Mechanism of photoisomerization of optically pure trans-2,3-diphenylcyclopropane-1-carboxylic acid derivatives†
J. Sivagurua, Takehiko Wadab, Yumi Origanec, Yoshihisa Inouebc and V. Ramamurthy*a
aDepartment of Chemistry, Tulane University, New Orleans, LA 70118. E-mail: murthy@tulane.edu
bDepartment of Molecular Chemistry and PRESTO (JST), Osaka University, 2-1 Yamada-oka, Suita, 565-0871, Japan
cICORP Entropy Control Project, JST, 4-6-3 Kamishinden, Toyonaka, 560-0085, Japan
First published on 4th October 2004
Abstract
The photochemistry of optically pure isomers of α-methylbenzylamide of trans-2,3-diphenylcyclopropane-1-carboxylic acid has been examined in isotropic solution and within zeolites. The results suggest that these isomerize through cleavage of C2–C3 bond. The direct excitation in solution leads to non-equilibrating 1,3-singlet diradical intermediates whereas triplet sensitization results in equilibrating 1,3-triplet diradical intermediates. The direct excitation within NaY zeolite seems to result in equilibrating zwitterionic intermediates. Studies on the optically pure trans isomers allow one to understand the mechanism of chiral induction during the photoisomerization of mesocis-2,3-diphenylcyclopropane-1-carboxylic acid. The current study has clarified the nature of the excited states involved during the classic (R)-N-acetyl-1-naphthylethylamine sensitized isomerization of 1,2-diphenylcyclopropane.
Introduction
The optically inactive cis-1,2-diphenylcyclopropane that can be transformed into its chiral trans isomer by both singlet and triplet-photosensitized irradiations1–5 is a benchmark system to examine the influence of the zeolite medium on the chiral induction process. We have recently demonstrated with several examples including 2,3-diphenylcyclopropane-1-carboxylic acid derivatives that a chiral auxiliary functions more effectively as a chiral inducer within a zeolite than in a solution.3 To comprehend this phenomenon one should have a good understanding of the intermediates involved in the photoprocess. The reversible photoisomerization between optically inactive cis-diphenylcyclopropane and optically active (R,R)- and (S,S)-trans isomers, in principle, can occur via cleavage of either C2–C3 bond or C1–C2/C1–C3 bonds (Scheme 1).4–6 If the reaction proceeds via cleavage of C2–C3 bond the chiral induction must occur at the stage of ring closure. On the other hand, if the cleavage occurs via C1–C2/C1–C3 bond the chiral discrimination must take place at the primary bond cleavage stage. One can distinguish between the above two possibilities by monitoring the photoisomerization of optically pure trans isomers. This report presents results of such an investigation.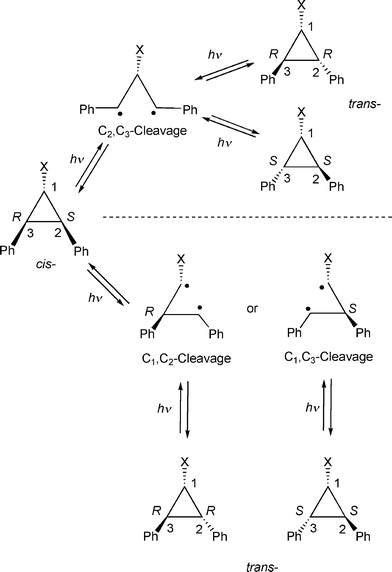 | ||
| Scheme 1 Photoisomerization of diphenylcyclopropane derivatives. | ||
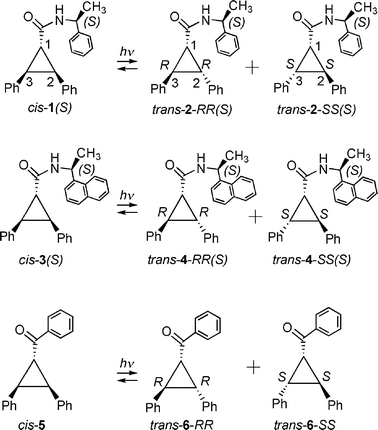
We have investigated the photochemistry of α-methylbenzylamide of trans-2,3-diphenylcyclopropane-1-carboxylic acid3,7 (trans-2-RRS and trans-2-SSS) in solution and within zeolites (Scheme 2). Recently we have shown that 2,3-diphenyl-1-benzoylcyclopropane derivatives (Scheme 2; cis-5, trans-6) isomerizes via C1–C2/C1–C3 bond cleavage leading to diradical intermediates.8 Results presented in this report on α-methylbenzylamide of trans-2,3-diphenylcyclopropane-1-carboxylic acid derivatives3 (Scheme 2; cis-1, trans-2) provide a complementary information on the isomerization behavior of 2,3-diphenyl-1-benzoylcyclopropane derivatives. Direct excitation of optically pure trans-2 (RRS or SSS) leads to reaction from S1 and triplet sensitization from T1. Results presented below show that the product distributions upon isomerization from excited singlet and triplet states of trans-2-RRS (and trans-2-SSS) are different and the isomerization occur via cleavage of C2–C3 bond cleavage.
 | ||
| Scheme 2 | ||
In the first example of asymmetric induction in photochemical reactions of 1,2-diphenylcyclopropane using (R)-N-acetyl-1-naphthylethylamine as the chiral sensitizer the authors suggested the naphthyl derivative to function as a triplet sensitizer.1a However, further studies demonstrated that the naphthalene sensitized photoisomerization could be due to charge transfer interaction between the diphenylcyclopropane and the naphthalene chromophores.2 To ascertain the reactive state in the above system we have investigated the photochemistry of trans-4-RRS and trans-4-SSS where the sensitizer used by Hammond and Cole is covalently linked as the chiral auxiliary.1a By comparing the product distribution upon excitation of pure diastereomer (RRS or SSS) of trans-4 with those from S1 and T1 of trans-2-RRS and trans-2-SSS we are able to conclude that (R)-N-acetyl-1-naphthylethylamine acts as a singlet and not as a triplet sensitizer.
Results
The structures of the reactants investigated and their photoproducts are provided in Scheme 2. The compounds are identified based on their geometrical relationship (cis and trans) between the two phenyl groups at C2 and C3 carbons and their stereochemical relationship (R and S) between them taking C1 carbon into account. The structures on the left side (Scheme 2) are the cis isomers and the two structures on the right side, trans isomers, are chiral. These are identified as RR and SS. In the case of 1–4 the the absolute configuration at the chiral auxiliary (labelled within parantheses; Scheme 2). For clarity of discussion trans-2-RR(S) is labelled as trans-2-RR and cis-1(S) as cis-1. This nomenclature is used throughout the text.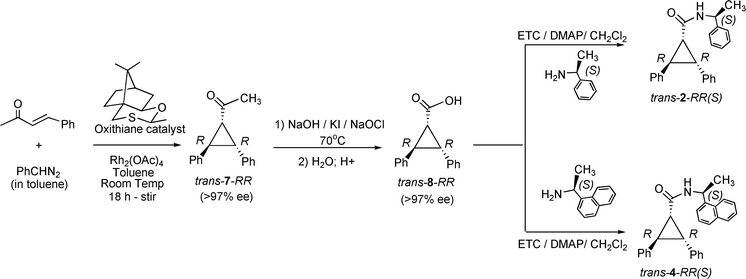 | ||
| Scheme 3 Asymmetric syntheses of optically pure trans-2-RR and trans-4-RR isomer. | ||
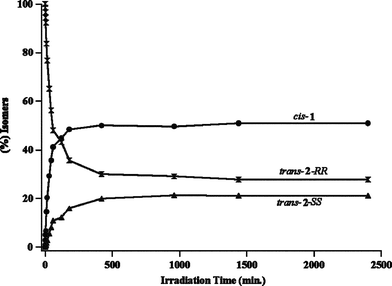 | ||
| Fig. 1 Product distribution upon triplet sensitization of trans-2-RR in acetone solution (acetone as solvent and sensitizer). | ||
| Time | Irradiation of trans-2-RR isomer in solution | ||
|---|---|---|---|
| cis-1 | trans-2-RR | trans-2-SS | |
| 0 min | 0 | 100 | 0 |
| 30 min | 7.7 | 92.3 | 0 |
| 1 h | 13 | 87 | 0 |
| 1 h 30 min | 21.3 | 78.7 | 0 |
| 2 h | 26.2 | 73.8 | 0 |
| 2 h 30 min | 32.2 | 67.8 | Trace |
| Irradiation of trans-4-RR isomer in solution | |||
|---|---|---|---|
| cis-3 | trans-4-RR | trans-4-SS | |
| a Solution irradiation done by dissolving ≈5 mg trans-2-RR (or trans-4-RR) in 0.5 mL dichloromethane and 15 mL hexane. Irradiation by dissolving the trans isomers in 12 mL acetonitrile also gave similar results. | |||
| 0 min | 0 | 100 | 0 |
| 1 h | 15.7 | 84.3 | 0 |
| 2 h | 36.8 | 63.2 | 0 |
| 3 h | 55.7 | 35.1 | 9.2 |
| 6 h | 60.0 | 20.4 | 17.6 |
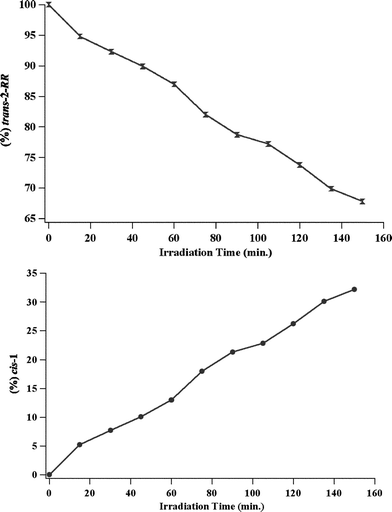 | ||
| Fig. 2 Product distribution with respect to time of direct excitation of trans-2-RR in methylene chloride/hexane solution. Top: disappearance of trans-2-RR. Bottom: appearance of cis-1. | ||
Discussion
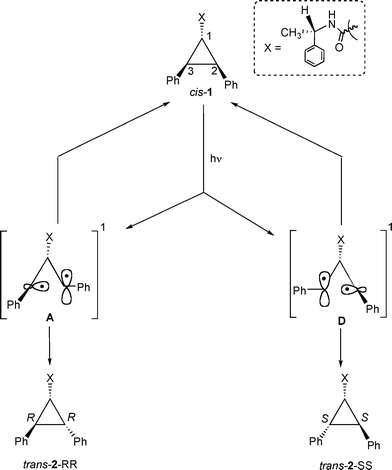
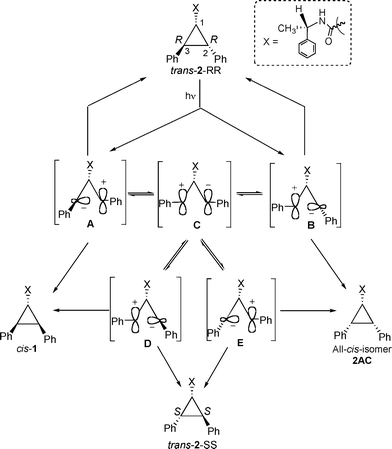
In order to probe the reactivity of intermediates involved during direct and sensitized excitations of 2,3-diphenylcyclopropane-1-carboxylic acid derivatives, product distributions from pure stereoisomers of trans-2 (RR or SS) were monitored with respect to duration of irradiation. The model that is built based on the photobehavior of a pure isomer (RR and SS) of trans-2 is utilized to understand the diastereoselective photoisomerization of cis-1 in solution and within zeolites.13The isomerization of 1,2-diarylcyclopropane has been examined previously and is suggested to proceed through a diradical intermediate.1,2,5,14 Formation of trans-2-SS and cis-1 from trans-2-RR, even at very early times, during sensitized irradiation in solution (Fig. 1) and within zeolites (Table 2) suggests that 1,3-diradicals A–E establish equilibrium prior to ring closure to the products (Scheme 4). If A and B establish an equilibrium via the diradical C, trans-2-SS would be formed as the primary photoproduct. Such equilibrium would yield cis-1 and all-cis-isomer 2AC as photoproducts, in addition to trans-2-SS. However, under our condition the all-cis isomer 2AC was not observed as a photoproduct. Formation of trans-2-SS from trans-2-RR is plausible only if the absolute configurations at both C2 and C3 carbons are reversed and this can occur only from the diradical C. The above mechanistic model would also be valid during the photoisomerization of cis-1. As per this model, the triplet 1,3-diradicals A, C and D (Scheme 5) formed upon triplet sensitization of cis-1, would be in equilibrium before they intersystem-cross and close to stereoisomeric products trans-2-RR and trans-2-SS. Therefore, any diastereoselectivity during triplet sensitized photoisomerization of cis-1 (difference in the amounts of trans-2-RR and trans-2-SS formed) must derive from alteration of the equilibrium in favor of one of the two diradicals (A or D).
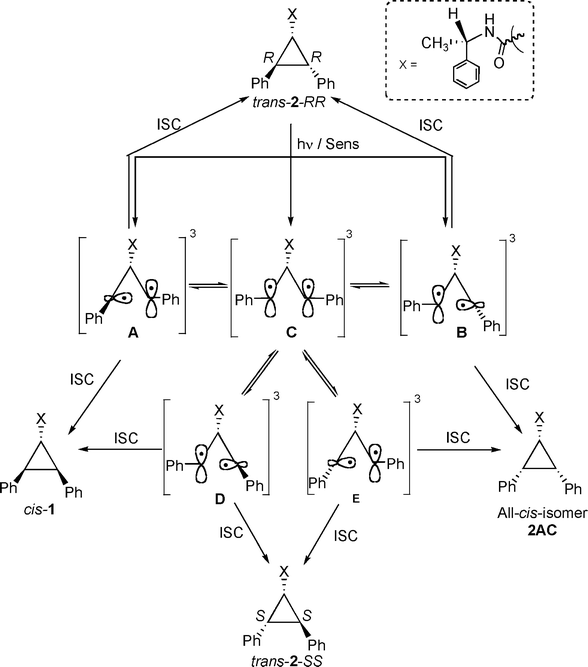 | ||
| Scheme 4 | ||
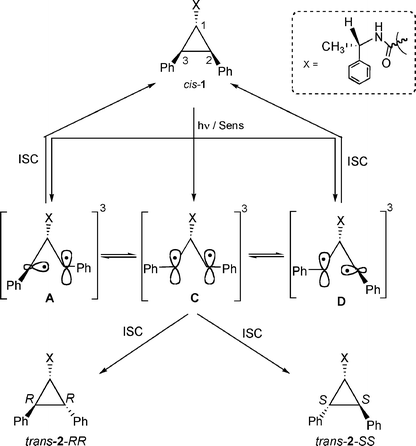 | ||
| Scheme 5 | ||
Isomer cis-1 was obtained as the exclusive photoproduct upon direct excitation of trans-2-RR in solution (Table 1). An important point to note is that during direct excitation, unlike in the case of the triplet sensitized irradiation, there is no interconversion between trans-2-RR and trans-2-SS isomers, suggesting that the half open singlet diradicals A and B are not in equilibrium with each other (Scheme 6). The diradical B, in principle, could produce all-cis isomer 2AC. However, all-cis isomer was not observed as a photoproduct. By extending the above model to cis-1, the singlet 1,3-diradicals A and D formed upon direct irradiation of cis-1 would be expected to close to the corresponding diastereomeric products trans-2-RR and trans-2-SS without establishing an equilibrium between them (Scheme 7). Accordingly, the observed diastereoselectivity during direct irradiation in solution would depend on the rates of formation of the two singlet diradicals A and D directly from the excited singlet state S1. In the absence of chiral influence, both diradicals would be formed in equal amounts and the diastereoselectivity would be zero. However presence of a chiral auxiliary could influence the rates of formation of the two diradicals A and D leading to diastereoselective isomerization.
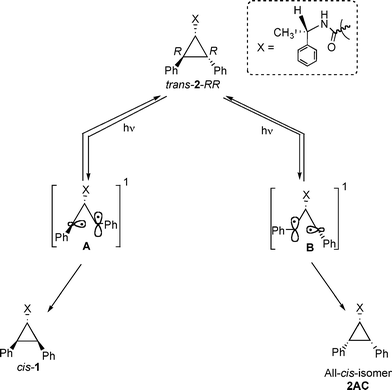 | ||
| Scheme 6 | ||
 | ||
| Scheme 7 | ||
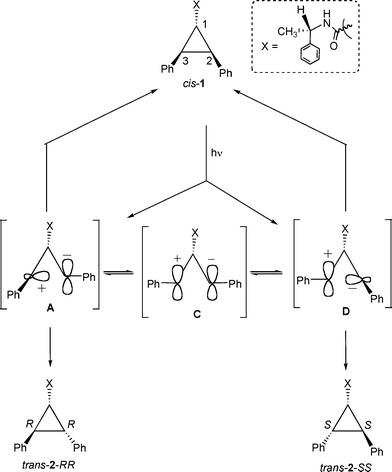
Initial product distributions upon direct irradiation of trans-2-RR included within alkali ion exchanged zeolites (Table 3) were different from that in solution. In polar zeolites15 such as LiY and NaY even after 2 h irradiation the major product is trans-2-SS (Table 3). As discussed above, in solution during direct excitation only cis-1 was obtained and during triplet sensitization both cis-1 and trans-2-SS were obtained as primary photoproducts. The observed behavior within zeolites is different from the above two. Diphenylcyclopropane derivatives, upon direct excitation, are known to undergo homolytic cleavage in non-polar media and heterolytic cleavage in polar media.14 We speculate that within the highly polarizable environment of a zeolite15 photoisomerization occurs via a heterolytic pathway. We believe that within LiY and NaY zeolites, the character of the intermediates A–E (Scheme 8) would be zwitterionic rather than diradical. Mutation at both chiral centers of trans-2-RR to form trans-2-SS suggests that the zwitterionic intermediate C (where the chiral information is lost) is most likely involved in the isomerization process. It is probable that the anionic walls and the charge compensating cations of zeolites stabilize the ionic intermediates. Under such conditions, the zwitterionic intermediates are sufficiently long lived to establish equilibrium between various structures. By extending the above observations, we speculate that the zwitterionic intermediates (A, C and D) formed upon direct excitation of cis-1 would be in equilibrium before they form the diastereomeric products trans-2-RR and trans-2-SS (Scheme 9). The above studies with the pure stereoisomers clearly suggest that the mechanism of photoisomerization and the type of intermediates involved in (a) sensitized irradiation in solution and within zeolite; (b) direct excitation in solution; and (c) direct excitation within zeolites are different.
 | ||
| Scheme 8 | ||
 | ||
| Scheme 9 | ||
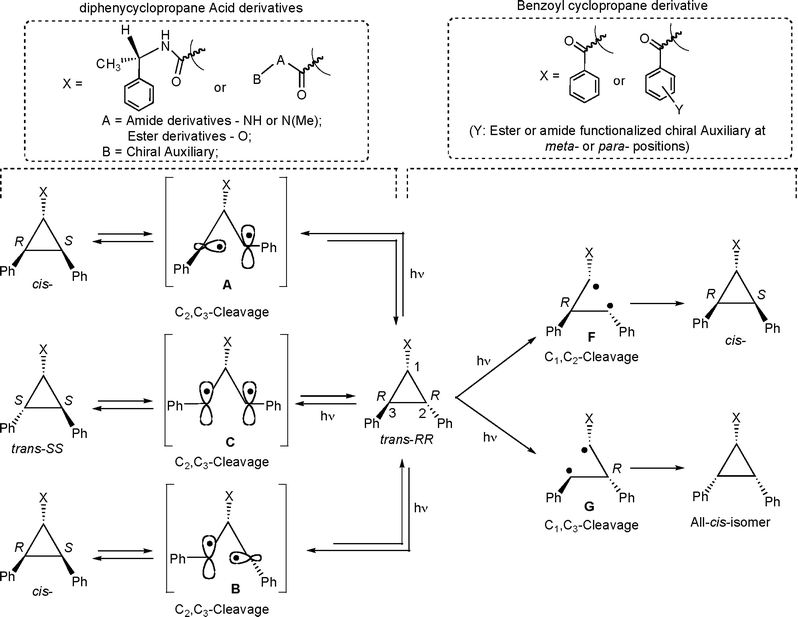
The above discussion assumes that photoisomerization occurs via the cleavage of the C2–C3 σ bond.4,5 In principle photoisomerization could also result by the cleavage of the C1–C2 or the C1–C3 bond (Scheme 1).6,8 However, the cleavage of the C1–C2 or C1–C3 bond would not allow interconversion between the trans-stereoisomers RR and SS (Scheme 10). The occurrence of interconversion between diastereomers during sensitized (in solution and within zeolite) and direct irradiations (within zeolite) suggests that isomerization occurs via cleavage of C2–C3 σ bond. The cleavage of the C1–C2 or C1–C3 β-bond has been postulated during the photoisomerization of cis-5 (Schemes 1 and 2). This is discussed in one of our recent publication8 in which we showed that there was no interconversion between trans-6-RR and trans-6-SS upon irradiation of trans-6-RR (or trans-6-SS) and the photoisomerization proceeds most likely through the cleavage of the C1–C2 or the C1–C3 β-bond (Scheme 10).
 | ||
| Scheme 10 | ||
The final aspect of this presentation concerns with the role of (R)-N-acetyl-1-naphthylethylamine during isomerization of 1,2-diphenylcyclopropane.1a,2 As indicated in the introduction the exact function (electron, singlet energy or triplet energy transfer) of the naphthalene chromophore during the above photoisomerization process is not clear and has been debated for the last five decades.1,2 We believed that the role of the naphthalene derivative in the photoisomerization process could be uncovered by comparing the initial product distribution upon excitation of the pure stereoisomer trans-4-RR and trans-4-SS with that of trans-2-RR and trans-2-SS, respectively. As illustrated in Table 1, irradiation of trans-4-RR (naphthalene chromophore was excited) in solution exclusively gave cis-3 at early irradiation times. There was no interconversion between trans-4-RR to trans-4-SS even after 2 h. On prolonged irradiation (>3 h) trans-4-SS was formed due to the substantial presence of cis-3. Exclusive formation of cis-3 from trans-4-RR suggests that the mechanism of photoisomerization is similar to the results observed during the direct irradiation of trans-2-RR in solution. Comparing the initial product distribution upon direct irradiation of trans-2-RR and trans-4-RR in solution, it could be concluded that the naphthalene assisted photoisomerization in solution proceeds via excited singlet state and not via triplet state of naphthalene as originally suggested. We have recently estimated the triplet energy of 1,2-diphenylcyclopropane to be ca. 301 kJ mol−1, well above that of naphthalene.16 Thus the results of photochemical study reported here is consistent with the photophysics of 1,2-diphenylcyclopropanes.16
Conclusion
Studies with pure stereoisomers were invaluable in understating the mechanism of photoisomerization of diphenylcyclopropane derivatives both in solution and within zeolite. The mechanism of photoisomerization and the type of intermediates involved in the photoisomerization processes depend on the mode of excitation (sensitized or direct irradiation) and the medium in which the reaction is carried out (solution or zeolite). Results of this study have clarified the nature of excited state and intermediates involved during the photoisomerization of diphenylcyclopropane sensitized by (+)-1-(1-N-acetylaminoethyl) naphthalene. The mechanistic details unraveled in this study should help in understanding the diastereoselective photoisomerization of diphenylcyclopropane derivatives.Experimental
Materials and methods
NaY zeolite was obtained from Zeolyst International, The Netherlands. Monovalent cation-exchanged zeolites (LiY, KY, RbY and CsY) were prepared by refluxing 10 g of NaY with 100 mL of a 10% solution of the corresponding metal nitrate in water for 24 h. The cation exchanged MY zeolite was filtered and washed thoroughly with distilled water. This procedure was repeated three times. Subsequently, the cation-exchanged zeolite was dried at 120 °C for about 3 h.(i) Synthesis of cis-2,3-diphenylcyclopropane-1-carboxylic acid (cis-8) and trans-2,3-diphenylcyclopropane-1-carboxylic acid (trans-8). These were prepared following reported literature procedures17 and their spectral data follow.
cis-2,3-Diphenylcyclopropane-1-carboxylic acid (cis-8). 1H NMR (CDCl3, 400 MHz, δ): 2.49 (t, 1H, J = 5.2 Hz), 3.07 (d, 2H, J = 5.2 Hz), 6.83–7.00 (m, 4H), 7.05–7.2 (m, 6H). 1H–1H COSY (CDCl3, 400 MHz, δ): Cross-peak between δ 2.49 and 3.07. 13C NMR (CDCl3, 100 MHz, δ): 27.57 (s), 34.17 (s), 126.72 (s) 127.02 (s), 120.09 (s), 128.4 (s), 129.05 (s), 129.15 (s), 135.35 (s), 179.94 (s). Mass spectral data: m/z (relative intensity): 238 (M+, 22), 220 (5), 192 (83), 178 (32), 152 (8), 131 (4), 115 (100), 91 (33), 65 (17), 45 (22).
(Racemic)-trans-2,3-diphenylcyclopropane-1-carboxylic acid (trans-8). 1H NMR (CDCl3, 400 MHz, δ): 2.36 (dd, 1H, J = 5.2, 9.6 Hz), 2.97 (dd, 1H, J = 7.2, 9.6 Hz), 3.17 (dd, 1H, J = 5.2, 7.2 Hz), 7.21–7.32 (m, 10H). 1H–1H COSY (CDCl3, 400 MHz, δ): Cross-peaks between δ (a) 2.36 and 2.97; (b) 2.36 and 3.17; (c) 2.97 and 3.17. 13C NMR (CDCl3, 100 MHz, δ): 30.4 (s), 30.97 (s), 35.41 (s), 126.83 (s) 127.1 (s), 127.29 (s), 128.33 (s), 128.8 (s), 129.36 (s), 135.57 (s), 139.91 (s), 175.3 (s). Mass spectral data: m/z (relative intensity): 238 (M+, 16.3), 220 (5.8), 192 (81.4), 178 (27.9), 165 (17.4), 152 (9.3), 131 (4.6), 115 (100), 91 (34.9), 77 (23.3), 45 (29), 39 (16.2).
(ii) Conversion of acids to amides (1–4). 2,3-Diphenylcyclopropane-1-carboxylic acid (≈150 mg; cis-8 used for cis-1, cis-3 and trans-8 used for trans-2, trans-4) was dissolved in dichloromethane in a dry 100 mL round bottom flask. 1.1 eq. of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (ETC)12 and 0.1 eq. of dimethylamino pyridine (DMAP) was added followed by the addition of 1.1 eq. of the chiral amine [S-(−)-1-phenylethyl amine for 1 and 2 and S-(−)-1-naphthylethyl amine for 3 and 4]. The mixture was stirred overnight. The solution was washed with de-ionized water (3 × 5 mL), dried over Na2SO4, concentrated and purified by column chromatography (silica) using hexane–ethyl acetate as eluent. The trans-derivatives were synthesized to verify the photoproducts and used for separation on of pure isomers by preparative HPLC.
S(−)-1-Phenylethyl amide of cis-2,3-diphenylcyclopropane-1-carboxylic acid (cis-1). 1H NMR (CDCl3, 400 MHz, δ): 1.45 (d, 3H, J = 6.8 Hz), 2.19 (t, 1H, J = 5.6 Hz), 3.06 (d, 2H, J = 5.6 Hz), 5.12 (merged dq, 1H, J = 6.8, 8.2 Hz), 6.03 (br d, 1H, J = 8.2 Hz), 6.88–6.96 (m, 4H), 7.05–7.17 (m, 6H), 7.23–7.34 (m, 5H). 1H–1H COSY (CDCl3, 400 MHz, δ): Cross-peaks between δ (a) 1.45 and 5.12; (b) 2.19 and 3.06; (c) 5.12 and 6.03. 13C NMR (CDCl3, 100 MHz, δ): 22.16 (s), 29.9 (s), 32.5 (d), 49.5 (s), 126.5 (s), 127.7 (d), 128.2 (d), 128.9 (d), 129.1 (d), 129.2 (d), 136.4 (d), 143.4 (s), 171.1 (s). Mass spectral data: m/z (relative intensity): 341 (M+, 1.1), 220 (2.3), 193 (100), 178 (23), 152 (3.4), 115 (94.2), 105 (75.9), 91 (8), 77 (29.9), 51 (10.3).
S(−)-1-Phenylethyl amide of trans-2,3-diphenylcyclopropane-1-carboxylic acid (trans-2). 1H NMR (CDCl3, 400 MHz, δ): 1.24 and 1.35 (two d, 3H, J = 7.4, 6.8 Hz), 2.21 (m, 1H), 2.79 (dd, 1H, J = 6.0 Hz and 8.0 Hz), 3.20 (dd, 1H, J = 6.0 Hz and 8.0 Hz), 4.97 (m, 1H), 5.59 and 5.65 (two br d, 1H, J = 7.6, 8.0 Hz), 6.98-7.32 (m, 15H). 1H–1H COSY (CDCl3, 400 MHz, δ): Cross-peaks between δ (a) 1.24 and 4.97; (b) 1.35 and 4.97; (c) 2.21 and 2.79; (d) 2.21 and 3.20; (e) 2.79 and 3.20; (f) 4.97 and 5.59; (g) 4.97 and 5.65. 13C NMR (CDCl3, 100 MHz, δ): 21.7 (d), 28.2 (s), 33.7 (d), 33.9 (d), 48.8 (d), 126.3 (s), 126.4 (s), 126.7 (d), 126.9 (d), 127.07 (d), 127.3 (s), 127.5 (s), 128.4 (s), 128.7 (s), 128.8 (d), 129.1 (d), 136.6 (d), 140.4 (d), 143.3 (d), 167.7 (d). Mass spectral data: m/z (relative intensity): 341 (M+, 1.1), 219 (2.3), 193 (100), 178 (20.7), 165 (10.3), 130 (8.0), 115 (97.7), 105 (94.3), 91 (33.3), 77 (31), 65 (8), 51 (9.2).
S(−)-1-Naphthylethyl amide of cis-2,3-diphenylcyclopropane-1-carboxylic acid (cis-3). 1H NMR (CDCl3, 400 MHz, δ): 1.69 (d, 3H, J = 6.4 Hz), 2.24 (t, 1H, J = 5.2 Hz), 3.10 (d, 2H, J = 5.2 Hz), 6.02 (merged dq, 1H, J = 6.4, 8.0 Hz), 6.3 (br d, 1H, J = 8.0 Hz), 6.8–8.2 (m, 17H). 1H–1H COSY (CDCl3, 400 MHz, δ): Cross-peaks between δ (a) 1.69 and 6.02; (b) 6.02 and 6.3; (c) 2.24 and 3.10. 13C NMR (CDCl3, 100 MHz, δ): 21.3 (s), 29.9 (s), 32.45 (d), 45.4 (s), 122.8 (s), 123.6 (s), 125.5 (s), 126.1 (s), 126.5 (d), 126.9 (s), 128.1 (d), 128.6 (s), 129.0 (d), 129.3 (s), 131.3 (s), 134.2 (s), 136.3 (s), 136.6 (s), 138.6 (s), 170.9 (s).
S(−)-1-Naphthylethyl amide of trans-2,3-diphenylcyclopropane-1-carboxylic acid (trans-4). 1H NMR (CDCl3, 400 MHz, δ): 1.4 (d, 3H, J = 6.8 Hz), 1.98 (m, 1H), 2.56 (dd, 1H, J = 7.2, 11.8 Hz), 3.12 (dd, 1H, J = 5.6, 7.2 Hz), 5.6 (m, 1H), 5.9 (br d, 1H), 6.7–8.1 (m, 17H).
(iii) Determination of absolute configuration of trans-2, and 4.
Synthesis of oxithiane (sulfide catalyst) (ref. 9, 10, 18). Chloroform (5 mL), followed by 792 µL (6.44 mmol) of BF3 etherate and 1.34 mL (23.4 mmol) of glacial acetic acid were added to a clean dry three neck round-bottom flask. The above mixture was refluxed for 15 min at 65 °C under N2 atmosphere. 1S-(−)-Mercaptoisoborneol (100 mg; 5.8 mmol) and acetaldehyde dimethyl acetal (742 µL) dissolved in 5 mL of chloroform were added to the flask via a pressure adjusting funnel over a period of 20 min. Slowly the colorless solution turned brown and then dark brown. The mixture was further stirred for 30 min and washed with water (3 × 5 mL), brine (3 × 5 mL) and 10% KOH solution (3 × 5 mL) and dried over K2CO3. The compound was then purified using column chromatography (silica) with ethyl acetate/hexane as eluent and characterized by NMR. The NMR data are consistent with literature reports (yield: 67%).9,10,181H NMR (CDCl3, 400 MHz, δ): 0.76-1.90 (m, 7H), 0.87 (s, 3H), 1.27 (s, 3H), 1.40 (d, 3H, J = 6.4 Hz), 2.64 (d, 1H, J = 14.4 Hz), 3.03 (d, 1H, J = 14.4 Hz), 3.55 (dd, 1H, J = 3.2, 4.8 Hz), 4.70 (q, 1H, J = 6.0 Hz). 13C NMR (CDCl3, 100 MHz, δ): 20.7 (s), 21.9 (s), 23.4 (s), 27.4 (s), 28.9 (s), 34.7 (s), 38.0 (s), 41.9 (s), 45.7 (s), 46.8 (s), 77.8 (s), 85.4 (s). Specific rotation (C 0.1 in CHCl3): [α]20D: −135°.
Synthesis of phenyldiazomethane (ref. 19). Benzaldehyde tosylhydrazone (5 g in 50 mL of toluene) was added slowly to a solution containing 0.5 g of benzyl triethyl ammonium chloride in 50 mL of 14 wt.% aqueous NaOH. The reaction was warmed and held at 65–70 °C for 2 h with stirring. After cooling, the organic layer was separated and dried over sodium sulfate. Using p-toluic acid as the calibrant, phenyldiazomethane content in the above solution was estimated by 1H NMR.
Synthesis of 1-acetyl-2R,3R-diphenylcyclopropane (trans-7-RR) (ref. 9). A mixture of Rh2(OAc)4 (1 mole%), oxithiane catalyst (1.0 mmol) and trans-4-phenyl-but-3-ene-2-one (1.0 mmol) were stirred in 5 mL of toluene under nitrogen atmosphere at room temperature. Phenyldiazomethane (1.3 mmol) was added via a pressure-adjusting funnel over a period of 18 h and the reaction was further stirred for an hour. The crude reaction mixture was concentrated and purified by column chromatography and characterized by GC (yield: 25%). Formation of 1-acetyl-2,3-diphenylcyclopropane was confirmed by NMR which matched the literature report. The retention time in HPLC (column OD) matched with the elution order reported in the literature. 1H NMR (CDCl3, 400 MHz, δ): 2.12 (s, 3H), 2.72 (dd, 1H, J = 5.6, 9.6 Hz), 3.08 (dd, 1H, J = 9.6, 7.6 Hz), 3.32 (dd, 1H, J = 5.6, 7.6 Hz), 7.15-7.40 (m, 10H). 1H–1H COSY (CDCl3, 400 MHz, δ): Cross-peaks between δ (a) 2.72 and 3.08; (b) 2.72 and 3.32; (c) 3.08 and 3.32. 13C NMR (CDCl3, 100 MHz, δ): 29.9 (s), 31.6 (s), 37.1 (s), 39.8 (s), 126.6 (s), 226.7 (s), 127.0 (s), 128.2 (s), 128.6 (s), 129.1 (s), 135.5 (s), 139.8 (s) 203.0 (s).
Synthesis of 2R,3R-diphenylcyclopropane-1-carboxamide 2 and 4 (trans-2-RR and trans-4-RR) (ref. 11, 12). 1-Acetyl-2R,3R-diphenylcyclopropane trans-7-RR was dispersed in a solution of 10 mL of potassium iodide (10% solution), 3 mL of 1 M sodium hydroxide and 3 mL of sodium hypochlorite. The mixture was warmed to 70 °C and stirred for 30 min. Precipitation of the yellow solid (iodoform) confirmed the formation of oxidized product. The above mixture was brought to room temperature and 5 mL of chloroform was added to dissolve the iodoform. The aqueous layer was separated and acidified with 10% HCl. The 2R,3R-diphenylcyclopropane-1-carboxylic acid trans-8-RR was extracted using dichloromethane (5 × 5 mL) and dried over sodium sulfate. trans-2-RR and trans-4-RR amides were synthesized from 2R,3Rdiphenylcyclopropane- 1-carboxylic acid trans-8-RR and chiral amine (S(−)-1-phenylethyl for trans-2-RR; S(−)-1-naphthylethyl amine for trans-4-RR) by ETC/DMAP coupling described before.
Identification of the absolute configurations of trans-2-RR and trans-4-RR isomers. The retention time of the trans-2-RR and trans-4-RR isomers were compared with that of the injection done with the racemic trans-2 and racemic trans-4, respectively. The peak whose retention time matched with authentic sample was assigned the RR isomer and the unmatched peak was assigned the trans-SS isomer.
Isolation of RR and SS isomers of trans- 2 and 4. Pure optical isomers were collected by preparative HPLC using racemic trans-2 (Chiralcel OD) and racemic trans-4 (chiralcel OJ-R) and the authenticity of the absolute configurations at the two chiral centers was confirmed by comparing the HPLC peaks with authentic samples prepared above. Thus isolated pure optical isomers were used for photochemical studies.
(iv) General procedure for photochemical studies.
Solution: direct irradiation pure isomers of trans-2 and trans-4. The pure stereoisomer (RR or SS isomer) of the amides of 2,3-diphenylcyclopropane-1-carboxylic acid was dissolved in 0.5 mL of dichloromethane in a quartz test-tube (Pyrex in the case of trans-4) followed by the addition of 15 mL of hexane. The testtube was stoppered with a rubber septum and wound with parafilm. The solution was then irradiated (unfiltered output from a 450 W medium pressure mercury lamp) for different time intervals, concentrated and analyzed by HPLC.
Zeolite: Direct irradiation pure isomer of trans-2. The pure stereoisomer (RR or SS isomer) of the amides of 2,3-diphenylcyclopropane-1-carboxylic acid (2–3 mg) was dissolved in 0.5 mL of dichloromethane in a quartz test-tube. 15 mL of hexane was added to the quartz test-tube and stirred. MY (M = Li+, Na+, K+, Rb+ and Cs+) zeolite (≈300 mg) activated at 500 °C was added with stirring. The quartz test-tube was stoppered with rubber septum and wound with parafilm and stirred for 8 h. The hexane slurry was irradiated (unfiltered output from a medium pressure of Hg lamp) and filtered. Analysis of the hexane supernatant after irradiation did not show the presence of reactant/photoproducts. The reactant and the photoproducts were extracted from the zeolite by stirring with acetonitrile for 8–10 h. The acetonitrile extract was concentrated and analyzed on HPLC.
Solution–triplet (acetone) sensitization of pure isomer of trans-2. The pure stereoisomer (RR or SS isomer) of trans-2 was dissolved in 12 mL of acetone in a test-tube, stoppered with a rubber septum and wound with parafilm. The solution was degassed with N2 for 30 min and irradiated (unfiltered output from a 450 W medium pressure mercury lamp) for different time intervals. The solution was concentrated and analyzed by HPLC.
Zeolite–triplet sensitization of the pure isomer of trans-2. The pure stereoisomer (RR or SS isomer) of trans-2 (2-3 mg) and the triplet sensitizer (4′-methoxyacetophenone; 15-22 mg) were dissolved in 0.5 mL of dichloromethane in a test-tube. 15 mL of hexane was added to the test-tube and stirred. MY (M = Li+, Na+, K+, Rb+ and Cs+) zeolite (≈300 mg) activated at 500 °C was added with stirring. The test-tube was stoppered with a rubber septum and wound with parafilm and stirred for 12 h. The zeolite slurry was then degassed with N2 for 30 min and then irradiated (unfiltered output from a 450 W medium pressure mercury lamp), filtered and washed again with fresh hexane. The supernatant was analyzed for the presence of reactant and photoproducts (reactant / photoproducts were not observed). The reactant and the photoproducts were extracted from the zeolite by stirring with acetonitrile. The extract was concentrated and analyzed by HPLC.
(v) Analysis conditions.
(S)-(−)-1-Phenylethyl amide of 2,3-diphenylcyclopropane-1-carboxylic acid (cis-1 and trans-2). GC: Shimadzu 17, a gas chromatograph; column: SE-30; injector port: 250 °C; detector: 250 °C; 250 °C; program: 100 °C 1 min, 7 °C min−1, 240 °C 10 min, 1 °C min−1, 270 °C 1 min; chart speed 5; retention times: cis-1: ≈44.9 min; trans-2: ≈46.1 and 47.8 min. HPLC (chiral stationary phase): Rainin HPLC; XPXL pump; column AD-RH; eluent hexane–2-propanol 95 ∶ 5; monitoring wavelength, 230 nm; flow 0.7 mL min−1; retention times: cis-1: 24.7 min; trans-2-SS isomer: 34.1 min; trans-2-RR isomer: 74.0 min.
(S)-(−)-1-Naphthylethylamide of 2,3-diphenylcyclopropane-1-carboxylic acid (cis-3 and trans-4). HPLC conditions (chiral stationary phase): Rainin HPLC; XPXL pump; column AD-RH; eluent hexane–2-propanol 95 ∶ 5; monitoring wavelength, 282 nm; flow 0.7 mL min−1; retention times: cis-3: ≈39.7 min; trans-4-RR isomer: ≈49.3 min; trans-4-SS isomer: ≈61.1 min.
Acknowledgements
VR thanks the National Science Foundation for support of the research (CHE-9904187 and CHE-0212042).References
- (a) G. S. Hammond and R. S. Cole, Asymmetric induction during energy transfer, J. Am. Chem. Soc., 1965, 87(14), 3256–3257 CrossRef CAS; (b) S.L. Murov, R. S. Cole and G. S. Hammond, Mechanisms of photochemical reactions in solution. LIV. A new mechanism of photosensitization, J. Am. Chem. Soc., 1968, 90(11), 2957–2958 CrossRef CAS.
- S. S. Hixson, J. Boyer and C. Gallucci, Arylcyclopropane photochemistry. Naphthalene-sensitized isomerization of 1,2- diarylcyclopropanes, J. Chem. Soc., Chem. Commun., 1974, 540–542 RSC.
- (a) J. Sivaguru, A. Natarajan, L. S. Kannumalle, J. Shailaja, S. Uppili, A. Joy and V. Ramamurthy, Asymmetric photoreactions within zeolites: role of confinement and alkali metal ions, Acc. Chem. Res., 2003, 36(7), 509–521 CrossRef CAS; (b) J. Sivaguru, J. Shailaja, S. Uppili, K. Ponchot, A. Joy, N. Arunkumar and V. Ramamurthy, Achieving Enantio and Diastereoselectivities in Photoreactions through the Use of a Confined Space, in Organic Solid State Reactions, ed. F. Toda; Kluwer Academic Press, Dordrecht, The Netherlands, 2002, pp. 159–188; Search PubMed; (c) A. Joy and V. Ramamurthy, Chiral Photochemistry within Zeolites, Chem. Eur. J., 2000, 6, 1287–1293 CrossRef CAS.
- E. W. Valyocsik and P. J. Sigal, Photoisomerization and related processes in 1,2-diphenylcyclopropane, J. Org. Chem., 1971, 36(1), 66–72 CrossRef.
- G. W. Griffin, J. Covell, R. C. Petterson, R. M. Dodson and G. Klose, Photoisomerization of cyclopropane derivatives. Photointerconversion of propenes and cyclopropanes, J. Am. Chem. Soc., 1965, 87(6), 1410–1411 CrossRef CAS.
- (a) H. E. Zimmerman and T. W. Flechtner, Excited-state three-ring bond opening in cyclopropyl ketones. Mechanistic organic photochemistry. LX, J. Am. Chem. Soc.,, 1970, 92(23), 6931–6935 CrossRef CAS; (b) H. E. Zimmerman, S. S. Hixson and E. F. McBride, Excited state bond weakening in photochemical rearrangements of cyclopropyl ketones. Exploratory and mechanistic organic photochemistry. XLVIII, J. Am. Chem. Soc.,, 1970, 92(7), 2000–2014 CrossRef CAS; (c) H. E. Zimmerman and C. M. Moore, Excited state electron distribution in cyclopropyl ketone rearrangements. Mechanistic and exploratory organic photochemistry. LI, J. Am. Chem. Soc., 1970, 92(7), 2023–2031 CrossRef CAS.
- (a) J. Sivaguru, J. R. Scheffer, J. Chandarasekhar and V. Ramamurthy, Confined space and cations enhance the power of a chiral auxiliary: photochemistry of 1,2-diphenylcyclopropane derivatives, Chem. Commun., 2002, 830–831 RSC; (b) L. S. Kaanumalle, J. Sivaguru, N. Arunkumar, S. Karthikeyan and V. Ramamurthy, Cation–π interactions as a tool to enhance the power of a chiral auxiliary during asymmetric photoreactions within zeolites, Chem. Commun., 2003, 116–117 RSC; (c) S. Uppili and V. Ramamurthy, Enhanced Enantio- and Diastereoselectivities via Confinement: Photorearrangement of 2,4-Cyclohexadienones Included in Zeolites, Org. Lett., 2002, 4(1), 87–90 CrossRef CAS; (d) E. Cheung, K. C. W. Chong, S. Jayaraman, V. Ramamurthy, J. R. Scheffer and J. Trotter, Enantio- and Diastereodifferentiating cis,trans-Photoisomerization of 2β,3β-Diphenylcyclopropane-1α-carboxylic Acid Derivatives in Organized Media, Org. Lett., 2000, 2(18), 2801–2804 CrossRef CAS; (e) L. S. Kaanumalle, J. Sivaguru, R. B. Sunoj, P. Lakshminarasimhan, J. Chandrasekhar and V. Ramamurthy, Light-Induced Geometric Isomerization of 1,2-Diphenylcyclopropanes Included within Y Zeolites: Role of Cation–Guest Binding, J. Org. Chem., 2002, 67(25), 8711–8720 CrossRef CAS; (f) J. Sivaguru, T. Shichi and V. Ramamurthy, Reactive-State Spin-Dependent Diastereoselective Photoisomerization of trans,trans-2,3-Diphenylcyclopropane-1-carboxylic Acid Derivatives Included in Zeolites, Org. Lett., 2002, 4(24), 4221–4224 CrossRef CAS.
- J. Sivaguru, R. B. Sunoj, T. Wada, Y. Origane, Y. Inoue and V. Ramamurthy, Enhanced Diastereoselectivity via Confinement: Diastereoselective Photoisomerization of 2,3-Diphenyl-1-benzoylcyclopropane Derivatives within Zeolites, J. Org. Chem., 2004, 69, 5528–5536 CrossRef CAS.
- V. K. Aggarwal, H. W. Smith, G. Hynd, R. V. H. Jones, R. Fieldhouse and S. E. Spey, Catalytic cyclopropanation of electrondeficient alkenes mediated by chiral and achiral sulfides: scope and limitations in reactions involving phenyldiazomethane and ethyl diazoacetate, J. Chem. Soc., Perkin Trans., 2000, 1, 3267–3276 Search PubMed.
- V. K. Aggarwal, A. Thompson, R. V. H. Jones and M. C. H. Standen, Novel Catalytic and Asymmetric Process for Aziridination Mediated by Sulfur Ylides, J. Org. Chem., 1996, 61, 8368–8369 CrossRef CAS.
- (a) R. C. Fuson and B. A. Bull, The Haloform Reaction, Chem. Rev., 1934, 15, 275 CrossRef CAS; (b) H. O. House, in Modern Synthetic Reactions, W. A. Benjamin, Menlo Park, CA, 2nd edn., 1972, pp. 464–465 Search PubMed; (c) J. March, in Advanced Organic Chemistry, Wiley-Interscience, New York, 4th edn., 1992, p. 632 Search PubMed; (d) VOGEL’s Textbook of Practical Organic Chemistry, rev. B. S. Furniss, A. J. Hannaford, V. Rogers, P. W. G. Smith and A. R. Tatchell, Longman Scientific & Technical, John Wiley & Sons, Chichester, 1978 Search PubMed.
- (a) A. Hassner and V. Alexanian, Direct room temperature esterification of carboxylic acids, Tetrahedron Lett., 1978, 46, 4475–4478 CrossRef; (b) J. Kress, A. Rosner and A. Hirsch, Depsipeptide Dendrimers, Chem Eur J., 2000, 6(2), 247–257 CrossRef CAS; (c) T. S. M. Taha and S. Ferguson-Miller, Interaction of cytochrome c with cytochrome c oxidase studied by monoclonal antibodies and a protein modifying reagent, Biochemistry, 1992, 31, 9090–9097 CrossRef CAS.
- J. Sivaguru, R. B. Sunoj, T. Wada, Y. Origane, Y. Inoue and V. Ramamurthy, Enhanced Diastereoselectivity via Confinement: Diastereoselective Photoisomerization of 2,3-Diphenylcyclopropane-1-carboxylic Acid Derivatives within Zeolites, J. Org. Chem., 2004, 69(20), 6533–6547 CrossRef CAS.
- (a) S. S. Hixson, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1979, vol. 4, pp. 191–260 and selected references therein Search PubMed; (b) S. S. Hixson and D. W. Garrett, Arylcyclopropane photochemistry. Photochemical addition of hydroxylic compounds to 1,2-diarylcyclopropanes, J. Am. Chem. Soc., 1974, 96, 4872–4879 CrossRef CAS; (c) S. S. Hixson, Arylcyclopropane photochemistry. Photochemical addition of amines to 1,2-diarylcyclopropanes, J. Am. Chem. Soc., 1974, 96, 4866–4871 CrossRef CAS.
- S. Uppili, K. J. Thomas, E. M. Crompton and V. Ramamurthy, Probing Zeolites with Organic Molecules: Supercages of X and Y Zeolites Are Superpolar, Langmuir, 2000, 16(1), 265–274 CrossRef CAS.
- J. Sivaguru, S. Jockusch, N. J. Turro and V. Ramamurthy, Photoisomerization of 2,3-diphenylcyclopropane-1-carboxylic acid derivatives, Photochem. Photobiol. Sci., 2003, 2, 1101–1106 RSC.
- (a) J. K. Blatchford and M. Orchin, The Synthesis of Some 2,3-Diarylcyclopropane-1-carboxylic Acids, J. Org. Chem., 1964, 29, 839–843 CAS; (b) I. A. D’Yakanov, M. I. Komendantev, F. -S. Gui and G. L. Korichev, “About: Reaction of aliphatic diazo compounds with unsaturated compounds”. Catalytic condensation from ethyl diazoacetate with cis- and trans-stilbene. Synthesis of new derivatives of cyclopropanes, J. Gen. Chem. USSR, 1962, 32, 928; (c) D. E. Applequist and R. D. Gdanski, Kinetic study of the homolytic brominolysis of 1,2-diarylcyclopropanes, J. Org. Chem., 1981, 46, 2502–2510 CrossRef CAS; (d) Y. Inoue, N. Yamasaki, H. Shimoyama and A. Tai, Enantiodifferentiating cis–trans photoisomerizations of 1,2-diarylcyclopropanes and 2,3-diphenyloxirane sensitized by chiral aromatic esters, J. Org. Chem., 1993, 58, 1785–1793 CrossRef CAS.
- (a) V. K. Aggarwal, J. G. Ford, A. Thompson, R. V. H. Jones and M. C. H. Standen, Direct Asymmetric Epoxidation of Aldehydes Using Catalytic Amounts of Enantiomerically Pure Sulfides, J. Am. Chem. Soc., 1996, 118, 7004–7005 CrossRef CAS; (b) V. K. Aggarwal, A. Thompson, R. V. H. Jones and M. C. H. Standen, The use of chiral sulfides in catalytic asymmetric epoxidation, Tetrahedron: Asymmetry, 1995, 6(10), 2557–2564 CrossRef CAS.
- (a) D. S. Wulfman, S. Yousefian and J. M. White, The Synthesis of aryldiazomethanes, Synth. Commun., 1988, 18(18), 2349–2352 CAS; (b) X. Creary, Tosylhydrazone Salt Pyrolyses: Phenyldiazomethanes, Org. Synth., 1986, 64, 207–216 CAS; (c) J. I. Finneman and J. C. Fishbein, Mechanisms of Benzyl Group Transfer in the Decay of (E)-Arylmethanediazoates and Aryldiazomethanes in Aqueous Solutions, J. Am. Chem. Soc., 1995, 117, 4228–423 CrossRef CAS.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |