Protic solvent effects on the photophysical properties of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) TiIVTSPP: photoinduced electron transfer†
TiIVTSPP: photoinduced electron transfer†
Su Young
Ryu
a,
Minjoong
Yoon
*b,
Sae Chae
Jeoung
c and
Namwoong
Song
c
aDepartment of Chemistry, Chungnam National University, Daejeon, 305-764, Korea
bDepartment of Chemistry, Chungnam National University, Daejeon, 305-764, Korea. E-mail: mjyoon@cnu.ac.kr; Fax: 82-42-823-7008; Tel: 82-42-821-6546
cSpectroscopy Laboratory, Korea Research Institute of Standards and Science, Daejeon, 305-764, Korea
First published on 21st October 2004
Abstract
The photophysical properties of oxotitanium(IV)
meso-tetra(4-sulfonatophenyl) porphyrin (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) TiIVTSPP) have been investigated in water and methanol by laser spectroscopic techniques. The fluorescence emission spectrum of OTiIVTSPP in methanol exhibits two strong emission bands at 610 and 670 nm at room temperature with the decay time of ca. 310 ± 10 ps and the rise time shorter than 30ps, in contrast to the extremely weak emission with the decay time of ca. 27 ± 4 ps in water, indicating that the fluorescence emissive states are different in the two solvents as supported by the solvent dependences of the excitation spectrum. The transient Raman spectra of OTiIVTSPP in water has been observed to exhibit a remarkable enhancement of phenyl-related mode at 1599 cm−1, while in methanol, the Raman frequencies of the porphyrin skeletal modes (υ2 and υ4) are down-shifted without any apparent enhancement of the phenyl-related mode, indicating different interactions of the two solvents with the excited OTiIVTSPP. These Raman studies reveal that methanol molecule interacts with the photoexcited OTiIVTSPP more strongly than water, forming the exciplex, OTiIVTSPP(MeOH)*, suggesting that the two different emissive states are the singlet Franck–Condon state and the exciplex state in methanol and water, respectively. A broad triplet transient absorption of OTiIVTSPP has been also observed at 480 nm in water as well as in methanol, which is decreased upon addition of methyl viologen (MV2+) with appearance of a new absorption band at 620 nm. This indicates that the photoinduced electron transfer (PET) takes place from the porphyrin to MV2+in both solvents. The kinetic analysis of the transient absorption band exhibits the PET rate constants of 4.76 × 105 s−1 and 3,03 × 104 s−1 in methanol and water, respectively. All these results infer that the PET takes place from the (d,π) CT state and the triplet state of the excited porphyrin in methanol and water, respectively.
TiIVTSPP) have been investigated in water and methanol by laser spectroscopic techniques. The fluorescence emission spectrum of OTiIVTSPP in methanol exhibits two strong emission bands at 610 and 670 nm at room temperature with the decay time of ca. 310 ± 10 ps and the rise time shorter than 30ps, in contrast to the extremely weak emission with the decay time of ca. 27 ± 4 ps in water, indicating that the fluorescence emissive states are different in the two solvents as supported by the solvent dependences of the excitation spectrum. The transient Raman spectra of OTiIVTSPP in water has been observed to exhibit a remarkable enhancement of phenyl-related mode at 1599 cm−1, while in methanol, the Raman frequencies of the porphyrin skeletal modes (υ2 and υ4) are down-shifted without any apparent enhancement of the phenyl-related mode, indicating different interactions of the two solvents with the excited OTiIVTSPP. These Raman studies reveal that methanol molecule interacts with the photoexcited OTiIVTSPP more strongly than water, forming the exciplex, OTiIVTSPP(MeOH)*, suggesting that the two different emissive states are the singlet Franck–Condon state and the exciplex state in methanol and water, respectively. A broad triplet transient absorption of OTiIVTSPP has been also observed at 480 nm in water as well as in methanol, which is decreased upon addition of methyl viologen (MV2+) with appearance of a new absorption band at 620 nm. This indicates that the photoinduced electron transfer (PET) takes place from the porphyrin to MV2+in both solvents. The kinetic analysis of the transient absorption band exhibits the PET rate constants of 4.76 × 105 s−1 and 3,03 × 104 s−1 in methanol and water, respectively. All these results infer that the PET takes place from the (d,π) CT state and the triplet state of the excited porphyrin in methanol and water, respectively.
Introduction
The excited state properties and photoinduced electron transfer (PET) reactions of donor-acceptor systems have been extensively studied to mimic photosynthetic electron transfer.1 The most extensively studied donor-acceptor systems are metalloporphyrins-linked systems, which can be used as an electron donor due to their efficient absorption of sunlight. Thus, many studies on the photophysical properties of metalloporphyrins have been performed for the development of solar energy conversion and storage systems.2–5 The photophysical properties of metalloporphyrins are known to depend on the central metal and the peripheral substituents as well as solvent nature.6,7 So far, the most metalloporphyrins used for these studies were water-insoluble, even though water-soluble porphyrins have attracted much attention as a possible photosensitizer, generating H2 from water. However, the photophysical and photochemical properties of water-soluble porphyrins have not been systematically studied. Thus, it is necessary to investigate the photophysical properties of water-soluble metalloporphyrin derivatives. Therefore, in this work we have attempted to investigate the photophysical properties of the water-soluble OTiIVTSPP by using laser spectroscopy. Our primary motive was aimed at establishment of a new possible porphyrin system for solar energy conversion or other photofunctional material.
The titanyl porphyrins are very rare mononuclear complexes, consisting of the titanyl ion, TiO2+, and it is classified as fluorescence porphyrin, 0.2 > ϕf > 10−3.3 Usually, the fluorescence quantum yield of metalloporphyrin tends to be lower than that of free-base porphyrins based on fast nonradiative transitions, such as intersystem crossing due to heavy atomic effect by a central metal. In addition to the heavy atomic effect, interaction of solvent with external phenyl substituent may play an important role in the fluorescence quenching.8 Furthermore, the fluorescence quenching of water-soluble metalloporphyrins can be caused by formation of a complex or dimer in water or other protic solvents. In this study the protic solvent effects on the photophysical properties of OTiIVTSPP have been investigated by using ps-time resolved fluorescence techniques, as well as ns-transient absorption and Raman spectroscopic techniques, being related to solvent effects on the dynamics and photoinduced electron transfer from OTiIVTSPP to MV2+. To our knowledge, this is the first report on the protic solvent effects on the photophysical properties of OTiIVTSPP including the photoinduced electron transfer.
Experimental
Materials
OTiIVmeso-tetrakis(4-sulfonatophenyl)porphyrin and methyl viologen were purchased from Porphyrin Products Inc. and Aldrich Chem. Co., and used without further purification. The methanol was HPLC grade. The water was triply distilled. The porphyrin concentration was adjusted to ≈1 × 10−4 M, and the samples for the flash photolysis were prepared by five freeze-pump-thaw cycles. The transient Raman experiments were performed by flowing the sample solutions (ca. 1 × 10−4 M) through a glass capillary (0.8 mm id) at a sufficient rate to ensure that each laser pulse encountered a fresh volume of the sample.
Spectroscopic measurements
The photophysical properties of OTiIVTSPP have been investigated by using CW and time resolved emission, transient Raman, and transient absorption spectroscopy. The absorption spectra were recorded on a UV-visible spectrophotometer (Cary 3E). The corrected fluorescence emission spectra were measured on a scanning SLM-AMINCO 4800 Spectrofluorometer by using Rhodamin as a quantum counter.
Temporal profiles of the fluorescence decays were measured by using time-correlated single photon counting method (TCSPC). The excitation source is a self mode-rocked picosecond Ti:sapphire laser (Coherent co.) pumped by an Nd:YVO4 laser. Laser output has a ≈3 ps pulse width, and it can span the excitation wavelength in the range of 235–300 and 350–490 nm by second- and third-harmonic generations, respectively. All the standard electronics for the TCSPC were from the Edinburgh Instruments. The instrumental response function was measured by detecting the scattered laser pulse of ca. 3 ps with quartz crystal. The resultant FWHM is 60 ps. This method allows a time resolution of about 20 ps after deconvolution.
The nanosecond transient resonance Raman spectra were measured by using the both pump and probe pulses at 416 nm generated by H2 Raman shifting of 3rd harmonics (355 nm) from a nanosecond Q-switched Nd:YAG laser. The Raman scattering signal was dispersed with a spectrograph (Jobin-Yvon) and detected with a gated intensified photodiode array detector (Princeton Instrument IRY 700). The photoexcitation pulse was power-controlled and synchronized to the gating electronics of the image detector with a pulse generator, and the triplet-state transient Raman spectra can be obtained by subtracting the low-power-induced Raman signal from the high-power-induced Raman signal.
For ns-transient absorption spectroscopic measurements, the 416 nm pulses generated by hydrogen Raman shifting of the third harmonic-generated beam (355 nm) from a nanosecond Q-switched Nd : YAG laser were used as the excitation light source. A CW Xe-arc lamp (ORIEL 68811) was used as a probe light source. The probe light, encountered with 90 ° to the excitation beam onto the reaction cell, was focused on the entrance slit of monochromator (Grand Junction, CO. 81505). A photomultiplier tube (PMT, Hamamatzu R 955) was attached to the exit slit for the signal detection. The temporal profiles of the transient absorption signal were monitored by using a 500 MHz digital storage oscilloscope (DSO, Hewlett Packard HP54503A). Average output from DSO was digitized and stored in a personal computer by using a GPIB interface board.
For irradiation experiments, we used the 380 nm cut off filter to prevent direct photoreaction of MV2+.
Results
Spectroscopic properties
The absorption spectra of OTiIVTSPP in both methanol and water exhibit identical spectral features at room temperature. They consist of the Soret band at 429 nm and two weak Q-bands at 563 and 602 nm (not shown). These observations are consistent with the previous reports. 9,10 It has been frequently reported that the water-soluble porphyrin aggregates significantly in moderate-to-high concentrations, exhibiting a drastic change in the region between the Soret and Q-bands in its absorption spectrum. However, no apparent change in the absorption spectra were observed for OTiIVTSPP in the concentration range from 10−4 to 10−6 M was observed. Also we have confirmed that the concentration dependence of changes in the absorbance at 563 nm or 602 nm follows the Beer–Lambert law in the concentration range below 10−4 M.9 Thus, it is evident that OTiIVTSPP is present as a monomer in the concentration range lower than 10−4 M, and the experiments were performed with the porphyrin concentration lower than 10−4 M.
Fig. 1 shows the fluorescence spectra of OTiIVTSPP in methanol and water observed at room temperature. The fluorescence spectrum in methanol exhibits two strong emission bands centered at 610 and 660nm, which is similar to that observed from the normal metal-free porphyrin. However, in water it exhibits an extremely weak emission as shown in Fig. 1, while the absorption spectrum is similar to that observed in methanol. These results indicate that the emission quantum yields are strongly dependent on the nature of the protic solvents.
 | ||
| Fig. 1 The fluorescence emission spectra of OTiIVTSPP in methanol and water measured at room temperature. The fluorescence spectra of OTiIVTSPP in methanol/water mixture solution, from neat methanol to neat water, are shown in the insert. The excitation wavelength was 430 nm. | ||
The fluorescence emission decay profiles are also strongly dependent on the solvent nature as shown in Fig. 2. The decay times are summarized in Table 1. In methanol solution, the fluorescence decay observed at both 610 and 670 nm are well fitted into a single exponential function, exhibiting a decay-time of 310 ± 10 ps. It is also noteworthy that there is an apparent rise component with time constant shorter than 30 ps (reliable time limit for our apparatus), indicating that an emissive state in methanol is formed by relaxation from the Franck–Condon state. On the other hand, in aqueous solution the fluorescence decay is much faster than that observed in methanol, exhibiting a decay-time of ca. 27 ± 4 ps without rise time. This result indicates that the fluorescence emission in water is originated from the Franck–Condon S1 state in contrast to that observed in methanol. Considering the emission decay times (τf) and the quantum yields (ϕf), the rate constants for the radiative (kr) and the nonradiative (knr) processes in the excited-state OTiIVTSPP were calculated by using the relations kr
=
ϕf
/τf and knr
=
(1 −
ϕf)/τf. According to this calculation, it was found that knr
(≈4.2 × 1010 s−1) is much larger than kr
(≈4.2 × 106 s−1) in water, while in methanol knr
(≈3.2 × 109 s−1) is not so much different from kr
(≈5.2 × 108 s−1), indicating that nonradiative processes are dominant in water in contrast to the competitive nonradiative and radiative processes in methanol.
| Solvent | Decay times (τ) | |
|---|---|---|
| 610 nm | 670 nm | |
| a Excitation wavelength is 430 nm. Measurement error limits are less than 10% of the listed values. | ||
| Water | 27 ps | 30 ps |
| Methanol | 307 ps (rise ≈ 30 ps) | 309 ps (rise ≈ 30 ps) |
| Ethanol | 797 ps (rise ≈ 30 ps) | 794 ps (rise ≈ 30 ps) |
 | ||
| Fig. 2 The fluorescence emission decay profiles of OTiIVTSPP in methanol (top) and water (bottom). The excitation wavelength was 430 nm. | ||
In order to further confirm the substantial difference in the fluorescent properties in water and methanol, we also observed the fluorescence spectra of OTiIVTSPP in both solvents at 77 K (Fig. 3). The extremely weak emission in water at room temperature gains its intensity drastically upon lowering the temperature to 77 K, exhibiting the spectral feature similar to that observed in methanol except the slight blue-shift, indicating the energy levels of the emissive states are similar in both water and methanol. The fluorescence excitation spectra were also measured at 77 K. As shown in Fig. 4, the fluorescence excitation spectrum for the 610 nm emission band in water is similar to the ground state absorption spectrum in its Soret and Q bands, whereas in methanol it exhibits different spectral feature with a remarkable red shift as compared to that observed in the aqueous solution. This supports again that the fluorescence emissions in water and methanol are originated from different emissive states.
 | ||
| Fig. 3 The fluorescence emission spectra of OTiIVTSPP in methanol and water at 77 K. The excitation wavelength was 430 nm. | ||
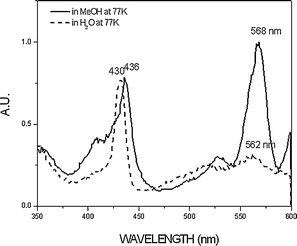 | ||
| Fig. 4 The fluorescence excitation spectra of OTiIVTSPP in methanol and water recorded at the emission band of 610 nm at 77 K. | ||
In order to clarify the different interactions of the two solvents with the excited OTiIVTSPP, the transient Raman spectroscopic measurements were performed. Figures 5 illustrates the different transient Raman spectral features of OTiIVTSPP in water and methanol, respectively, supporting that the nature of transient species participating in photophysical and photochemical processes is evidently affected by the solvent nature. The ground-state Raman spectra (Fig. 5(a)) were obtained in water and methanol with low-power laser excitation. This low-power Raman spectral features are found to be almost identical in both solvents, exhibiting a relatively weak phenyl mode at 1598 cm−1 observed in addition to the moderate intense υ2 mode at 1545 cm−1, υ4 mode at 1356 cm−1, and υ1 mode at 1238 cm−1, indicating that the vibrational structure of OTiIVTSPP is not affected by the solvent nature in the ground-state. To obtain the transient Raman spectrum (Fig. 5(c)) contributed purely by the excited triplet states of OTiIVTSPP, the ground-state Raman spectral features were subtracted from the high-power Raman spectrum (Fig. 5(b)). Comparing the Fig. 5(c) with the Fig. 5(a), in water the apparent enhancement of the Raman peak intensity of the phenyl mode at 1596 cm−1 is shown without any observable change in other modes, while in methanol an apparent down-shifts in the Raman frequency of the porphyrin skeletal modes for υ2 and υ4 modes are exhibited without an apparent enhancement of phenyl mode at 1598 cm−1
(Table 2). This indicates different interactions of the two solvents with the excited OTiIVTSPP.11
TiIVTSPP in methanol
 | ||
| Fig. 5 The transient resonance Raman spectra of OTiIVTSPP in methanol and water with both pump and probe pulses at 416 nm: (a) the low power (≈0.01 mJ pulse−1), (b) high power (≈0.2 mJ pulse−1), and (c) the difference spectra (b − a) with a proper scaling factor. The solution was flowed so that a fresh sample was exposed to every laser pulse. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) TiIVTSPP and MV2+.
Fig. 6 shows the absorption spectra of OTiIVTSPP with MV2+ in methanol as a function of irradiation time. Because a low-pass filter at 380 nm was used in the experiment, there is no contamination of the absorption spectrum due to the direct photoreaction of MV2+. The new absorption bands at 394 and 600 nm are characteristic of the reduced methyl viologen, MV+1,12 indicating that the photoinduced electron transfer (PET) takes place from the excited OTiIVTSPP to MV2+,. The irradiation experiments were also conducted in water under the same condition as in methanol. However, no apparent changes in the absorption bands were observed except the slight broadening of the Soret and Q-bands.
TiIVTSPP and MV2+.
Fig. 6 shows the absorption spectra of OTiIVTSPP with MV2+ in methanol as a function of irradiation time. Because a low-pass filter at 380 nm was used in the experiment, there is no contamination of the absorption spectrum due to the direct photoreaction of MV2+. The new absorption bands at 394 and 600 nm are characteristic of the reduced methyl viologen, MV+1,12 indicating that the photoinduced electron transfer (PET) takes place from the excited OTiIVTSPP to MV2+,. The irradiation experiments were also conducted in water under the same condition as in methanol. However, no apparent changes in the absorption bands were observed except the slight broadening of the Soret and Q-bands.
 | ||
| Fig. 6 The absorption spectral feature of OTiIVTSPP with MV2+ in methanol as a function of irradiation time. Irradiation was performed at wavelengths ≥ 380 nm to prevent a direct photoreaction of MV2+. | ||
The fluorescence intensity of OTiIVTSPP in methanol and its decay time were also observed to be decreased with an addition of MV2+ as shown in Fig. 7, indicating again that the PET is involved in the fluorescence quenching by MV2+. However, in water, due to the extremely low emission intensity of OTiIVTSPP, the reliable fluorescence quenching process was not observed in the presence of MV2+, and the involvement of the PET could not be confirmed in water by the fluorescence quenching method.
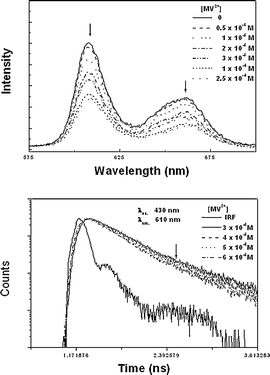 | ||
| Fig. 7 The fluorescence emission spectra (top) and their decay profiles (bottom) of OTiIVTSPP in methanol depends on concentration of MV2+: 0.5, 1, 2, 3, 10, 25 (×10−5 M). The excitation wavelength was 430 nm. | ||
Thus, in order to directly confirm the PET both in methanol and water, the nanosecond time resolved transient absorption experiment was performed. The transient absorption spectrum of OTiIVTSPP in methanol was measured immediately just after photoexcitation at 416, and it revealed the broad transient absorption band at 480 nm corresponding to T-T absorption as shown in Fig. 8.13 Upon addition of MV2+ to the methanol solution of OTiIVTSPP, the 480 nm transient band disappears with appearance of new band at 620 nm, which is attributed to the cation radical of the porphyrin. The similar spectral changes are also observed in water, indicating that the PET takes place even in water. 14
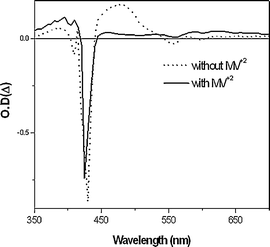 | ||
| Fig. 8 Transient absorption spectra of OTiIVTSPP in methanol with and without MV2+ after excitation at 416 nm. | ||
The temporal decay profiles of the transient absorption band of OTiIVTSPP at 480 nm can be well fitted with single exponential functions (not shown) with the decay times of ca. 109 ± 5 µs in methanol and ca. 233 ± 10 µs in water. Upon addition of MV2+ the transient absorption at 480 nm in methanol exhibits much shorter bi-exponential decay components of ca. 2.3 ± 0.1 µs and ca. 47 ± 1 µs, indicating the equilibrium between the reactive excited state of OTiIVTSPP and the primary product of the PET, probably the cation radical. Meanwhile, the transient absorption band at 620 nm exhibits a single exponential decay (decay time; ca. 51 ± 1)µs with the rise time of ca. 2.1 ± 1 µs. It is interesting to note that the rise time observed at 620 nm is almost identical to the decay time of the transient absorption band at 480 nm, and the rise time corresponds to the formation rate of the OTiIVTSPP cation radical by the electron transfer to MV2+. Thus, the rise time (τρ) observed in the presence of MV2+ would be the same as the time of the PET from OTiIVTSPP to MV2+, and the PET rate constant (k =1/τρ) in methanol is calculated to be 4.76 × 105 s−1. Also the transient absorption band at 480 nm observed in water could be fitted into a bi-exponential decay with a decay time ca. 30 ± 3 µs and ca. 62 µs. The temporal profiles of the transient absorption at 620 nm is fitted into the single decay component of ca. 63 ± 2 µs with a rise time of ca. 33 ± 3 µs (See Fig. 9). Following the same manner conducted in the case of methanol solution, the PET rate in water could be determined to be 3.03 × 104 s−1. This transient absorption studies demonstrate that the electron transfer between the excited OTiIVTSPP and MV2+ in methanol proceeds much faster (16 fold) than in water.

Discussion
All the observations of the fluorescence emission and excitation spectra measured in methanol and water reveal that the relaxation of photoexcited OTiIVTSPP is strongly dependent on the protic solvent nature. It has been known that transient Raman is valuable for obtaining information of the nature of photoexcited metalloporphyrin. The enhancement of phenyl mode at 1596 cm−1 for the photoexcited metalloporphyrins in water (Fig. 5) has been unambiguously characterized as typical Raman spectral feature of porphyrin triplet T (π, π*) state,15,16 and it is reasonable to suppose that the photoexcited OTiIVTSPP in water is populated mostly in the triplet state. Therefore, the weak fluorescence emission in water should be just due to the efficient intersystem crossing from the singlet to the triplet state of the OTiIVTSPP. However, the down shifts of the porphyrin skeletal frequencies observed in methanol (Fig. 5
(c)) are attributed to formation of a triplet state of the six-coordinated exciplex, OTiIVTSPP(MeOH)*. Actually, in the previous transient Raman studies of the CuII porphyrins such as CuIITSPP17–20 and CuIITMPyP421–24 released that the triplet exciplex is formed by interacting with the oxygen atom of solvents. Therefore, this Raman study also supports that the relaxation process from the singlet Franck–Condon state in methanol is to form the exciplex whereas intersystem crossing only in water, being consistent with the fact that the rise time for the emission was observed in methanol but not in water. Once the exciplex is formed in methanol, either (d,d) or (π,d) CT states is known to be easily formed and located slightly below the triplet state25,26 as shown in the schematic diagram of the relaxation channels of the excited OTiIVTSPP (Fig. 10). However, because the oxygen atoms of solvents are known to act as an axial ligand, it is intriguing why in water no evidence was observed for the six-coordinated exciplex OTiIVTSPP(H2O)*, except for the triplet state in the transient Raman spectrum. This may be because the electron density on the oxygen atom of methanol tends to be larger than that of water due to an electron-donating effect by the methyl group. Therefore, it is reasonable that methanol molecule acts as a strong ligand to the photoexcited OTiIVTSPP, while water molecule acts as a very weak ligand. If this is true, the ligation effect of the protic solvents (ROH) might be increased as alkyl chain increases. Actually, the increasing fluorescence lifetime was observed in the order of ethanol > methanol > water (see Table 1). This infers that the fast nonradiative processes due to the vibronic coupling are less dominant as the ligation effect of the solvent increases.

Based on the proposed relaxation processes of the photoexcited OTiIVTSPP in the two different solvents, the observations on the relaxation processes could be rationalized as follows: the negligible fluorescence observed in the aqueous solution of OTiIVTSPP at room temperature can be attributed to the fast nonradiative processes including intersystem crossing to the triplet state. The resultant triplet state was found to be relaxed with a decay time of 233 ± 10 µs as observed from the transient absorption experiments. Meanwhile, the rise component of fluorescence decay profiles observed in the methanol solution might be due to the equilibration between the five-coordinate species and the six-coordinate species, which is partly consistent with the results of the excitation spectral feature. The moderately strong intensity, with its long decay time of the fluorescence observed in methanol solution, led us to suppose that the resultant state is either emissive or at least thermally equilibrated with an emissive excited state. This rather fast relaxation process is followed by a relaxation to the ground state with a decay time of 109 ± 5 µs as shown in the transient absorption experiment. The relaxation dynamics of the photoexcited OTiIVTSPP in methanol and water are also summarized in Fig. 10.
The difference in the nature of the excited states in methanol and water might result in the difference in the PET from OTiIVTSPP to MV2+. As described above, the transient absorption demonstrates that the electron transfer between OTiIVTSPP and MV2+ in methanol proceeds much faster (16 fold) than in water. Considering that the solvent effects on the formation of exciplex, the faster PET rate in methanol must be due to the formation of the six-coordinate OTiIVTSPP(MeOH) which enhances the electron donating ability toward MV2+.27–29
In order to further characterize the nature of the excited state, it is worthwhile to discuss the quenching of fluorescence of OTiIVTSPP in the presence of MV2+. First, we attempted to use the Stern–Volmer equation, which is given below,
 TiIVTSPP fluorescence by MV2+ in methanol. The resulting plot is not linear, indicating that the fluorescence quenching is not simply dynamic. Such a downward curving Stern–Volmer plot was reported to appear in iodide quenching of tryptophan fluorescence,30 indicating existence of two types of fluorophores in this system, which are not equally accessible to the quencher. Among the two emissive species, one must be accessible to the quencher, but another must be inaccessible to the quencher, i.e. MV2+.30,31 According to the fluorescence spectral analysis of OTiIVTSPP in methanol, the fluorophore species, which is accessible or inaccessible to the quencher, is probably OTiIVTSPP–(MeOH) complex.
TiIVTSPP fluorescence by MV2+ in methanol. The resulting plot is not linear, indicating that the fluorescence quenching is not simply dynamic. Such a downward curving Stern–Volmer plot was reported to appear in iodide quenching of tryptophan fluorescence,30 indicating existence of two types of fluorophores in this system, which are not equally accessible to the quencher. Among the two emissive species, one must be accessible to the quencher, but another must be inaccessible to the quencher, i.e. MV2+.30,31 According to the fluorescence spectral analysis of OTiIVTSPP in methanol, the fluorophore species, which is accessible or inaccessible to the quencher, is probably OTiIVTSPP–(MeOH) complex.
![Stern–Volmer (a) and modified Stern–Volmer (b) plots showing quenching of OTiIVTSPP fluorescence in methanol as determined by fluorescence intensity as a function of [MV2+].](/image/article/2005/PP/b409751c/b409751c-f11.gif) | ||
| Fig. 11 Stern–Volmer (a) and modified Stern–Volmer (b) plots showing quenching of OTiIVTSPP fluorescence in methanol as determined by fluorescence intensity as a function of [MV2+]. | ||
The Stern–Volmer equation can be modified as shown in the following equation,
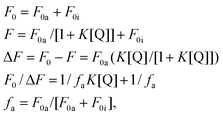
A plot of F0/ΔFvs. 1/[Q] yields fa−1 as an intercept, and (faK)−1 as a slope. The intercept on the y axis (fa−1) is 1.40, and the slope is 5.18 × 10−5. Hence the fa−1 is 1.40, indicating that 71% of the total fluorescence of OTiIVTSPP is accessible to MV2+. The quenching constant K is calculated as 2.70 × 104 M−1 s−1 from (1.40/5.18 × 10−5) M−1 s−1
(see Fig. 11(b)). This value is extremely small as compared to other collisional quenching constant, ≈1 × 1010 M−1 s−1 in the S1 emissive state. Thus, the fluorescence is not quenched directly by collision of S1 state of OTiIVTSPP. The fluorescence quenching is rather induced indirectly by the PET from the relaxed state such as CT state formed through formation of hexadentate OTiIVTSPP–(MeOH) exciplex as evidenced by the transient Raman studies. This is consistent with the fact that the small quenching constant is about the same as the electron transfer rate constant determined by the transient absorption experiment. However, in water the triplet state is a dominant relaxed state, and the PET occurs simply from the triplet state. This might be why the PET rate is 16 times slower in water than in methanol.
Conclusions
The PET between OTiIVTSPP and MV2+ strongly depends on the solvent nature. OTiIVTSPP in methanol seems to form the six-coordinate OTiIVTSPP(MeOH) on photoexcitation. The lowest excited state of OTiIVTSPP in methanol are the quenching states such as the (d,d) or (π,d) CT states located below that of the triplet T(π,π*) states, while the lowest excited state of OTiIVTSPP in water are the triplet T(π,π*) states. The lowest excited states of OTiIVTSPP (CT state in methanol and triplet state in water) are probably the major reactive states for the PET reaction from OTiIVTSPP to MV2+. The rate constant for the electron transfer from the lowest excited states of OTiIVTSPP to MV2+ was calculated to be 4.76 × 105 s−1 in methanol, while it was 3.03 × 104 s−1 in water. Furthermore, the CT state seems to bring an enhancement in the electron donating ability of OTiIVTSPP toward MV2+.
Acknowledgements
This work was financially supported by the Korea Research Institute of Bioscience and Biotechnology through 2004 KISTEP National R&D Program.References
- (a) M. R. Wasielewski, Photoinduced electron transfer in supramolecular systems for artificial photosynthesis, Chem. Rev., 1992, 92, 435–461 CrossRef CAS; (b) D. Gust, T. A. Moore and A. L. Moore, Molecular mimicry of photosynthetic energy and electron transfer, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS; (c) M. N. Paddon-Row, Investigating long-range electron-transfer processes with rigid, covalently linked donor-(norbornylogous bridge)–acceptor systems, Acc. Chem. Res., 1994, 27, 18–25 CrossRef CAS; (d) A. Osuka, N. Mataga and T. Okada, A chemical approach towards the photosynthetic reaction center, Pure. Appl. Chem., 1997, 69, 797–802 CrossRef CAS; (e) M. J. Blanco, J. M. Consuelo, J.-C. Chambron, V. Heitz, M. Linke and J.-P. Sauvage, Rotaxanes as new architectures for photoinduced electron transfer and molecular motions, Chem. Soc. Rev., 1999, 28, 293–305 RSC.
- (a) D. E. Dolphin, The porphyrins V, Academic Press, ed. David Dolphin, New York, 1979, ch. 9, pp. 402–458 Search PubMed; (b) K. Sauer, A role for manganese in oxygen evolution in photosynthesis, Acc. Chem. Res., 1980, 13, 249–256 CrossRef CAS; (c) M. Calvin, Simulating photosynthetic quantum conversion, Acc. Chem. Res., 1978, 11, 369–374 CrossRef CAS; (d) G. R. Seely, The energetics of electron-transfer reactions of chlorophyll and other compounds, Photochem. Photobiol., 1978, 27, 639–654 CAS.
- (a) D. G. Whitten, Photoinduced electron transfer reactions of metal complexes in solution, Acc. Chem. Res., 1980, 13, 83–90 CrossRef CAS; (b) M. Graetzel, Artificial photosynthesis: water cleavage into hydrogen and oxygen by visible light, Acc. Chem. Res., 1981, 14, 376–384 CrossRef; (c) K. Kalyanasun-daram and M. Neumann-Spallart, Photophysical and redox properties of water-soluble porphyrins in aqueous media, J. Phys. Chem., 1982, 86, 5163–5169 CrossRef CAS.
- M. E. Perez-Bernal, R. Ruano-Casero and T. J. Pinnavaia, Catalytic autoxidation of 1-decanethiol by cobalt(II) phthalocyaninetetrasulfonate intercalated in a layered double hydroxide, Catal. Lett., 1991, 11, 55–61 CrossRef.
- J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M. C. Richoux, Metal phthalocyanines and porphyrins as photosensitizers for reduction of water to hydrogen, Coord. Chem. Rev., 1982, 44, 83–126 CrossRef CAS.
- S. C. Jeoung, D. Kim, D. W. Cho and M. Yoon, Time-Resolved Resonance Raman and Transient Absorption Studies on Water-Soluble Copper(II) Porphyrins, J. Phys. Chem., 1996, 100, 3075–3083 CrossRef CAS.
- J. B. Allison and P. S. Becker, Effect of metal atom perturbations on the luminescent spectra of porphyrins, J. Chem. Phys., 1960, 32, 1410–1417.
- D. E. Dolphin, The porphyrins III, Academic Press, ed. David Dolphin, New York, 1979, ch. 1 Search PubMed.
- D. E. Dolphin, The porphyrins IV, Academic Press, ed. David Dolphin, New York, 1979, ch. 7, pp. 355–378 Search PubMed.
- K. M. Kadish, G. B. Maiya, C. Araullo and R. Guilard, Micellar effects on the aggregation of tetraanionic porphyrins. Spectroscopic characterization of free-base meso-tetrakis(4-sulfonatophenyl)porphyrin, (TPPS)H2, and (TPPS)M (M = zinc(II), copper(II), and vanadyl) in aqueous micellar media, Inorg. Chem., 1989, 28, 2725–2731 CrossRef CAS.
- M. Hoshino, M. Imamura, S. Watanabe and Y. Hama, Photoexcited triplet state of oxotitanium(IV) tetraphenylporphyrin studied by ESR and laser photolysis. Proton-induced quenching of the triplet state, J. Phys. Chem., 1984, 88, 45–49 CrossRef CAS.
- T. Watanabe and K. Honda, Measurement of the extinction coefficient of the methyl viologen cation radical and the efficiency of its formation by semiconductor photocatalysis, J. Phys. Chem., 1982, 86, 2617–2619 CrossRef CAS.
- J. Rodriguez, C. Kirmaier and D. Holten, Optical properties of metalloporphyrin excited states, J. Am. Chem. Soc., 1989, 111, 6500–6506 CrossRef CAS.
- J. Peon, X. Tan, J. D. Hoerner, C. Xia, Y. F. Luk and B. Kohler, Excited State Dynamics of Methyl Viologen. Ultrafast Photoreduction in Methanol and Fluorescence in Acetonitrile, J. Phys. Chem. A, 2001, 105, 5768–5777 CrossRef CAS.
- S. E. J. Bell, A. H. R. Al-Obaidi, M. Hegarty, R. E. Hester and J. J. McGarvey, Time-resolved resonance Raman spectroscopy of excited singlet and triplet states of free-base meso-tetraphenylporphyrin, J. Phys. Chem., 1993, 97, 11599–11602 CrossRef CAS.
- R. A. Reed, R. Purrello, K. Prendergast and T. G. Spiro, Resonance Raman characterization of the radical anion and triplet states of zinc tetraphenylporphine, J. Phys. Chem., 1991, 95, 9720–9727 CrossRef CAS.
- E. D. A. Stemp, M. R. Arkin and J. K. Barton, Oxidation of Guanine in DNA by Ru(phen)2(dppz)3+ using the flash-quench technique, J. Am. Chem. Soc., 1997, 119, 2921–2925 CrossRef CAS.
- P. Y. Turpin, L. Chinsky, A. Laigle, M. Tsuboi, J. R. Kincaid and K. Nakamoto, A porphyrin–DNA exciplex: resonance Raman spectra of electronically excited Cu(TMpy-P4) bound to poly(dA-dT).poly(dA-dT), Photochem. Photobiol., 1990, 51, 519–525 CAS.
- L. Chinsky, P. Y. Turpin, A. H. R Al-Obaidi, S. E. J. Bell and R. E. Hester, Contribution of resonance Raman excitation spectroscopy for probing electronically excited states: nature of a porphyrin–DNA exciplex, J. Phys. Chem., 1991, 95, 5754–5756 CrossRef CAS.
- G. D. Strahan, D. Lu, M. Tsuboi and K. Nakamoto, Resonance Raman spectra of electronically excited, water-soluble copper(II) porphyrins bound to oligonucleotides: possibility of translocation from a GC to an AT site, J. Phys. Chem., 1992, 96, 6450–6457 CrossRef CAS.
- S. C. Jeoung, D. Kim, D. W. Cho and M. Yoon, Time-resolved resonance Raman spectroscopic study on copper(II) porphyrins in various solvents: Solvent effects on the charge transfer states, J. Phys. Chem., 1995, 99, 5826–5833 CrossRef CAS.
- S. G. Kruglik, V. A. Galievsky, V. S. Chirvony, P. A. Apanasevich, V. V. Ermolenkov, V. A. Orlovich, L. Chinsky and P. Y. Turpin, Dynamics and mechanism of the exciplex formation between Cu(TMpy-P4) and DNA model compounds revealed by time-resolved transient absorption and resonance Raman spectroscopies, J. Phys. Chem., 1995, 99, 5732–5741 CrossRef CAS.
- P. A. Apanaseivich, V. V. Ermolenkov, S. G. Kruglik, V. A. Kvach and V. A. Orlovich, in Proceedings of 6th International Conference on Time resolved Vibrational Spectroscopy, ed. A. Lau, F. Siebert and W. Werncke, Springer verlag, Berlin–Heidelberg, 1994, vol. 74, p. 120 Search PubMed.
- S. G. Kruglik, P. A. Apanasevich, V. S. Chirvony, V. V. Kvach and V. A. Orlovich, Resonance Raman, CARS, and picosecond absorption spectroscopy of copper porphyrins: The evidence for the exciplex formation with oxygen-containing solvent molecules, J. Phys. Chem., 1995, 99, 2978–2995 CrossRef CAS.
- P. Neta, One-electron transfer reactions involving zinc and cobalt porphyrins in aqueous solutions, J. Phys. Chem., 1981, 85, 3678–3684 CrossRef CAS.
- J. C. de Paula, V. A. Walters, B. A. Jackson and K. Cardozo, Transient resonance Raman spectroscopy of copper(II) complexes of meso-tetraphenylporphine and meso-tetraphenyl-chlorin, J. Phys. Chem., 1995, 99, 4373–4379 CrossRef CAS.
- M. Atamian, R. J. Donohoe, J. S. Lindsey and D. F. Bocian, Resonance Raman spectra and normal-coordinate analysis of reduced porphyrins. 1. Zinc(II) tetraphenyl-porphyrin anion, J. Phys. Chem., 1989, 93, 2236–2243 CrossRef CAS.
- P. Neta, A. Scherz and H. Levanon, Electron transfer reactions involving porphyrins and chlorophyll a, J. Am. Chem. Soc., 1979, 101, 3624–3629 CrossRef CAS.
- P. Neta, V. Grebel and H. Levanon, One-electron oxidation of chlorophyll a and (tetraphenylporphyrinato)cobalt(II) by various metalloporphyrin cation radicals; Kinetic spectrophotometric studies, J. Phys. Chem., 1981, 85, 2117–2119 CrossRef CAS.
- S. S. Lehrer, Solute perturbation of protein fluorescence. Quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion, Biochemistry, 1971, 10, 3254–3263 CrossRef CAS.
- N. Chitose, J. A. LaVerne and Y. Katsumura, Effect of formate concentration on radical formation in the radiolysis of aqueous methyl viologen solutions, J. Phys. Chem. A, 1998, 102, 2087–2090 CrossRef CAS.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |