The photorelease of nitrogen monoxide (NO) from pentacyanonitrosyl coordination compounds of group 8 metals†
Mariela Videlaab, Silvia E. Braslavsky*a and José A. Olabe*b
aMax-Planck-Institut für Bioanorganische Chemie (formerly Strahlenchemie), Postfach 101365, D-45413, Mülheim an der Ruhr, Germany. E-mail: braslavskys@mpi-muelheim.mpg.de; Fax: +49 208 306 3951
bINQUIMAE and Departamento de Química Inorgánica, Analítica y Química Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pab. II, Ciudad Universitaria, Buenos Aires, Argentina C1428EHA
First published on 13th October 2004
Abstract
The photodetachment of NO from [MII(CN)5NO]2− with M = Fe, Ru, and Os, upon laser excitation at various wavelengths (355, 420, and 480 nm) was followed by various techniques. The three complexes showed a wavelength-dependent quantum yield of NO production Φ(NO), as measured with an NO-sensitive electrode, the highest values corresponding to the larger photon energies. For the same excitation wavelength the decrease of Φ(NO) at 20 °C in the order Fe > Ru ≫ Os, is explained by the increasing M–N bond strength and inertness of the heavier metals. Transient absorption data at 420 nm indicate the formation of the [MIII(CN)5H2O]2− species in less than ca. 1 μs for M = Fe and Ru. The enthalpy content of [FeIII(CN)5H2O]2− with respect to the parent [FeII(CN)5NO]2− state is (190 ± 20) kJ mol−1, as measured by laser-induced optoacoustic spectroscopy (LIOAS) upon excitation at 480 nm. The production of [FeIII(CN)5H2O]2− is concomitant with an expansion of (8 ± 3) ml mol−1 consistent with an expansion of the water bound through hydrogen bonds to the CN ligands plus the difference between NO release into the bulk and water entrance into the first coordination sphere. The activated process, as indicated by the relatively strong temperature dependence of the Φ(NO) values and by the temperature dependence of the appearance of the [FeIII(CN)5H2O]2− species, as determined by LIOAS, is attributed to NO detachment in less than ca. 100 ns from the isonitrosyl (ON) ligand (MS1 state).
Introduction
Nitrogen monoxide (NO) has been implicated in many physiological processes, including cardiovascular control, neuronal signalling, and as an agent for defence against microorganisms and tumours.1,2 These discoveries have stimulated interest in the chemistry and biochemistry of NO and derivatives such as metallonitrosyl complexes,3,4 especially in their potential capability of acting as carriers, scavengers, or deliverers of NO.5,6 Both the chemical7,8 and the photochemical9,10 aspects of this reactivity are subjects of recent studies.Reports with metallonitrosyls containing classical co-ligands X have been published for the series [MIIX5–NO]n, where M is Fe, Ru, or Os and X is NH3 and amines, CN−, polypyridines, or mixed type X4L.10–12 In these complexes, NO may be formally considered a three electron donor (i.e., NO+), with strong back donation from the low-spin d6 MII centre to the π*(NO) orbital. This is an arbitrary formalism, because the valence orbitals of the [MNO] moiety are in fact highly delocalised.13 Chemical reactions of coordinated NO+ with bases (with potential release of nitrite or nitrosothiols) and light-induced M–NO electron transfer are the most important paths of NO delivery from these complexes. Emerging results with compounds of the above mentioned series provide information on the nature of the metal and coligands, as well as medium conditions that would favour or impair the trapping14 or the photodetachment of NO.15–18
In this work we focus on the pentacyanonitrosyl complexes of Fe, Ru, and Os.11 Work with the iron derivative has been a classic in photochemical studies on coordination compounds19–22 but key questions still remain as to the detailed mechanism of the processes following photoexcitation up to the NO-release. The Ru and Os analogues of sodium nitroprusside {[Fe(CN)5NO]2−, NP} have been prepared and further studied during the last two decades.23,24 We present here a study of their photoreactivity, in comparison with that of NP, with the aim of better understanding the factors controlling NO release in these complexes, given the scarce amount of systematic studies with analogue compounds comprising the elements pertaining to the three transition series.
On the basis of the generally accepted conclusion that a photoredox process operates upon primary excitation (i.e., formally an FeII–NO+ → FeIII–NO photoconversion), and that NO is subsequently released, we analyse the conditions influencing both processes. The studies with the three metal centres comprise measurements of quantum yields at different wavelengths and temperatures, and for NP in the presence of an electron acceptor macrocyclic ligand. Transient absorption after laser pulse excitation and laser-induced optoacoustic techniques are also used, in an effort to disclose the detailed mechanistic issues associated with the NO release. Comparisons are made with previous work with NP and other relevant metallonitrosyls.
Experimental
Materials
Na2[FeII(CN)5NO]·2H2O, sodium nitroprusside (NP, 99.99%, Aldrich), was used as supplied. Sodium or potassium salts of the [RuII(CN)5NO]2− and [OsII(CN)5NO]2− analogue compounds were synthesized according to published procedures.23,24 The product (either the Ru- or Os-compound) was purified by chromatography (Sephadex LH-20). In order to eliminate remaining nitrite, the solution was acidified to pH 4 with trifluoroacetic acid and an excess of H2O2 was added. One hour later the complex was precipitated as a silver salt by addition of AgNO3 and it was filtered through a fine glass frit, washed with water, then methanol, and finally with ether. After drying in vacuum for 12 h, the complex Ag2[M(CN)5NO] was suspended in approximately 100 ml water, KBr was added (less that 1 ∶ 1 molar ratio), and the solution was stirred overnight. The AgBr formed was filtered and the remaining solution was evaporated to dryness in a rotary evaporator. The solid was dried over silica gel in vacuum for 12 h. Only sodium salts were used in the present work, after cation exchange with a DOWEX exchange column when needed. Purity was checked by UV-visible and IR absorption spectroscopies.The azamacrocycles 24[ane]-N8H8·8HCl and 32[ane]-N8H8·8HCl25 were provided by Professor Fernando Pina (Universidade Nova de Lisboa, Portugal).
Buffer solutions were at pH 6.5 (NaH2PO4-Na2HPO4) and pH 4.5 (acetic acid-NaOH). For the concentrations see Methods. Water was always distilled and deionized. NaNO2, NaI, and all other common reagents were used as provided without further purification.
Methods and signal handling
UV-vis spectra were recorded with a Shimadzu UV-2102 PC or a Shimadzu UV-2401 PC spectrophotometer. IR spectra were taken in KBr pellets with a Nicolet 510P FTIR instrument.Fluorescence quantum yields were determined with a corrected spectrofluorimeter (Spex-Fluorolog) using tryptophan in water as reference, Φf = 0.14 at 25 °C.26 The absorbances of the sample solutions in buffer at 308 nm were A308 = 0.11, 0.12, and 0.16 for M = Os, Ru, and Fe, respectively. Excitation was at 308 nm in order to obtain a full emission spectrum. For the evaluation of the Φf values, the areas under the emission curves of the corrected spectra for the sample solutions between 360 and 600 nm were compared to those of the tryptophan solution. A correction was made for the absorbance differences. No effect on the emission yield or position of the maximum was observed upon changing excitation wavelength or upon addition of NaCl (ca. 0.1 M). An approximation had to be made for the tryptophan emission under 380 nm, because no correction was available for the spectrofluorimeter for λ < 380 nm. Thus, the yields reported are more accurate in their relative than in their absolute values.
All spectral and photochemical experiments were performed with solutions of the complexes at pH 6.5 (0.1 M buffer), after Ar bubbling and kept protected from room light before irradiation.
Nitrogen monoxide quantum yield [Φ(NO)] measurements were carried out with a commercial NO detector (inNO, Nitric Oxide Measuring System, Harvard Apparatus GmbH). The system consists of an 850 mV potentiostat and a combined electrode (700 μm diameter, amiNO Series of nitrogen monoxide sensors), covered with a series of permeable membranes allowing only NO to diffuse through and react at the electrode surface, where it is oxidized. The exchange of electrons results in an electrical current directly proportional to the amount of NO diffused through the membrane, in turn proportional to the NO concentration in the solution. Calibration of the electrode in the range approximately (10–200) × 10−9M was performed by generating NO according to reaction (1), under Ar atmosphere.
| H2SO4 + NaNO2 + NaI → NO + ½ I2 + H2O + Na2SO4 | (1) |
 | ||
| Fig. 1 Signals and plot for calibration of the 700 μm-amiNO electrode. (a) Developed current vs. time, after zeroing the equipment; (b) linear response of the electrode (sensitivity in this case: 137 pA nM−1). | ||
The slopes [b/(pA J−1)] of the linear regression of the developed current (Δi) vs. absorbed energy (ΔEabs, varied by accumulating several pulses at wavelength λ of low fluence each, Fig. 2), together with the calibration slope [Cal/(pA M−1)], and the volume of solution irradiated, Vsol, were used to calculate the NO quantum yields according to eqn. (2), with E(λ) the molar energy of the laser beam at the excitation wavelength.
 | (2) |
![Chronoamperograms of [M(CN)5NO]2− (M = Fe, Ru, Os) at 20 °C, in 0.1 m NaH2PO4/Na2HPO4 buffer (pH 6.5) after irradiation at λexc = 355 nm. Each step in the chronoamperogram corresponds to an increasing number of laser pulses. See text for calculations.](/image/article/2005/PP/b408367a/b408367a-f2.gif) | ||
Fig. 2 Chronoamperograms of [M(CN)5NO]2− (M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =Fe, Ru, Os) at 20 °C, in 0.1 M NaH2PO4/Na2HPO4 buffer (pH 6.5) after irradiation at λexc=355 nm. Each step in the chronoamperogram corresponds to an increasing number of laser pulses. See text for calculations. =Fe, Ru, Os) at 20 °C, in 0.1 M NaH2PO4/Na2HPO4 buffer (pH 6.5) after irradiation at λexc=355 nm. Each step in the chronoamperogram corresponds to an increasing number of laser pulses. See text for calculations. | ||
Nanosecond laser flash photolysis experiments were carried out with standard equipment, similar to the one used in previous work,27 consisting of two beams (one for excitation and the other for analysis) crossing each other at 90°, a holder with a 1 cm quartz cuvette and a photomultiplier for detection. A XeCl excimer laser pulse at 308 nm (EMG 101 MSC Lambda Physik) pumped a dye laser (FL 2000 Lambda Physik, pulse length ≈15 ns, 1 Hz repetition rate) in order to have excitation wavelengths near 400 nm. The third harmonic of the Nd:YAG laser at 355 nm was used in some cases. The analysing beam source was a pulsed 150 W Xe arc lamp for the μs range and a continuum halogen lamp for the ms range. Two matched energy heads (Laser Precision RJP 735) connected to a Laser Precision RJ-7100 energy meter or Rm6600 energy ratio meter were used for the monitoring of the laser beam energy. The signal detected by the photomultiplier was fed through a 50 ohm resistor into a Tektronix TDS 520A oscilloscope. The signal, the baseline (no laser), and the scattering signal (no analysing beam) were taken and combined in order to obtain the ΔA values at the various observation wavelengths.
The set-up used for laser-induced optoacoustic spectroscopy (LIOAS) experiments was similar to that previously described.28–31 The pulses from the OPO system described above were used for excitation. The fluence of the pulses was measured with energy meters as described above. The laser beam width was set with a slit (0.5 × 5 height) mm, which allows a time resolution from ca. 20 ns (using deconvolution) up to 5 μs. The sound wave was detected with a PZT ceramic transducer pressed against the sidewall of the cuvette, 100 times amplified, and fed into a signal digitiser (Tektronix 520 A or TDS 744 A). The absorbance of the samples (approximately 0.15, the exact value depending on the experiment) were matched at room temperature within ±0.005 units to those of the calorimetric reference solution (brilliant black). No temperature dependence was detected for the absorption spectra of sample and reference solutions.
The LIOAS signal handling using deconvolution was discussed in several publications.28,29,32 A sum of single-exponential decays fitting function was used for the time-resolved pressure evolution in the medium upon excitation of the sample. The recovered amplitudes (φi) of the fitting function, normalized with respect to the amplitude of the LIOAS signal for the calorimetric reference, are given by eqn. (3):
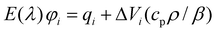 | (3) |
The thermoelastic parameter ratio, (cpρ/β)T, for the solutions in 0.1 M buffer, was determined by comparing the signal amplitudes of the reference (Hr) in the buffered solution (sol) and in neat water (w), at the same temperature T, according to eqn. (4), as already described.33
 | (4) |
 | (5) |
 | (6) |
Results and discussion
Quantum yields
The photochemistry of NP has been studied under various conditions, mainly through steady-state methods.22,35,36 A time resolved study shows that the [FeIII(CN)5H2O]2− complex is formed immediately after excitation with μs light pulses.37 By irradiation at 366 and 436 nm in buffered solution (pH 6.5), reaction (7) is operative for NP, and a similar process is presently proposed for the Ru and Os analogues when irradiated at the appropriate wavelength.| [MII(CN)5NO]2−
+
hν
→
[MIII(CN)5(H2O)]2−
+ NO (M = Fe, Ru, Os) | (7) |
In previous work with NP, the quantum yields of the reaction measured through the production of the pentacyanoaquaferrate(III), Φ(FeIII), were shown to be wavelength dependent (0.35 and 0.18 at 366 and 435 nm, respectively).22 Irradiation at 313 nm afforded Φ(FeIII) = 0.37.35 The absorption maxima of NP appear at ca. 510, 400, and 320 nm (Fig. 3).38 A wavelength dependent Φ(NO) (0.20 at 420 nm and 0.32 at 320 nm) was also determined from the rate of decay of nitronyl nitroxide (monitored by ESR spectrometry) produced upon reaction of the photogenerated NO with it.36
![Absorption spectra of [MII(CN)5NO]2− ions in 0.1 m NaH2PO4/Na2HPO4 buffer. Solid line: M = Fe(ii); dotted line: M = Ru(ii); dotted-dashed line: M = Os(ii).](/image/article/2005/PP/b408367a/b408367a-f3.gif) | ||
| Fig. 3 Absorption spectra of [MII(CN)5NO]2− ions in 0.1 M NaH2PO4/Na2HPO4 buffer. Solid line: M=Fe(II); dotted line: M=Ru(II); dotted-dashed line: M=Os(II). | ||
In the present work, the quantum yields for NO-formation, Φ(NO), for the three [M(CN)5NO]2− complexes were obtained by electrochemically sensing the NO produced upon irradiation at various wavelengths, a technique already employed for the determination of the stoichiometry of the photodecomposition reaction of [Fe(CN)5NO]2− . This is a simple and fast method for the detection of the gas product in the 10−9M scale, allowing measurements at low conversion.39
The three complexes showed a wavelength-dependent value of Φ(NO) (Table 1), the highest values corresponding to the larger photon energies (lower λexc). The yields for NP in Table 1 are closely similar to those reported earlier at similar wavelengths.22 The coincidence of our Φ(NO) values and the Φ(FeIII) as previously determined (see two paragraphs above), in agreement with the stoichiometry in eqn. (7), is remarkable, indicating that the quantum yield for the detachment of NO (as observed by transient absorbance)22,35 is identical to the final time-integrated quantum yield, and supporting the evaluation of Φ(NO) for the Ru- and Os-analogues, particularly for the latter, which displays a very small photoconversion yield.
=Fe, Ru, Os, at various excitation wavelengths; pH 6.5 (0.1 M phosphate buffer)
| Sample | T/°C | Φ(NO) 355 nm | Φ(NO) 435 nm |
|---|---|---|---|
| [Fe(CN)5NO]2− | 20.0 | 0.28 ± 0.04 | 0.18 ± 0.02 |
| [Ru(CN)5NO]2− | 20.0 | 0.10 ± 0.01 | 0.03 ± 0.02 |
| [Os(CN)5NO]2− | 20.0 | 0.01 ± 0.005 | ≪ 0.01 |
The quantum yields (for each of the used wavelengths) decrease in the order: Φ(NO)(Fe) > Φ(NO)(Ru) ≫ Φ(NO)(Os). These results should be analysed in terms of the nature of the photoactivation process and the subsequent events. By assuming that the ground state in NP can be described as a {FeIINO+} species,22 the initial excited state should contain an electron in a predominantly π*(NO) orbital, with the metal formally as FeIII. According to the spectral assignments for NP, the main photoactive band for NO-release should be that with maximum at 400 nm, assigned to a transition from the strongly bonding dxz and dyz orbitals to the π*(NO) orbital.22 No significant photoactivity was detected by irradiating above 480 nm,22,37 consistent with the assignment of the band at 510 nm to a transition from the nearly non-bonding dxy orbital to the same π*(NO) orbital. This transition should not produce a significant loss in the M–N bond strength upon excitation. The lower value of Φ(NO) at 436 nm may be traced to the simultaneous excitation of both the overlapping photoactive and inactive bands with maxima at 400 and 510 nm, respectively. On the other hand, the larger value of Φ(NO) on irradiation at 366 or 355 nm should be due to the simultaneous excitation of the band with maximum at 400 nm, as well as that with maximum at 320 nm (mainly a d–d transition). As the same Φ(FeIII)35 and a similar Φ(NO)36 were observed for NP on irradiation at 320 nm, we conclude that an efficient population of the species absorbing at 400 nm is achieved also when exciting at shorter wavelengths.
The trends in the quantum yields correlate with the increasing M–N bond strengths and inertness of the heavier metals, as measured by the relative dissociation rates for the three [MII(CN)5L]n− complexes with a given L ligand, namely k−L(Os) ≪ k−L(Ru) < k−L(Fe).40 In the M(III) complexes, the MIII–NO bond is mainly of σ type,41 and then we should also expect an increasing σ-bond strength when going from the Fe to the Os derivative. Thus, the decreasing quantum yields in Table 1 can be rationalized by considering that a rate-controlling M(III)–nitrosyl bond reorganization is the key factor for NO release (see below). However, it must be recognized that the same trend in the quantum yields should also be expected considering the stronger spin–orbit effects when going from Fe to Os. These should favour the non-radiative deactivation routes in competition with the photodetachment of NO.19
Upon excitation at 308 nm the three [M(CN)5NO]2− ions showed a broad emission in the range 300–600 nm, centred around 500 nm, with a half-bandwidth of ca. 5000 cm−1. Some structure could be observed in the emission of [Ru(CN)5NO]2− with shoulders at 470 and 500 nm.
Similar emissions, both in energies and bandwidths, have been detected upon excitation of [Ru(CN)6]4−,42 and of [Fe(CN)5CO]3−.43 These emissions have been assigned to the deactivation of the corresponding d–d states (3T1g and 3A2, respectively). An analogous population of 3A2 states can be also envisioned for [Fe(CN)5NO]2− upon the competitive deactivation of the initially accessible CT state, in view of the similar binding properties of the isoelectronic CO and NO+ ligands.
The trend in the fluorescence quantum yield (Table 2) is opposite to that determined for the Φ(NO) values, suggesting that the primary CT state deactivates competitively (at least upon excitation at 308 nm) into the photochemically active ground state potential energy surface and the emissive d–d state.
=308 nm
| Sample | (Φf ± 2) × 103 |
|---|---|
| [Os(CN)5NO]2− | 8.5 |
| [Ru(CN)5NO]2− | 7 |
| [Fe(CN)5NO]2− | 3 |
Two additional aspects of the photoreactivity were tested with the Fe(II) complex presenting the highest NO production yield, i.e., changes in temperature and in the medium. Φ(NO) decreased to one third of the original value upon decreasing the temperature from 20 to 0.8 °C (Table 3, Fig. 4). This is consistent with an activated process for an event subsequent to photoexcitation.
| Sample | T/°C | Φ(NO) | ||
|---|---|---|---|---|
| 355 nm | 428 nm | 480 nm | ||
| [Fe(CN)5NO]2− | 20.0 | 0.28 ± 0.04 | 0.18 ± 0.02 | 0.10 ± 0.02 |
| [Fe(CN)5NO]2− | 4.8 | 0.10 ± 0.02 | 0.19 ± 0.02 | 0.04 ± 0.02 |
| [Fe(CN)5NO]2− | 0.8 | 0.10 ± 0.02 | 0.14 ± 0.02 | 0.03 ± 0.02 |
| [Fe(CN)5NO]2−/[24]ane-N8+ | 20.0 | — | 0.11 ± 0.02 | — |
| [Fe(CN)5NO]2−/[32]ane-N8+ | 20.0 | 0.16 ± 0.02 | — | — |
![NO photodelivery upon excitation of [FeII(CN)5NO]2− at 355 nm in 0.1 m NaH2PO4/Na2HPO4 (pH = 6.5) at (●) 20 and (○) 0.8 °C.](/image/article/2005/PP/b408367a/b408367a-f4.gif) | ||
| Fig. 4 NO photodelivery upon excitation of [FeII(CN)5NO]2− at 355 nm in 0.1 M NaH2PO4/Na2HPO4 (pH=6.5) at (●) 20 and (○) 0.8 °C. | ||
Regarding the medium changes, the Φ(NO) values proved to be independent of pH (in the 1–7 range).
Nitrogen macrocycles influenced the reactivity. Addition of an excess of 24[ane]-N8H8·8HCl (0.1 M HCl, I = 0.8 M, NaCl) and of 32[ane]-N8H8·HCl (0.1 M HAcO/AcO− buffer, pH 4.5, I = 0.5 M, NaCl) to an NP solution did not change the absorption spectra, but decreased Φ(NO) to almost half of the value in the absence of the macrocycle. This is interpreted in terms of specific (electrostatic and hydrogen bond) interactions between the macrocycle and the cyano groups of the caged compound.44 Proton donation to cyanides should decrease the metal electron density, thus strengthening the σ Fe(III)–nitrosyl bond. In a recent work with a series of cis-[RuL(bpy)2NO]3+ complexes,16 the values of Φ(NO) were shown to decrease for L = 4-picoline > py > 4-acetylpyridine. This is consistent with our argument that the more donor L ligands favour the M(III)–NO bond cleavage.
Transient absorbance after laser excitation
Some examples of traces from transient absorption experiments are shown in Fig. 5. Excitation for these experiments was performed at wavelengths where Φ(NO) reached at least 0.1 for each compound. The detection of [MIII(CN)5H2O]2− was performed at 400 nm for M = Fe (cf. the maximum for [FeIII(CN)5H2O]2− at 394 nm)22,45 and at 420 nm for M = Ru.![Transient absorption signals for [M(CN)5NO]2−; λexc = 355 nm, (a) M = FeII, λobs = 400 nm, (b) M = RuII, λobs = 420 nm. Each signal is the average of 50 transient traces, already corrected for variations in base line and scattering. No transient absorption was observed for M = OsII.](/image/article/2005/PP/b408367a/b408367a-f5.gif) | ||
| Fig. 5 Transient absorption signals for [M(CN)5NO]2−; λexc=355 nm, (a) M=FeII, λobs=400 nm, (b) M=RuII, λobs=420 nm. Each signal is the average of 50 transient traces, already corrected for variations in base line and scattering. No transient absorption was observed for M=OsII. | ||
In each case a stable product is formed in less than 1 μs (Fig. 5). The small values of Φ(NO), the spectral overlap between reactant and product, the low absorption coefficients of the product, and the photodecomposition of the parent compound, which hindered the averaging of a large number of traces, determined a very bad signal/noise ratio, and, consequently, impaired a good time resolution. This is probably also the reason why no clear transient absorbance signal was detected for the Os aqua-derivative [electrochemically detected Φ(NO) = 0.16 ± 0.02 upon excitation at 308 nm]. [FeIII(CN)5H2O]2− was already shown to be produced within the resolution of the equipment in the microsecond time range upon pulsed excitation of NP.37
Laser-induced optoacoustic spectroscopy (LIOAS)
The experiments were performed only with NP by exciting the solutions at 355 and 480 nm at various temperatures. See Fig. 6 for an example of a LIOAS signal. The LIOAS signals obtained upon excitation at 428 nm (especially at Tβ=0) were too small to allow a quantitative evaluation. | ||
| Fig. 6 LIOAS signals after excitation at 480 nm of solutions of the calorimetric reference and NP in 0.1 M NaH2PO4/Na2HPO4 buffer (pH 6.5) at 4.8 °C, together with the simulation curve (overlapping the thick sample signal) and the residuals distribution. One exponential was used for the fitting with a time constant of 44 ns. | ||
Upon excitation at 355 nm, deconvolution with one exponential always resulted in a very short production time of < 1 fs, which essentially means that the pressure wave is generated in a time well below the observation window of the experiment. However, since the amplitude of the signal was extremely small at this temperature and wavelength, the lifetimes are not very precise, and should be taken only as an indication of the occurrence of the process.
The LIOAS signals obtained upon excitation at 480 nm were well fitted with a single exponential function with a time constant of (120 ± 40) ns at 0.8 °C and (45 ± 15) ns at 4.8 °C (Table 4). The very small and noisy transient absorption signals (see above) impaired the analysis of the initial formation of [Fe(CN)5H2O]2−.
=0.8 °C and T=4.8 °C, and calculated enthalpy [ΔH, eqn. (5) and (6)] and structural volume change per absorbed einstein (ΔVe) and per mol of [Fe(CN)5H2O]2− (ΔVstr), after excitation of [Fe(CN)5NO]2− with 8 ns laser pulses at 480 nm
| λexc/nm | T/°C | φ | τ/ns | ΔVe/ml mol−1 | ΔVstr/ml mol−1 | ΔH/kJ−1 mol−1 |
|---|---|---|---|---|---|---|
| 480 | 0.8 | 0.12 ± 0.02 | 120 ± 40 | 0.27 | 8 ± 3 | 190 ± 20 |
| 4.8 | 1.09 ± 0.05 | 45 ± 15 |
The LIOAS signals after excitation at 480 nm could be analysed in a better manner precisely because the quantum yield for NO release is smaller at this wavelength, i.e., there is a larger heat release from the excited species and the LIOAS signals are larger. Therefore, especially the calculation of the enthalpy change is more reliable with the data obtained after excitation at 480 nm. Additionally, one should consider that at lower wavelengths more heat evolves from radiationless deactivation from upper excited levels, which masks other effects.
The above results tell us that at both temperatures and excitation wavelengths the product formed in less than at most 120 ns (at 0.8 °C, as observed upon excitation at 480 nm) lives longer than about 5 times the pressure integration time (i.e. longer than ca. 600 ns). All data indicate that the product is [Fe(CN)5H2O]2−.
A two-exponential function (not shown) also fitted the data, but produced results non-compatible with the transient absorbance observations, since the shorter lifetime was much longer than the rising time of the transient absorption changes, and the amplitude afforded an enthalpy level much larger than the molar laser energy. The distribution of residuals and the χ2 values with the two-exponential function were not better than with the one-exponential fitting function.
The data taken upon excitation at 480 nm indicates that the production time decreases with temperature upon excitation, i.e., there is an activation process on the way from the excited state to the final product.
The reaction volume change per einstein absorbed calculated with eqn. (5) together with the determined value of (β/cpρ) = 9.2 10−3 ml kJ−1 at 4.8 °C and E(480 nm) = 249.1 kJ mol−1, is ΔVe = (0.27 ± 0.05) ml mol−1.
The LIOAS signals upon excitation at 355 nm do not afford reliable quantitative data (see above), especially because at 0.8 °C they are too small.
The calculated fraction of heat released is α(480) = (0.97 ± 0.07) [eqn. (6)]. In view of the fact that NP shows a fluorescence quantum yield of 3 × 10−3 (Table 3), energy balance considerations readily lead to eqn. (8). The energy provided by excitation (Eλ) should be equal to the sum of the prompt heat released (q) and the energy stored by the produced [Fe(CN)5H2O]2−, i.e., the product of its formation quantum yield [(Φ = Φ(NO), vide supra] and its enthalpy content with respect to the parent compound (ΔH).
| E(λ) = q + ΦΔH = αE(λ) + Φ(NO)ΔH | (8) |
Considering the molar excitation wavelength E(480 nm) = 249.1 kJ mol−1, the heat delivered is q = αE(λ) = (241.6 ± 100) kJ mol−1. Thus, with Φ(NO) = 0.04 at 480 nm and 4.8 °C (Table 3) the enthalpy difference between [Fe(CN)5NO]2− and [Fe(CN)5H2O]2− is ΔH = [E(λ) − q]/Φ(NO) = (190 ± 20) kJ mol−1.
The structural volume change upon excitation at 480 nm is ΔVstr = ΔVe/Φ(NO) = (0.27 ± 0.05)/ 0.04 = (8 ± 3) ml mol−1. This structural volume change is concomitant with the appearance in ca. 50 ns at 4.8 °C of the long-lived species. Thus, it should correspond to the volume difference between [Fe(CN)5NO]2− and [Fe(CN)5H2O]2−and is consistent with the sum of an expansion due to the loosening of the water molecules linked via hydrogen bonds to the CN groups upon oxidation of Fe(II) to Fe(III), plus the difference in the partial molar volumes of the released NO from the first coordination sphere into the bulk and the entering water replacing the NO. An expansion due to loosening of the H2O molecules was found for the production of the 3MLCT state upon excitation of ruthenium(II)–bipyridine cyano complexes.28 The result is in quite satisfactory agreement with the recently measured activation volume of 7 ml mol−1 for the dissociation of NO from the [Fe(CN)5NO]3− ion.46 This reaction occurs through a dissociative mechanism, in which the structure of the transition state nearly equals the structure of the products; therefore the comparison between an activation volume and the presently reported reaction volume appears as reasonable.
At this point, it seems appropriate to discuss a work on excited state Raman spectroscopy with the potassium salt of NP.47 By pumping at 406 nm after excitation with 9 ns pulses, a new peak appeared at 1835 cm−1, largely down-shifted with respect to that for the parent compound (N–O stretching in NP, ca. 1940 cm−1). The shift was traced to NO-bending upon excitation, but the consideration of more recent evidence on the formation of NO-linkage isomers upon low-temperature irradiation of NP48 allows us to suggest that similar phenomena could be present within the conditions of the irradiation process at ambient temperature. Thus, the primary excitation product, {M(III)–NO} is likely to rapidly deactivate to the ground state potential energy surface,48 affording the η1-M–ON isomer (the so-called MS1 state, ON = isonitrosyl), probably with an intermediate formation of the η2-side-bound isomer (the MS2 state). The MS1 state should be labile for NO release, consistent with the short times observed for the [Fe(CN)5H2O]2− appearance. This process is most likely activated as demonstrated by the temperature dependence of the [Fe(CN)5H2O]2− appearance times determined by LIOAS, and of the NO quantum yields. Quite relevant to this reinterpretation is the fact that the same value for the N–O stretching mode at 1835 cm−1 has been reported for the now well characterized MS1 linkage isomer of NP.49
Conclusions
A consistent picture emerges on the factors determining the changes in the quantum yields for NO release (at different wavelengths) from the cyanometallates of group 8 metals, covering the metals of the three transition series. The major decrease when going from the Fe to the Os compound is traced to the MIII–nitrosyl bond reorganization process and further cleavage with NO ejection in few ns. The reorganization is probably a MIII–NO → MIII–ON isomerization, according to previous evidence. The influence of macrocycle-addition reported here, as well as the comparative analysis of the quantum yields previously measured for other {X5MNO} complexes agree with the expected influence of the coligands or of the second-sphere interactions on the metal electron density, thus determining the metal–nitrosyl bond strength. The temperature dependence of the Φ(NO) values as well as of the production of the [FeIII(CN)5H2O]2− species, as determined by LIOAS, demonstrate the presence of an activated state in the way from excitation to the final product. This activated step could be the NO loss from the Fe–ON isomer (the so-called MS1 state, ON = isonitrosyl). The volume expansion of 8 ml mol−1 upon production of [FeIII(CN)5H2O]2− is attributed mainly to the expansion of the water molecules attached to the cyano-ligands, concomitant with the decrease in electron density on the FeIII.Acknowledgements
We thank Norbert Dickmann, Gudrun Klihm, and Simone Möhlenbeck for their able technical assistance, Alberto Rizzi for interesting discussions and help, and Professor Fernando Pina (Universidade de Lisboa) for providing the macrocycles. MV was supported by CONICET (Argentina) and DAAD (Germany) during her stay in Mülheim.References
- Nitric Oxide, Biology and Pathobiology, ed. J. L. Ignarro, Academic Press, San Diego, CA, 2000 Search PubMed.
- J. S. Stamler and M. Feelisch, Biochemistry of nitric oxide and redox-related species, in Methods in Nitric Oxide Research, ed. M. Feelisch and J. S. Stamler, Wiley, Chichester, 1996, pp. 19–27 Search PubMed.
- G. B. Richter-Addo and P. Legzdins, Metal Nitrosyls, Oxford University Press, New York, 1992 Search PubMed.
- J. A. Olabe and L. D. Slep, Reactivity and structure of complexes of small molecules: nitric and nitrous oxide, in Comprehensive Coordination Chemistry II, from Biology to Nanotechnology, ed. J. A. Mc Cleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 1, section III, ch. 1.31, pp. 603–623 Search PubMed.
- M. J. Clarke and J. B. Gaul, Chemistry relevant to the biological effects of nitric oxide and metallonitrosyls, Struct. Bond., 1993, 81, 147–181 CAS.
- S. P. Fricker, E. Slade, N. A. Powell, O. J. Vaughan, G. R. Henderson, B. A. Murrer, I. L. Megson, S. K. Bisland and F. W. Flitney, Ruthenium complexes as nitric oxide scavengers: a potential therapeutic approach to nitric oxide-mediated diseases, Br. J. Pharmacol., 1997, 122, 1441–1449 CAS.
- (a) P. C. Ford and I. M. Lorkovic, Mechanistic aspects of the reactions of nitric oxide with transition-metal complexes, Chem. Rev., 2002, 102, 993–1017 CrossRef CAS; (b) P. C. Ford, L. E. Laverman and I. M. Lorkovic, Adv. Inorg. Chem, 2003, 54, 203–257 CrossRef CAS.
- T. W. Hayton, P. Legzdins and W. B. Sharp, Coordination and organometallic chemistry of metal–NO complexes, Chem. Rev., 2002, 102, 935–991 CrossRef CAS.
- P. C. Ford, J. Bourassa, K. Miranda, B. Lee, I. Lorkovic, S. Boggs, S. Kudo and L. Laverman, Photochemistry of metal nitrosyl complexes. Delivery of nitric oxide to biological targets, Coord. Chem. Rev., 1998, 171, 185–202 CrossRef CAS.
- E. Tfouni, M. Krieger, B. McGarvey and D. W. Franco, Structure, chemical and photochemical reactivity and biological activity of some ruthenium amine nitrosyl complexes, Coord. Chem. Rev., 2003, 236, 57–69 CrossRef CAS.
- L. M. Baraldo, P. Forlano, A. R. Parise, L. D. Slep and J. A. Olabe, Advances in the coordination chemistry of [M(CN)5L]n− ions (M= Fe, Ru, Os), Coord. Chem. Rev., 2001, 219, 881–921 CrossRef.
- R. W. Callahan and T. J. Meyer, Reversible electron transfer in ruthenium nitrosyl complexes, Inorg. Chem., 1977, 16, 574–581 CrossRef CAS.
- J. H. Enemark and R. D. Feltham, Principles of structure, bonding, and reactivity for metal nitrosyl complexes, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- B. R. Cameron, M. C. Darkes, H. Yee, M. Olsen, S. P. Fricker, R. T. Skerlj, G. J. Bridger, N. A. Davies, M. T. Wilson, D. J. Rose and J. Zubieta, J. Ruthenium(III) polyaminocarboxylate complexes: efficient and effective nitric oxide scavengers, Inorg. Chem., 2003, 42, 1868–1876 CrossRef CAS.
- M. Sauaia, R. G. de Lima, A. C. Tedesco and R. S. da Silva, Photoinduced NO release by visible light irradiation from pyrazine-bridged nitrosyl ruthenium complexes, J. Am. Chem. Soc., 2003, 125, 14718–14719 CrossRef CAS.
- M. Sauaia, F. de Souza Oliveira, A. C. Tedesco and R. S. da Silva, Control of NO release by light irradiation from nitrosyl-ruthenium complexes containing polypyridyl ligands, Inorg. Chim. Acta, 2003, 355, 191–196 CrossRef CAS.
- V. Togniolo, R. S. da Silva and A. C. Tedesco, Photo-induced nitric oxide release from chlorobis(2,2′-bipyridine)nitrosylruthenium(II) in aqueous solution, Inorg. Chim. Acta, 2001, 316, 7–12 CrossRef CAS.
- K. Szacilowski, M. Wojciech, G. Stochel, Z. Stasicka, S. Sostero and O. Traverso, Ligand and medium controlled photochemistry of iron and ruthenium mixed-ligand complexes: prospecting for versatile systems, Coord. Chem. Rev., 2000, 208, 277–297 CrossRef CAS.
- V. Balzani and V. Carassiti, Photochemistry of Coordination Compounds, Academic Press, San Diego, CA, 1970 Search PubMed.
- G. Stochel, High-pressure mechanistic studies on thermal and photochemical reactions of pentacyanoferrate complexes, Coord. Chem. Rev., 1992, 114, 269–295 CrossRef CAS.
- G. Stochel and R. van Eldik, Photochemical behaviour of metal complexes. Pressure effect versus mechanism, Coord. Chem. Rev., 1997, 159, 153–170 CrossRef CAS.
- S. K. Wolfe and J. H. Swinehart, Photochemistry of pentacyanonitrosylferrate(2-), nitroprusside, Inorg. Chem., 1975, 14, 1049–1053 CrossRef CAS.
- J. A. Olabe, L. A. Gentil, G. Rigotti and A. Navaza, Crystal and molecular structure of sodium pentacyanonitrosylruthenate(II) dihydrate and its spectroscopic properties and reactivity, Inorg. Chem., 1984, 23, 4297–4302 CrossRef CAS.
- L. M. Baraldo, M. S. Bessega, G. E. Rigotti and J. A. Olabe, Crystal and molecular structure, spectroscopic properties, and electrophilic reactivity of sodium pentacyanonitrosylosmate(II) dihydrate, Inorg. Chem., 1994, 33, 5890–5896 CrossRef CAS.
- B. Dietrich, M. W. Hosseini, J.-M. Lehn and R. B. Sessions, Synthesis and protonation features of 24-, 27-, and 32-membered macrocyclic polyamines, Helv. Chim. Acta, 1983, 66, 1262–1278 CrossRef CAS.
- D. F. Eaton, Reference Materials for Fluorescence Measurements, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS.
- P. Schmidt, T. Gensch, A. Remberg, W. Gärtner, S. E. Braslavsky and K. Schaffner, The complexity of the Pr to Pfr phototransformation kinetics is an intrinsic property of native phytochrome, Photochem. Photobiol., 1998, 68, 754–761 CAS.
- C. D. Borsarelli and S. E. Braslavsky, Volume changes correlate with enthalpy changes during the photoinduced formation of the 3MLCT state of ruthenium(II) bipyridine cyanocomplexes in the presence of salts. A case of the entropy–enthalpy compensation effect, J. Phys. Chem. B, 1998, 102, 6231–6238 CrossRef CAS.
- C. D. Borsarelli and S. E. Braslavsky, Enthalpy, volume and entropy changes associated with the electron transfer reaction between the 3MLCT state of Ru(bpy)32+ and methyl viologen cation in aqueous solutions, J. Phys. Chem. A, 1999, 103, 1719–1727 CrossRef CAS.
- I. Yruela, M. S. Churio, T. Gensch, S. E. Braslavsky and A. R. Holzwarth, Optoacoustic and singlet oxygen near-IR-emission study of the isolated D1-D2-Cyt b559 reaction center complex of photosystem II. Protein movement associated with charge separation, J. Phys. Chem., 1994, 98, 12789–12795 CrossRef CAS.
- C. D. Borsarelli and S. E. Braslavsky, Nature of the water structure inside the pools of reverse micelles sensed by laser-induced optoacoustic spectroscopy, J. Phys. Chem. B, 1997, 101, 6036–6042 CrossRef CAS.
- P. R. Crippa, A. Vecli and C. Viappiani, Time-resolved photoacoustic spectroscopy: new developments of an old idea, J. Photochem. Photobiol., B: Biol., 1994, 24, 3–15 CrossRef CAS.
- M. S. Churio, K. P. Angermund and S. E. Braslavsky, Combination of laser-induced optoacoustic spectroscopy (LIOAS) and semiempirical calculations for the determination of molecular volume changes: the photoisomerization of carbocyanines, J. Phys. Chem., 1994, 98, 1776–1782 CrossRef CAS.
- T. Gensch, C. Viappiani and S. E. Braslavsky, Structural volume changes upon photoexcitation of porphyrins: role of the nitrogen–water interactions, J. Am. Chem. Soc., 1999, 121, 10573–10582 CrossRef CAS.
- G. Stochel, R. van Eldik and Z. Stasicka, Mechanistic information from medium and high-pressure effects on the photooxidation of nitrosylpentacyanoferrate(II), Inorg. Chem., 1986, 25, 3663–3666 CrossRef CAS.
- R. J. Singh, N. Hogg, F. Neese, J. Joseph and B. Kalyanaraman, Trapping of nitric oxide formed during photolysis of sodium nitroprusside in aqueous and lipid phases: an electron spin resonance study, Photochem. Photobiol., 1995, 61, 325–330 CAS.
- T. Jarzynowski, T. Senkowski and Z. Stasicka, Flash photolysis of nitrosylpentacyanoferrate(II) complex, Pol. J. Chem., 1981, 55, 3–10 CAS.
- P. T. Manoharan and H. B. Gray, Electronic structure of nitroprusside ion, J. Am. Chem. Soc., 1965, 87, 3340–3348 CrossRef CAS.
- S. Kudo, J. L. Bourassa, S. E. Boggs, Y. Sato and P. C. Ford, In situ nitric oxide (NO) measurement by modified electrodes: NO labilized by photolysis of metal nitrosyl complexes, Anal. Biochem., 1997, 247, 193–202 CrossRef CAS.
- L. D. Slep, P. Alborés, L. M. Baraldo and J. A. Olabe, Kinetics and mechanism of ligand interchange in pentacyano-L-osmate(II) complexes (L = H2O, NH3, N-heterocyclic ligands), Inorg. Chem., 2002, 41, 114–120 CrossRef CAS.
- H. E. Toma and C. Creutz, Pentacyanoferrate(II) complexes: evaluaton of their formal potentials and mechanism of their quenching of tris(2,2′-bipyridine)ruthenium(II) luminescence, Inorg. Chem., 1977, 16, 545–550 CrossRef CAS.
- M. Mingardi and G. B. Porter, Spectra of K4Ru(CN)6, Spectrosc. Lett., 1968, 1, 293–310 CAS.
- L. Vera and F. Zuloaga, Triplet–singlet emission of Fe(CN)5CO3−, Inorg. Chem., 1978, 17, 765–766 CrossRef CAS.
- M. A. Rampi, M. T. Indelli, F. Scandola, F. Pina and A. J. Parola, Photophysics of supercomplexes. Adduct between Ru(bpy)(CN)42− and the [32]ane-N8H88+ polyaza macrocyclic, Inorg. Chem., 1996, 35, 3355–3361 CrossRef CAS.
- J. H. Espenson and S. G. Woneluk, Jr., Kinetics and mechanisms of some substitution reactions of pentacyanoferrate(III) complexes, Inorg. Chem., 1972, 11, 2034–2041 CrossRef CAS.
- F. Roncaroli, J. A. Olabe and R. van Eldik, Kinetics and mechanism of the interaction of nitric oxide with pentacyanoferrate(II). Formation and dissociation of [Fe(CN)5NO]3−, Inorg. Chem., 2003, 42, 4179–4189 CrossRef CAS.
- Y. Y. Yang and J. I. Zink, Excited-state nitrosyl bending and metal oxidation in K2[Fe(CN)5NO] determined by excited-state Raman spectroscopy, J. Am. Chem. Soc., 1985, 107, 4799–4800 CrossRef CAS.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Photoinduced linkage isomers of transition-metal nitrosyl compounds and related complexes, Chem. Rev., 2002, 102, 861–883 CrossRef CAS.
- M. E. Chacón Villalba, J. A. Güida, E. L. Varetti and P. J. Aymonino, Infrared evidence of NO linkage photoisomerization in Na2[Fe(CN)5NO]·2H2O at low temperature: experimental and theoretical (DFT) isotopic shifts from 15N(O), 18O and 54Fe species, Spectrochim. Acta, Part A, 2001, 57, 367–373 CrossRef CAS.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |