5–10 ps timescale following population of the MLCT state.37The distortion parameter can be determined by Franck–Condon analysis of the emission spectra of each of the complexes involved. Unfortunately, both complexes have broad emission completely lacking in vibronic structure at room temperature. In frozen matrices at 77 K both complexes emit and the spectra are shown in Fig. 3. The spectra can be fit in a relatively simple way by assuming that excited state relaxation is promoted through one medium-frequency, ℏωm, and one low-frequency vibrational mode, ℏωl, and their distortion parameters, Sm and Sl.10,14,38Eqn. (3), below, was used to simulate the 77 K spectra of [(bpy)3Os]2+ and [(bpy)2Os(bpy-AN)]2+.
 | (3) |
![Emission spectra of [Os(bpy)2(bpy-AN)]2+ (dashed line) and [Os(bpy)3]2+ (solid line) obtained at 77 K in a 4 ∶ 1 ethanol–methanol matrix.](/image/article/2005/PP/b410472m/b410472m-f3.gif) | ||
| Fig. 3 Emission spectra of [Os(bpy)2(bpy-AN)]2+ (dashed line) and [Os(bpy)3]2+ (solid line) obtained at 77 K in a 4 ∶ 1 ethanol–methanol matrix. | ||
The remaining terms of the expression are the zero–zero emission energy, E0, and a term that reflects broadening of the individual vibronic contributions to the emission, vh. Parameters obtained from the fits of the spectra are given in Table 2. Both complexes have very similar values for each of the parameters and the Sm value is actually slightly lower for [(bpy)3Os]2+ than the bpy-AN complex. The results suggest that both the complexes should have very similar excited state lifetimes at 77 K. Unfortunately, this analysis may have little to do with the behavior observed in room-temperature solution since the conformational reorganization necessary to achieve the planarized excited state for the bpy-AN complex probably cannot occur in frozen solution.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =450 cm−1, Sl=0.4
=450 cm−1, Sl=0.4
| Complex | Eo/cm−1 | ℏω/cm−1 | Sm | νh/cm−1 |
|---|---|---|---|---|
| [(bpy)3Os]2+ | 14190 | 1350 | 0.25 | 700 |
| [(bpy)2Os(bpy-AN)]2+ | 14130 | 1300 | 0.28 | 670 |
Transient absorption spectra
Another possible explanation for the difference in the excited state lifetimes of [(bpy)3Os]2+ and [(bpy)2Os(bpy-AN)]2+ is reversible excitation energy transfer between the 3MLCT state of [(bpy)2Os(bpy-AN)]2+ and the 3(π–π*) state of the attached anthracene substituent. The anthracene triplet is not emissive in solution at room temperature, but is nearly isoenergetic with the Osdπ
→ bpyπ*
3MLCT state. If the two excited states were present as an equilibrium mixture, it might be possible to observe both states by transient absorption spectroscopy. Reversible energy transfer of this type has been noted for other covalently linked Os(II) diimine/anthracene derivatives.26,27Fig. 4 shows transient absorbance spectra obtained for both of the complexes having the bpy-AN ligand following excitation by a frequency-doubled Nd:YAG laser (532 nm). The spectrum of [(bpy)2Os(bpy-AN)]2+ has features that are very similar to those of the parent chromophores [(bpy)3Os]2+.39 The principal features of the spectrum are bleaching of the ground state absorption between 450 and 650 nm and excited state absorption with a maximum of 380 nm. Thus, from the perspective of reversible energy transfer between the 3MLCT state and the anthracene 3(π–π*) state, the transient spectrum of [(bpy)2Os(bpy-AN)]2+ appears to be largely MLCT in nature.
![Transient absorption spectra of (A) [Os(bpy)2(bpy-AN)]2+ and (B) [Os(bpy-AN)2(CO)Br]+ in nitrogen degassed CH3CN at room temperature.](/image/article/2005/PP/b410472m/b410472m-f4.gif) | ||
| Fig. 4 Transient absorption spectra of (A) [Os(bpy)2(bpy-AN)]2+ and (B) [Os(bpy-AN)2(CO)Br]+ in nitrogen degassed CH3CN at room temperature. | ||
The complex [(bpy-AN)2Os(CO)Br]+ has a spectrum that is entirely different from that of [(bpy)2Os(bpy-AN)]2+. The spectral features closely resemble those of the 3(π–π*) state of anthracene derivatives. The visible maxima in the spectra of anthracene, 9-phenylanthracene and 9,10-diphenylanthracene are at 430, 430 and 450 nm, respectively in benzene.40 The maximum for [(bpy-AN)2Os(CO)Br]+ is at approximately 430 nm with a shoulder at 475 nm.
Since the transient absorption data obtained for the two complexes strongly resemble spectra of isolated chromophores, it appears reasonable to assume the MLCT and 3(π–π*) states are at best weakly interacting. In order to rationalize the difference in behavior of the two Os(II) diimine complexes, it is important to have reasonable estimates for the energies of the 3MLCT states on both the complexes and the 3(π–π*) state of the covalently attached anthracene. From published work of Meyer and co-workers, the difference in energy of the 3MLCT emission between [(bpy)3Os]2+ and [(bpy)2Os(CO)Cl]+ is approximately 900 cm−1.11 Based on the transient absorption results, the 3(π–π*) state of the anthracene substituent in bpy-AN should be between the energy of the 3MLCT state of [(bpy)2Os(bpy-AN)]2+ and [(bpy-AN)2Os(CO)Br]+.
If it is assumed that the observed transient decays for the two complexes result from relaxation of the 3MLCT and anthracene 3(π–π*) states in rapid equilibrium, the decays can be fit to a model in which the variable parameters are the rate constants for forward and reverse energy transfer from the 3MLCT state to the 3(π–π*) state and the rate constant for relaxation of the anthracene triplet state (Scheme 1).41 From the resulting ratio of rate constants, the energy gap between the 3MLCT state and 3(π–π*) state of the anthracene is directly obtained. To fit the experimental decays of the two bpy-AN complexes the rate constant for energy transfer from the 3MLCT state to the 3(π–π*) state was assumed to be fixed at 109 s−1; this value was selected to be significantly faster than the 3MLCT lifetime of either complex and the anthracene triplet, thus assuring the excited state equilibrium. The lifetime of the triplet state of the anthracene (1/knrA) was fixed at 100 μs; this value is an approximation based upon reported lifetimes for the triplet excited state of various substituted anthracene derivatives.40 In addition, the lifetime of the 3MLCT state (1/kM) was fixed at 76 ns, the average of the lifetimes of [(bpy)3Os]2+ and [(bpy)2Os(CO)Cl]+. Using these parameters to fit the observed decay for [(bpy)2Os(bpy-AN)]2+, the anthracene triplet energy is found to be lower than the 3MLCT state by 105 cm−1
(KMA
= 1.7). This result leads to the conclusion that the excited state will consist of an excess of the anthracene triplet in equilibrium with the 3MLCT state, but that both should be observed in the transient absorption spectrum. As can be seen from Fig. 4, the 3MLCT state is the only species clearly observed in the TA spectrum. It may be that the excited state equilibrium assumption is not valid for this complex and the slightly lengthened excited state lifetime of 200 ns may be the result of delocalization of the 3MLCT state, as discussed above.
 | ||
| Scheme 1 | ||
Using the same fixed parameters to fit the decay of [(bpy-AN)2Os(CO)Br]+, the anthracene triplet state is found to be lower than the 3MLCT state by 980 cm1 (KMA = 114). The equilibrium constant obtained indicates that the anthracene triplet will be the dominant state present, as is clearly observed in the transient absorption spectrum.
From the calculated energy gaps for the two complexes, the energy difference between the 3MLCT states of [(bpy-AN)2Os(CO)Br]+ and [(bpy-AN)3Os]2+ can be estimated to be 875 cm−1, only slightly smaller than the gap of 890 cm−1 between the emission maxima of [(bpy)3Os]2+ and [(bpy)2Os(CO)Cl]+ measured by Meyer and co-workers.32 The relatively good agreement suggests that the equilibrium model is effective for analyzing excited state decay in these complexes where the Os complex donor and anthracene acceptor are linked by a single sigma bond. The striking conclusion from these results is that, despite the fact that there is no “bridge” between the energy donor and acceptor and the fact that the excited states of the donor and acceptor are nearly isoenergetic, the interaction between the two chromophores involved is small enough that an equilibrium approximation (assuming no electronic interaction) can be used to evaluate excited state relaxation.
Conclusion
This manuscript presents the photophysical behavior of two Os(II) complexes having bipyridine ligands with anthracene attached directly to the bipyridine. While the energies of the 3MLCT states of the two complexes studied differ by less than 800 cm−1, the observed photophysical behavior is entirely different. The complex with the higher energy 3MCLT state, [(bpy-AN)2Os(CO)Br]+, is nonemissive at room temperature, but has a long lived excited state that is localized on the 3(π–π*) state of the anthracene substituent. The other complex [(bpy)2Os(bpy-AN)]2+, exhibits emission at room temperature and has a transient absorption spectrum that is consistent with a localized 3MLCT state. The excited state decay behavior of the two complexes can be fit well assuming a model in which noninteracting 3MLCT and 3(π–π*) states are in equilibrium. The results suggest that there is a negligible degree of electronic interaction between the 3MLCT state and the anthracene 3IL state, despite the fact that the two chromophores are separated by only a single C–C bond.Acknowledgements
The authors wish to thank the U.S. Department of Energy, Office of Chemical Sciences (DE-FG-02-96ER14617) for support of this research.References
- N. S. Hush and J. R. Reimers, Solvent effects on metal to ligand charge transfer excitations, Coord. Chem. Rev., 1998, 177, 37–60 CrossRef CAS
.
- T. J. Meyer, Intramolecular control of excited state electron and energy transfer, Pure Appl. Chem., 1990, 62, 1003–1009 CrossRef CAS
- H. Riesen and E. Krausz, Dynamic processes in the lowest-excited 3MLCT states of [M(L)3−x(L′)x]2+
(L, L′
= diimine; M = Ru, Os), Comments Inorg. Chem., 1995, 18, 27–63 CAS
- J. A. Treadway, B. Loeb, R. Lopez, P. A. Anderson, F. R. Keene and T. J. Meyer, Effect of delocalization and rigidity in the acceptor ligand on MLCT excited-state decay, Inorg. Chem., 1996, 35, 2242–2246 CrossRef CAS
- D. Graff, J. P. Claude and T. J. Meyer, Calculation of rate constants from spectra. Nonradiative decay and electron transfer, Adv. Chem. Ser., 1997, 253, 183–198 CAS
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Ruthenium(II) polypyridine complexes: photophysics, photochemistry, electrochemistry and chemiluminescence, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS
- T. J. Meyer, Photochemistry of metal coordination complexes: metal to ligand charge transfer excited states, Pure Appl. Chem., 1986, 58, 1193–1206 CrossRef CAS
- E. M. Kober and T. J. Meyer, Concerning the electronic structure of the ions M(bpy)33+
(M = Fe, Ru, Os; bpy = 2,2′-bipyridine), Inorg. Chem., 1983, 22, 1614–1616 CrossRef CAS
- E. M. Kober and T. J. Meyer, An electronic structural model for the emitting MLCT excited states of Ru(bpy)32+ and Os(bpy)32+, Inorg. Chem., 1984, 23, 3877–3886 CrossRef CAS
- K. R. Barqawi, Z. Murtaza and T. J. Meyer, Calculation of relative nonradiative decay rate constants from emission spectral profiles: polypyridyl complexes of ruthenium(II), J. Phys. Chem., 1991, 95, 47–50 CrossRef CAS
- J. V. Caspar and T. J. Meyer, Application of the energy gap law to nonradiative, excited-state decay, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS
- E. M. Kober, J. V. Caspar, R. S. Lumpkin and T. J. Meyer, Application of the energy gap law to excited-state decay of osmium(II)-polypyridine complexes: calculation of relative nonradiative decay rates from emission spectral profiles, J. Phys. Chem., 1986, 90, 3722–3734 CrossRef CAS
- K. F. Freed, Radiationless transitions in molecules, Acc. Chem. Res., 1978, 11, 74–80 CrossRef CAS
- S. Boyde, G. F. Strouse, W. E. Jones, Jr. and T. J. Meyer, The effect on MLCT excited states of electronic delocalization in the acceptor ligand, J. Am. Chem. Soc., 1990, 112, 7395–7396 CrossRef CAS
- G. F. Strouse, J. R. Schoonover, R. Duesing, S. Boyde, W. E. Jones, Jr. and T. J. Meyer, Influence of electronic delocalization in metal-to-ligand charge transfer excited states, Inorg. Chem., 1995, 34, 473–487 CrossRef CAS
- J. V. Caspar and T. J. Meyer, Photochemistry of MLCT excited states. Effect of nonchromophoric ligand variations on photophysical properties in the series cis-Ru(bpy)2L22+, Inorg. Chem., 1983, 22, 2444–2453 CrossRef CAS
- X.-Y. Wang, A. Del Guerzo and R. H. Schmehl, Photophysical behavior of transition metal complexes having interacting ligand localized and metal-to-ligand charge transfer states, J. Photochem. Photobiol., C: Photochem. Rev., 2004, 5, 55–77 CrossRef CAS
- R. Schmehl, Something new in transition metal complex sensitizers: bringing metal diimine complexes and aromatic hydrocarbons together, Spectrum, Bowling Green, OH, US, 2000, vol. 13, pp. 17–21 Search PubMed
- A. I. Baba, J. R. Shaw, J. A. Simon, R. P. Thummel and R. H. Schmehl, The photophysical behavior of d6 complexes having nearly isoenergetic MLCT and ligand localized excited states, Coord. Chem. Rev., 1998, 171, 43–59 CrossRef CAS
- A. Del Guerzo, S. Leroy, F. Fages and R. H. Schmehl, Photophysics
of Re(I) and Ru(II) diimine complexes covalently linked to pyrene: contributions from intra-ligand charge transfer states, Inorg. Chem., 2002, 41, 359–366 CrossRef CAS
- D. S. Tyson, K. B. Henbest, J. Bialecki and F. N. Castellano, Excited state processes in ruthenium(II)/pyrenyl complexes displaying extended lifetimes, J. Phys. Chem. A, 2001, 105, 8154–8161 CrossRef CAS
- A. Harriman, M. Hissler, A. Khatyr and R. Ziessel, A ruthenium(II) tris(2,2′-bipyridine) derivative possessing a triplet lifetime of 42 μs, Chem. Commun., 1999, 735–736 RSC
- J. A. Simon, S. L. Curry, R. H. Schmehl, T. R. Schatz, P. Piotrowiak, X. Jin and R. P. Thummel, Intramolecular electronic energy transfer in ruthenium(II) diimine donor/pyrene acceptor complexes linked by a single C–C bond, J. Am. Chem. Soc., 1997, 119, 11012–11022 CrossRef CAS
- W. E. Ford and M. A. J. Rodgers, Reversible triplet–triplet energy transfer within a covalently linked bichromophoric molecule, J. Phys. Chem., 1992, 96, 2917–2920 CrossRef CAS
- Z. Murtaza, A. P. Zipp, L. A. Worl, D. Graff, W. E. Jones, Jr., W. D. Bates and T. J. Meyer, Energy transfer in the “inverted region”, J. Am. Chem. Soc., 1991, 113, 5113–5114 CrossRef CAS
- A. El-ghayoury, A. Harriman, A. Khatyr and R. Ziessel, Intramolecular triplet energy transfer in metal polypyridine complexes bearing ethynylated aromatic groups, J. Phys. Chem. A, 2000, 104, 1512–1523 CrossRef CAS
- B. Maubert, N. D. McClenaghan, M. T. Indelli and S. Campagna, Absorption spectra and photophysical properties of a series of polypyridine ligands containing appended pyrenyl and anthryl chromophores and of their ruthenium(II) and osmium(II) complexes, J. Phys. Chem. A, 2003, 107, 447–455 CrossRef CAS
- J. G. Cordaro, J. K. McCusker and R. G. Bergman, Synthesis of mono-substituted 2,2′-bipyridines, Chem. Commun., 2002, 1496–1497 RSC
- D. A. Buckingham, F. P. Dwyer, H. A. Goodwin and A. M. Sargeson, Mono- and bis(2,2′-bipyridine) and (1,10-phenanthroline) chelates of ruthenium and osmium. III. Monochelates of bivalent, trivalent and quadrivalent osmium, Aust. J. Chem., 1964, 17, 315–324 CAS
- S. L. C. Murov and I. G. L. Hug, Handbook of Photochemistry, Marcel Dekker Inc., New York, 1993 Search PubMed
- J. M. Kelly, C. M. O'Commell and J. G. Vos, Preparation, spectroscopic characterisation, electrochemical and photochemical properties of cis-bis(2,2′-bipyridyl)carbonylruthenium(II) complexes, J. Chem. Soc., Dalton Trans., 1986, 253–258 RSC
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, Application of the energy gap law to the decay of charge-transfer excited states, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS
- D. S. Tyson and F. N. Castellano, Intramolecular singlet and triplet energy transfer in a ruthenium(II) diimine complex containing multiple pyrenyl chromophores, J. Phys. Chem. A, 1999, 103, 10955–10960 CrossRef CAS
- D. S. Tyson, C. R. Luman, X. Zhou and F. N. Castellano, New Ru(II) chromophores with extended excited-state lifetimes, Inorg. Chem., 2001, 40, 4063–4071 CrossRef CAS
- A. Del Guerzo, C. Balazs, F. Fages and R. H. Schmehl, Long-lived metallic charge transfer and organic intraligand triplet states in Ru(II)-pyrene and Os(II)-anthracene complexes, Abstr. Pap. Am. Chem. Soc., 2000, 362
- M. Hissler, A. Harriman, A. Khatyr and R. Ziessel, Intramolecular triplet energy transfer in pyrene-metal polypyridine dyads: A strategy for extending the triplet lifetime of the metal complex, Chem. Eur. J., 1999, 5, 3366–3381 CrossRef CAS
- N. H. Damrauer and J. K. McCusker, Ultrafast dynamics in the metal-to-ligand charge transfer excited-state evolution of [Ru(4,4′-diphenyl-2,2′-bipyridine)3]2+, J. Phys. Chem. A, 1999, 103, 8440–8446 CrossRef CAS
- B. J. Coe, D. W. Thompson, C. T. Culbertson, J. R. Schoonover and T. J. Meyer, Synthesis and photophysical properties of mono(2,2′,2″-terpyridine) complexes of ruthenium(II), Inorg. Chem., 1995, 34, 3385–3395 CrossRef CAS
- M. A. Bergkamp, P. Guetlich, T. L. Netzel and N. Sutin, Lifetimes of the ligand-to-metal charge-transfer excited states of iron(III) and osmium(III) polypyridine complexes. Effects of isotopic substitution and temperature, J. Phys. Chem., 1983, 87, 3877–3883 CrossRef CAS
- I. Carmichael and G. Hug, Triplet-triplet absorption spectra of organic molecules in condensed phases, J. Phys. Chem. Ref. Data, 1986, 15, 1–250
- J. N. Demas, Excited State Lifetime Measurements, Dekker, New York, 1983 Search PubMed
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |