A spectroscopic and photophysical study on molecular recognition via hydrogen-bonding and π–π stacking interactions†
Noboru
Kitamura
*,
Yusuke
Suzuki
and
Shoji
Ishizaka
Division of Chemistry, Graduate School of Science, Hokkaido University, Sapporo, Japan 060-0810. E-mail: kitamura@sci.hokudai.ac.jp
First published on 16th September 2004
Abstract
Spectroscopic and photophysical properties of a Kemp's tricarboxylic acid derivative having an anthracene chromophore (I) upon recognition of 9-butyladenine (BA) in chloroform were studied in detail. Molecular recognition of BA by Ivia hydrogen-bonding and π–π stacking interactions were sensed successfully on the basis of absorption and fluorescence spectroscopies, by which the binding constant of the I:BA complex was determined to be 240 M−1. The fluorescence quantum yield and lifetime of I in the absence of BA were 0.24 and 5.6 ns, respectively, while those in the presence of an enough amount of BA increased to 0.35 and 13 ns, respectively. These values demonstrated that the nonradiative decay rate constant of I decreased from 13.6 × 107 to 5.0 × 107 s−1 upon binding with BA. Such changes in the photophysical properties of I before and after complexation with BA were discussed in terms of hydrogen-bonding and π–π stacking interactions between I and BA. In particular, intramolecular hydrogen-bonding between the amide and imide groups in I was shown to play important roles in determining the photophysical characteristics of I before complexation, while intermolecular hydrogen-bonding between I and BA governed the excited-state properties of the I:BA complex. The change in the hydrodynamic diameter of I before and after complexation with BA was also discussed on the basis of the results by fluorescence dynamic anisotropy measurements.
1 Introduction
Selective recognition of a molecule or ion is one of the important issues in various sciences: supramolecular chemistry, analytical science, biochemistry, protein chemistry, and so on.1–3 In particular, molecular recognition via hydrogen (H)-bonding is the fundamental bases for base pairing in DNA and also plays essential roles in various biological and artificial systems.4,5 For double-helix formation in DNA, furthermore, it is worth noting that the π–π stacking interaction between nucleic acids operates cooperatively with H-bonding, which is very characteristic to molecular recognition in biological systems. On the other hand, an introduction of such H-bonding and π–π stacking modes to molecular recognition in a synthetic system needs structural and functional auxiliaries in host and/or guest molecules.5 In practice, various synthetic approaches have been explored to mimic biological functions.1–5 One such example has been reported by Hamilton and Engen for recognition of thymine or uracyl by a macrocyclic receptor molecule, composed of a naphthyl group for π–π stacking and a diaminopyridine group for H-bonding: R in Scheme 1.6 Another class of the system has been demonstrated by Rebek and his co-workers.7–12 They introduced a unique molecule derived from a Kemp's tricarboxylic acid,13 in which the triaxial conformation of three carboxyl groups forces a U-shaped relationship between any two of them: Scheme 2. A typical example of the noncyclic receptor reported by Rebek and co-workers is I shown in Scheme 1,11,12 which is composed of an imide moiety for H-bonding and an aromatic π chromophore (i.e., anthryl group in I) for π–π stacking upon recognition of a guest molecule. The resulting conformation upon recognition via H-bonding features the parallel surface between the aromatic π chromophore and a guest molecule. For both R and I, the π–π and H-bonding interactions upon recognition of a guest have been demonstrated by 1H NMR measurements.11,12 Although these systems or molecules are certainly very interesting as a model for molecular recognition in biological systems, a detailed study on the systems other than 1H NMR has not been reported. Since both H-bonding and π–π stacking interactions upon molecular recognition will reflect on the electronic structures of host and/or guest molecules, spectroscopic and photophysical studies on such systems are worth exploring in detail.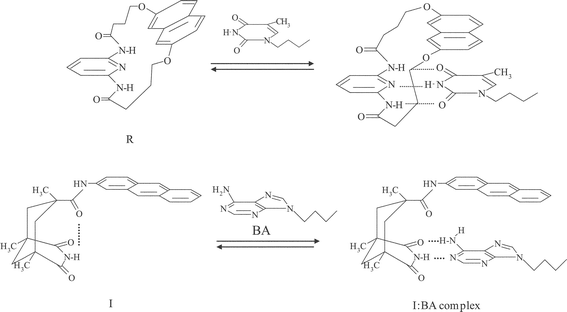 | ||
| Scheme 1 Hydrogen-bonding and π–π stacking interactions in host–guest complexes. | ||
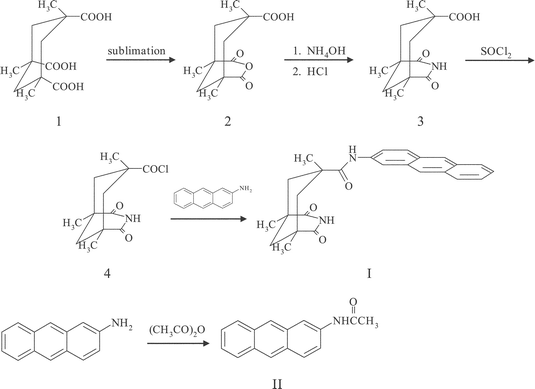 | ||
| Scheme 2 Synthetic routes to I and II. | ||
In the case of R, we showed recently direct evidence for the π–π interaction between a thymine derivative and the naphthyl group in R on the basis of absorption and steady-state/time-resolved fluorescence spectroscopies.14 Upon recognition of the guest, R showed new exciplex-like fluorescence and, the electronic interactions between R and a thymine derivative were shown to have an effect on the fluorescence dynamics of the host–guest complex. Excited-species involved in a system can be discriminated through the fluorescence decay time constant of each component (τi) and, in principle, quantitative discussion on the observed phenomena can be made on the basis of the amplitude value (Ai) of each τi component. Therefore, spectroscopic and photophysical studies on the systems shown in Scheme 1 are very promising to elucidate roles of π–π stacking and H-bonding interactions in molecular recognition.
We report in this paper spectroscopic and photophysical characteristics of the noncyclic receptor, I, before and after complexation with 9-butyladenine (BA) in chloroform. It is worth noting that, although molecular recognition of BA by I or analogous receptors possessing various aryl groups has been reported by Rebek and his co-workers,7–12 its information is limited to that by 1H and 13C NMR measurements as described above. On the basis of the photophysical data, including rotational relaxation times of I and the I:BA complex, we discuss the mechanisms and characteristics of molecular recognition in the I–BA system.
2 Experimental
2.1 Preparations of host and guest molecules
A Kemp's triacid derivative having an anthracene group (I) and 9-butyladenine (BA) were prepared according to the literatures with some modifications: Scheme 2.11,15,16 An anthracene derivative, II (2-acetaminoanthracene), was also prepared as a model compound for I without the Kemp's triacid structure.2.2 Spectroscopic measurements
1H NMR measurements were conducted by using a Varian Gemini-2000 (300 MHz) spectrometer. Steady-state absorption spectroscopy was conducted by using a Hitachi UV-3300 spectrophotometer and corrected fluorescence spectra were recorded on a Hitachi F-4500 spectrofluorometer. The fluorescence quantum yields (Φf) of I and II in chloroform were determined on the basis of that of 9,10-diphenylanthracene in cyclohexane as a standard (Φf = 0.91) after refractive index correction.17Fluorescence dynamic spectroscopy was conducted based on a time-correlated single photon-counting technique.18 Laser pulses (800 nm; repetition rate, 76 MHz; FWHM, 200 fs) from a mode-locked Ti:sapphire laser (Coherent, Mira 900-F), pumped by a diode laser (532 nm, 5W, Coherent, Verdi), were introduced to a pulse picker (Coherent, 9200) to modulate the repetition rate. The second harmonic of the laser pulses (400 nm, repetition rate; 3.8 MHz) was used as an excitation light source. By using a Glan-laser prism, the polarized direction of the excitation laser beam was set a vertical direction. A polarizer was set at the magic angle (54.7°) in front of a detector system, while it was arranged vertically or horizontally for fluorescence dynamic anisotropy measurements.19 A microchannel-plate photomultiplier tube (Hamamatsu Photonics, R3809U-50) equipped with a monochromator (Jobin Ybon, H-20) and a time-correlated single photon counting module (Edinburgh Instruments, SPC-300) was used as a detector. The fluorescence monitoring wavelength was set 430 nm throughout the study.
All of the experiments were conducted at ambient temperature (296 K) and sample solutions for fluorescence measurements were deaerated by purging with an Ar gas stream over 20 min.
3 Results and discussion
3.1 1H NMR study on hydrogen-bonding and aromatic π–π interactions upon molecular recognition
Before discussing spectroscopic and photophysical characteristics of I in the absence and presence of BA, we describe briefly the results of the 1H NMR experiments, which provide structural information on I upon complexation with BA. 1H NMR spectra of I in the absence and presence of BA in CDCl3 are shown in Fig. 1. In the absence of BA (Fig. 1a), the imide proton signal of I was observed at 7.6 ppm (shown by the arrow), while the amide proton signal appeared at around 7.3 ppm. In the presence of BA, on the other hand, large changes in the chemical shifts were observed as shown in Fig. 1b and c. The data showed clearly that the imide proton signal of I shifted gradually to the downfield side with an increase in the BA concentration (δ = 10.6 (Fig. 1b) or ≈13 ppm (c) at [BA] = 1.0 × 10−2 or 0.1 M, respectively), while the amide proton signal did not show any appreciable shift. The amino proton signal of BA observed at δ = 5.6 ppm in the absence of I (not shown in Fig. 1a) also shifted to δ = 5.9 (b) ≈6.1 ppm (c) in the presence of I. These results demonstrated explicitly intermolecular hydrogen (H)-bond formation between I and BA as shown in Scheme 1. Slight upfield shifts were also observed for the aromatic proton signals (δ = 7.9–8.3 ppm). The aromatic resonances show generally ≈0.3 ppm upfield shifts when aromatic rings approach in close contact. Therefore, these NMR spectral changes in Fig. 1 demonstrate that both H-bonding and π–π stacking interactions between I and BA take place simultaneously, which is in good accordance with the report by Askew et al.11 Although the data are not shown here, furthermore, we conducted 1H NMR titration experiments to evaluate the binding constant of the I:BA complex: Ka. The Ka value thus determined by the [BA] dependence of the chemical shift of the imide proton was 200 M−1 (discussed later again).![1H NMR spectra of I (5.0 × 10−3 M) in the absence (a) and presence of BA [1.0 × 10−2 (b), and 0.1 M (c)] in CDCl3.](/image/article/2005/PP/b409932j/b409932j-f1.gif) | ||
| Fig. 1 1H NMR spectra of I (5.0 × 10−3 M) in the absence (a) and presence of BA [1.0 × 10−2 (b), and 0.1 M (c)] in CDCl3. | ||
3.2 Absorption and fluorescence spectral changes upon molecular recognition
The absorption and fluorescence spectra of I and II in chloroform (1.0 × 10−4 M) are shown in Fig. 2. The Kemp's triacid derivative, I, exhibited structured absorption in the wavelength (λ) region of 300–420 nm, with the 0–0 absorption band position (λa) being 390 nm. Since II as a reference compound for I without a Kemp's triacid chromophore showed analogous spectrum with that of I except for the slight red shift of the spectrum (λa = 392 nm), the absorption spectrum observed for I or II in this wavelength region was assigned confidently to the π–π* transition of the anthryl group in the compound. However, the absorption spectrum of I or II was more structured as compared to that of anthracene itself,20 suggesting contributions of the vibrational modes of the amide group in I or II to the spectrum. Furthermore, the vibrational bands of I were broader than those of II. We suppose that intramolecular H-bond formation between the amide and imide groups could be the primary reason for the broad spectrum of I, since other data described later also support participation of intramolecular H-bonding in I. It is worth noting that the amide proton signal of I (7.3 ppm without BA) shows no appreciable shift even in the presence of BA (Fig. 1). Therefore, we conclude that the amide group in I acts as a hydrogen acceptor for the imide group as illustrated in Scheme 1.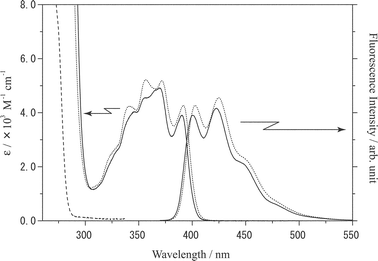 | ||
| Fig. 2 Absorption and fluorescence spectra of I (1.0 × 10−4 M, solid lines) and II (1.0 × 10−4 M, dotted lines) in chloroform. The absorption spectrum of BA is shown by the broken line. The excitation wavelength was 400 nm. | ||
On the other hand, both I and II exhibited structured fluorescence in λ = 400–520 nm, which was ascribed to the monomer excited-state of the anthryl group in I or II. The fluorescence band shapes of I and II were very similar with each other, while the spectrum of II shifted slightly to the longer wavelength as compared to that of I; the 0–0 fluorescence band positions (λf) of I and II were 402 and 406 nm, respectively (Table 1).
Absorption and fluorescence spectral responses of I (1.0 × 10−4 M in CHCl3) upon an addition of BA ((1.0–50) × 10−3 M) are shown in Fig. 3. An addition of BA resulted in a decrease in the absorbance at 390 nm and, this accompanied an appearance of a new absorption band at 394 nm and a decrease in the absorption intensity at λ < 370 nm. Clear isosbestic points were also observed at 373, 380 and 392 nm. It is worth noting that BA shows no absorption above 300 nm as the spectrum is included in Fig. 2. Furthermore, II did not exhibit any absorption spectral change even in the presence of BA up to 5.0 × 10−2 M (data are not shown here). Therefore, the absorption spectral changes of I are essentially due to intermolecular H-bond formation between I and BA, and probably to simultaneous π–π stacking between BA and the anthryl group in I as illustrated in Scheme 1. In the present experiments, there is no direct evidence for double H-bonds formation in the I:BA complex. Nonetheless, the thymine-type structure in the Kemp's triacid (i.e., imide moiety) will favor double H-bonds formation with BA, as in the case for the thymine–adenine base pair in DNA. On the basis of 1H NMR studies, in practice, Askew et al. reported that both Watson–Crick and Hoogsteen type H-bonding participated in analogous aryl substituted Kemp's triacid derivative:BA complexes.11 Therefore, we think that complex formation between I and BA takes place via double H-bonding.
![Absorption and fluorescence spectral responses of I (1.0 × 10−4 M) upon an addition of BA in chloroform: [BA] = 0 (a), 1.0 × 10−4 (b), 1.0 × 10−3 (c), 2.5 × 10−3 (d), 5.0 × 10−3 (e), 1.0 × 10−2 (f), 2.5 × 10−2 (g), and 5.0 × 10−2 M (h). The excitation wavelength was 400 nm.](/image/article/2005/PP/b409932j/b409932j-f3.gif) | ||
Fig. 3 Absorption and fluorescence spectral responses of I (1.0 × 10−4 M) upon an addition of BA in chloroform: [BA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =0 (a), 1.0 × 10−4 (b), 1.0 × 10−3 (c), 2.5 × 10−3 (d), 5.0 × 10−3 (e), 1.0 × 10−2 (f), 2.5 × 10−2 (g), and 5.0 × 10−2 M (h). The excitation wavelength was 400 nm. =0 (a), 1.0 × 10−4 (b), 1.0 × 10−3 (c), 2.5 × 10−3 (d), 5.0 × 10−3 (e), 1.0 × 10−2 (f), 2.5 × 10−2 (g), and 5.0 × 10−2 M (h). The excitation wavelength was 400 nm. | ||
It is worth pointing out, furthermore, that the vibrational structures of the absorption spectrum of I become more resolved with an increase in [BA] and, the spectrum in the presence of an enough amount of BA (5 × 10−2 M) resembles with that of II in Fig. 2. When intramolecular H-bonding between the amide and imide groups participates in I as discussed above, complexation between I and BAvia intermolecular H-bonding should break intramolecular H-bonding in I. If this is the case, the absorption spectrum of I in the presence of BA should be similar to that of II without an intramolecular H-bonding ability. Therefore, the spectral changes observed for I in Fig. 3 would support participation of intramolecular H-bonding in the absence of BA.
Fig. 3 also demonstrated that an addition of BA brought about an increase in the fluorescence intensity of I, and this accompanied a slight red shift of the spectrum: shift from λf = 402 to 406 nm ([BA] = 0.1 M). In the case of the macrocyclic receptor, R in Scheme 1, recognition of a thymine derivative by R brought about an appearance of exciplex-like fluorescence in addition to structured monomer fluorescence of the naphthyl group as mentioned before, and this proved directly π–π stacking between the two aromatic chromophores.14 However, such clear evidence for π–π stacking was not observed in the present I–BA system, while the 1H NMR spectra in Fig. 1 indicated participation of the π–π stacking interaction: discussed later again. On the other hand, the fluorescence quantum yield (Φf) of I increased appreciably from 0.24 in the absence of BA to 0.35 at [BA] = 0.1 M,21 while that of II was constant at 0.34–0.35 irrespective of the absence and presence of BA (up to 0.1 M, see Table 1). Therefore, molecular recognition of BA by Ivia H-bonding can be thus sensed as the fluorescence intensity change. Both absorption and fluorescence spectroscopies demonstrated that the presence of the imide moiety of the Kemp's triacid chromophore in I was essential to recognize BA in the present system.
3.3 Determination of the binding constant
The observation of the isosbestic points in Fig. 3 proves stoichiometric binding of BA by I. For quantitative discussion, therefore, we evaluated the binding constant of the I:BA complex (Ka) on the basis of the spectroscopic data in Fig. 3. In principle, Ka can be determined by the [BA] dependence of the absorption or fluorescence spectrum of I. In the present study, we determined Ka based on the fluorescence intensity change of I with [BA], since the relevant absorbance change was rather small. In the case of 1 ∶ 1 complex formation, the change in the fluorescence intensity of I (ΔI) as a function of [BA] can be analyzed by the following equation,22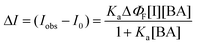 | (1) |
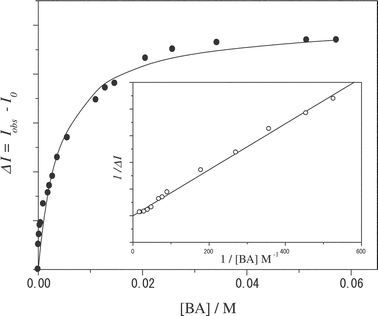 | ||
| Fig. 4 A relationship between ΔI (determined for I at 430 nm) and the BA concentration in chloroform. The solid curve represents the best fit of the data by eqn. (1). The inset represents the double-reciprocal plot of the data. | ||
3.4 Fluorescence dynamics
Fluorescence dynamics provide more detailed information about the characteristics of I:BA complexation. In the case of steady-state fluorescence spectroscopy, fluorescence from both I and the I:BA complex contributes to the observed spectrum, and spectral separation of the two species is in general very difficult. On the other hand, the excited-states of I and the I:BA complex can be discriminated through their fluorescence lifetimes (τf) when the values differ one another. This is worth exploring to elucidate the characteristics of the present system. Furthermore, the larger Φf value of the I:BA complex (0.35) as compared to that of I (0.24) indicates the change in the electronic structure of I upon host–guest complexation. In order to discuss such aspects, we studied fluorescence dynamics of I and II in chloroform.A typical example of the fluorescence dynamics of I in the absence and presence of BA is shown in Fig. 5. The fluorescence decay profiles of I and II in the absence of BA were fitted satisfactorily by single exponential functions and, the fluorescence lifetimes of I and II in chloroform were determined to be 5.6 and 8.0 ns, respectively (Table 2). Knowing the τf and Φf values, we calculated the radiative (kr) and nonradiative decay rate constants (knr) of I and II on the basis of the relation, Φf = kr/(kr + knr) = krτf. The parameters thus obtained are included in Table 1. Table 1 indicates that, since the kr values of I and II are comparable with each other ((4.3–4.4) × 107 s−1), the shorter τf and smaller Φf values of I as compared to those of II are ascribable to the variation of the nonradiative decay rate constant between the two compounds. In practice, the knr value of I (13.6 × 107 s−1) was larger than that of II (8.1 × 107 s−1) by a factor of ≈1.7. We suppose that this will be due to intramolecular H-bonding between the amide and imide groups in I. As discussed in the previous section, participation of intramolecular H-bonding was demonstrated by the relatively broad absorption spectrum of I in the absence of BA and sharpening of the spectrum with the increase in [BA] (Fig. 3). Generally, intra- or intermolecular H-bonding of a fluorophore induces more or less radiationless deactivation of the excited-state.23 Therefore, the knr value of I becomes larger than that of II without an intramolecular H-bonding ability. The intramolecular H-bonding thus plays an important role in determining the electronic state of I before complexation and, this should reflect on the photophysical characteristics of I upon binding with BA.
| [BA]/103 M | τ 1(A1)/ns | τ 2(A2)/ns | χ 2 a | DWa | |
|---|---|---|---|---|---|
| a χ 2 and DW represent chi-squared and Durbin–Watson parameters for the fitting, respectively. | |||||
| I | 0 | 5.6 | 0.97 | 1.81 | |
| 0.2 | 5.5 (0.89) | 13.0 (0.11) | 1.02 | 1.98 | |
| 2.0 | 5.6 (0.58) | 12.6 (0.42) | 1.03 | 1.92 | |
| 10 | 5.9 (0.34) | 12.4 (0.66) | 1.03 | 1.92 | |
| 20 | 5.9 (0.23) | 12.6 (0.77) | 1.09 | 2.04 | |
| II | 0 | 8.0 | 1.04 | 1.96 | |
| 2.0 | 8.3 | 1.03 | 1.88 | ||
| 10 | 8.5 | 1.05 | 2.02 | ||
![Fluorescence decay profiles of I (2.4 × 10−4 M) in the absence and presence of BA in chloroform: [BA] = 0 (a), 2.0 × 10−4 (b), 2.0 × 10−3 (c), 1.0 × 10−2 (d), 5.0 × 10−2 M (e). The excitation and monitoring wavelengths were 400 and 430 nm, respectively.](/image/article/2005/PP/b409932j/b409932j-f5.gif) | ||
| Fig. 5 Fluorescence decay profiles of I (2.4 × 10−4 M) in the absence and presence of BA in chloroform: [BA]=0 (a), 2.0 × 10−4 (b), 2.0 × 10−3 (c), 1.0 × 10−2 (d), 5.0 × 10−2 M (e). The excitation and monitoring wavelengths were 400 and 430 nm, respectively. | ||
In the presence of BA, on the other hand, the fluorescence decay profiles of I were best fitted by double-exponential functions with τ1 ≈ 6 (5.5–6.3) and τ2 ≈ 13 ns (12.4–13.0), with their amplitudes (Ai, i = 1 and 2) being varied with [BA]. As the data were summarized in Table 2, the τ1 value agreed very well with that observed for I without BA (5.6 ns) within an experimental error. The I:BA complex is produced in the ground state and, I and the I:BA complex are excited independently by incident light (excitation wavelength = 400 nm). Furthermore, the contribution of the excited-state equilibrium between I and the complex to the observed data will be neglected. This indicates that the excited-states of both I and the I:BA complex should decay independently via single exponential functions. When I and the complex are present in the system, therefore, the total fluorescence decay should obey with a double-exponential function. These discussions indicate that the τ2 component observed in the presence of BA (≈13 ns) is assigned confidently to the excited-state lifetime of the I:BA complex. An increase in [BA] thus gives rise to that in the A2 value without the change in the τ value (see Table 2). Under analogous conditions, fluorescence from II in the presence of BA showed always a single exponential decay with the τ value being similar to that in the absence of BA, but became longer slightly from τf = 8.0 ([BA] = 0) to 8.5 ns ([BA] = 1.0 × 10−2 M, Table 2). Since II did not show any fluorescence spectral and intensity changes in the presence of BA, we concluded that there was no interaction between II and BA. The longer fluorescence lifetime of II in the presence of BA as compared to that without BA will be due to a slight increase in the viscosity of the medium by the presence of BA.
Table 1 also includes the kr and knr values of the I:BA complex, which were determined at the I:BA concentration ratio of ≈1 ∶ 200 (2.4 × 10−4 ∶ 5.0 × 10−2 M). At the concentration ratio, the fraction of the I:BA complex to the total mole number of I is >90% as estimated from the Ka value (240 M−1). Therefore, the kr and knr values estimated here would reflect those of the I:BA complex. The radiative rate constant of the I:BA complex (2.7 × 107 s−1) was slightly smaller than that of I (4.3 × 107 s−1), which might suggest the change in the electronic structure of the anthryl group in I through the π–π stacking interaction with BA, as demonstrated by the 1H NMR spectra in Fig. 1.
On the other hand, the knr values of I in the presence and absence of BA were 5.0 × 107 and 13.6 × 107 s−1, respectively, indicating that nonradiative decay of the excited-state of I was suppressed upon complexation with BA. Upon complexation, intramolecular H-bonding between the amide and imide groups in I is replaced by intermolecular H-bonding between I and BA as described above. Although H-bonding of the amide group attached directly to the anthryl group acts as a nonradiative decay channel of the excited-state of I, that between the imide moiety in I and BA does not influence the excited-state decay, since the imide moiety is not conjugated with the anthryl group. Therefore, formation of intermolecular H-bonding reduces the knr value of the I:BA complex. Furthermore, the π–π stacking interaction between BA and the anthryl group in I implies suppression of the rotational motions of the anthryl group around the Canthryl–Namide bond, which should decrease the radiationless deactivation efficiency in the excited-state of I. It is concluded, therefore, that the hydrogen-bonding and π–π stacking interactions between I and BA in the ground-state take place cooperatively, and this leads to the change in the H-bonding mode from the intra- to intermolecular mode as well as to suppression of the rotational motions of the anthryl group in I. As a result, the fluorescence intensity of I increases upon recognition of BA.
3.5 Fluorescence dynamic anisotropy
Further information about the molecular motions of I and the I:BA complex can be obtained through fluorescence dynamic anisotropy measurements. Fluorescence dynamic anisotropy (r(t)) is defined by the following equation,19,24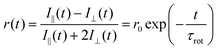 | (2) |
=0.514), ethanol (EtOH, η=1.078), and dimethyl sulfoxide (DMSO, η=1.996)a
| τ rot/ns | r 0 | χ 2 b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CHCl3 | EtOH | DMSO | CHCl3 | EtOH | DMSO | CHCl3 | EtOH | DMSO | |
| a The η values were taken from ref. 27. b Chi-squared parameter for the fitting. | |||||||||
| I | 0.06 | 0.12 | 0.24 | 0.27 | 0.26 | 0.26 | 1.00 | 1.49 | 1.36 |
| I:BA | 0.10 | 0.19 | 0.26 | 0.26 | 0.24 | 0.30 | 1.00 | 1.66 | 1.55 |
| II | 0.04 | 0.07 | 0.11 | 0.29 | 0.26 | 0.30 | 1.22 | 1.95 | 1.31 |
| II + BA | 0.03 | 0.06 | 0.11 | 0.28 | 0.27 | 0.30 | 1.48 | 1.17 | 1.21 |
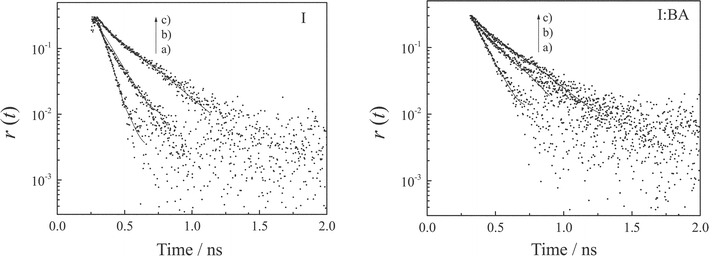 | ||
| Fig. 6 Fluorescence anisotropy decays of I (1.8 × 10−4 M) in the absence (left) and presence of BA (right, 2.0 × 10−2 M) in chloroform (a), ethanol (b), and dimethyl sulfoxide (c). The excitation and monitoring wavelengths were 400 and 430 nm, respectively. | ||
It is well known that the rotational correlation time of a molecule is related to its hydrodynamic volume (V) under the assumption of a spherical molecular shape: Debye–Stokes–Einstein (DSE) model.25 According to the DSE model, the rotational correlation time is given by the following equation,
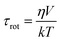 | (3) |
![Solvent viscosity dependences of the τrot values of I (1.8 × 10−4 M) in the absence (squares) and presence of BA (2.0 × 10−2 M, circles). The results observed for II (1.8 × 10−4 M) are also shown ([BA] = 2.0 × 10−2 M, triangles).](/image/article/2005/PP/b409932j/b409932j-f7.gif) | ||
| Fig. 7 Solvent viscosity dependences of the τrot values of I (1.8 × 10−4 M) in the absence (squares) and presence of BA (2.0 × 10−2 M, circles). The results observed for II (1.8 × 10−4 M) are also shown ([BA]=2.0 × 10−2 M, triangles). | ||
4 Conclusions
Recognition of the adenine derivative (BA) by the Kemp's triacid derivative having an anthracene chromophore (I) was elucidated on the basis of absorption and steady-state/time-resolved fluorescence spectroscopies. Hydrogen-bonding and π–π stacking interactions between I and BA in the ground-state took place cooperatively, and this gave rise to the enhancement of the fluorescence quantum yield and the decrease in the nonradiative decay rate constant of the anthryl group in I. In addition to intermolecular H-bonding between I and BA upon molecular recognition, the present photophysical study demonstrated the important role of intramolecular H-bonding between the amide and imide groups in I before complexation. Thus, intramolecular H-bonding in I was shown to be replaced by intermolecular H-bonding (Scheme 1), which governed the overall changes in the spectroscopic and photophysical characteristics of I before and after recognition. The photophysical tools are certainly powerful enough to elucidate the molecular mechanisms of recognition in the present system.References
- J.-M. Lehn, Supramolecular Chemistry, Concepts and Perspective, VCH, Weinheim, 1995 Search PubMed.
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Monographs in Supramolecular Chemistry, The Royal Society of Chemistry, Cambridge, 1997 Search PubMed.
- Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. Garcia-Espana, Wiley-VCH, New York, 1997 Search PubMed.
- For biological aspects, see a review article as an example: H. Gohlke and G. Klebe, Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors, Angew. Chem., Int. Ed., 2002, 41, 2644–2676 Search PubMed.
- For artificial systems, see this review article as an example: L. J. Prins, D. N. Reinhoudt and P. Timmerman, Noncovalent synthesis using hydrogen bonding, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 Search PubMed.
- A. D. Hamilton and D. V. Engen, Induced fit in synthetic receptors: nucleotide base recognition by a “molecular hinge”, J. Am. Chem. Soc., 1987, 109, 5035–5036 CrossRef CAS.
- J. Rebek, Jr, L. Marshall, R. Wolak, K. Parris, M. Killoran, B. Askew, D. Nemeth and N. Islam, Convergent functional groups: synthetic and structural studies, J. Am. Chem. Soc., 1985, 107, 7476–7481 CrossRef CAS.
- J. Rebek, Jr, B. Askew, M. Killoran, D. Nemeth and F.-T. Lin, Convergent functional groups. 3. A molecular cleft recognizes substrates of complementary size, shape, and functionality, J. Am. Chem. Soc., 1987, 109, 2426–2431 CrossRef.
- J. Rebek, Jr, B. Askew, D. Nemeth and K. Parris, Convergent functional groups. 4. Recognition and transport of amino acids across a liquid membrane, J. Am. Chem. Soc., 1987, 109, 2432–2434 CrossRef.
- J. Rebek, Jr, B. Askew, P. Ballester, C. Buhr, S. Jones, D. Nemeth and K. Williams, Molecular recognition: Hydrogen bonding and stacking interactions stabilize a model for nucleic acid structure, J. Am. Chem. Soc., 1987, 109, 5033–5035 CrossRef.
- B. Askew, P. Ballester, C. Buhr, K. S. Jeong, S. Jones, K. Parris, K. Williams and J. Rebek, Jr, Molecular recognition with convergent functional groups. 6. Synthetic and structural studies with a model receptor for nucleic acid components, J. Am. Chem. Soc., 1989, 111, 1082–1090 CrossRef CAS.
- K. Williams, B. Askew, P. Ballester, C. Buhr, K. S. Jeong, S. Jones and J. Rebek, Jr, Molecular recognition with convergent functional groups. 7. Energetics of adenine binding with model receptors, J. Am. Chem. Soc., 1989, 111, 1090–1094 CrossRef CAS.
- D. S. Kemp and K. S. Petrakis, Synthesis and conformational analysis of cis,cis-1,3,5-trimethylcyclohexane-1,3,5-tricarboxylic acid, J. Org. Chem., 1981, 46, 5140–5143 CrossRef CAS.
- N. Kitamura, Y. Suzuki, M. Chiba, N. Sakata and H.-B. Kim, Spectroscopic evidence for cooperative binding of a host in molecular hinge, Chem. Lett., 2001, 720–721 CrossRef CAS.
- K. C. Murdock and R. B. Angier, Alternative approaches to 1-substituted thymines, J. Org. Chem., 1962, 27, 3317–3319 CAS.
- T. D. Browne, J. Eisinger and N. J. Leonard, Synthetic spectroscopic models related to coenzymes and base pairs. II. Evidence for intramolecular base–base interactions in dinucleotide analogs, J. Am. Chem. Soc., 1968, 90, 7302–7323 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel-Dekker, New York, 1993 Search PubMed.
- H.-B. Kim, S. Habuchi and N. Kitamura, Dynamic fluorescence spectroscopic study on the microstructures in ion-exchange resin particles, Anal. Chem., 1999, 71, 842–848 CrossRef CAS.
- S. Ishizaka, K. Nakatani, S. Habuchi and N. Kitamura, Total internal reflection fluorescence dynamic anisotropy of sulforhodamine 101 at a liquid/liquid interface: Rotational reorientation times and interfacial structures, Anal. Chem., 1999, 71, 419–426 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970 Search PubMed.
- Under the conditions, more than 90 mol% of I is bound with BA, as estimated from the binding constant: Ka = 240 M−1.
- K. A. Connors, The Measurements of Molecular Complex Stability, John-Wiley & Sons, New York, 1987 Search PubMed.
- N. Mataga and T. Kubota, Molecular Interactions and Electronic Spectra, Marcel Dekker, New York, 1970 Search PubMed.
- R. L. Christensen, R. C. Drake and D. Phillips, Time-resolved fluorescence anisotropy of perylene, J. Phys. Chem., 1986, 90, 5960–5967 CrossRef CAS.
- R. S. Moog, M. D. Ediger, S. G. Boxer and M. D. Fayer, Viscosity dependence of the rotational reorientation of rhodamine B in mono- and polyalcohols. Picosecond transient grating experiments, J. Phys. Chem., 1982, 86, 4694-4700.
- S. Ishizaka, S. Kinoshita, Y. Nishijima and N. Kitamura, Direct observation of molecular recognition mediated by triple hydrogen bonds at a water/oil interface: Time-resolved total internal reflection fluorometry study, Anal. Chem., 2003, 75, 6035–6042 CrossRef CAS.
- Organic Solvent, Techniques of Chemistry, ed. J. A. Riddik and W. B. Bunger, Wiley-Interscience, New York, 1970, vol. 2 Search PubMed.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |