Photophysical study of photoinduced electron transfer in a bis-thiophene substituted peryleneimide†‡
Eduard
Fron
a,
Marc
Lor
a,
Roberto
Pilot
a,
Gerd
Schweitzer
a,
Haluk
Dincalp
a,
Steven
De Feyter
a,
Jens
Cremer
b,
Peter
Bäuerle
b,
Klaus
Müllen
c,
Mark
Van der Auweraer
a and
Frans C.
De Schryver
*a
aDepartment of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200 F, 3001, Heverlee, Belgium. E-mail: DeSchryver@Chem.kuleuven.ac.be
bDepartment Organic Chemistry II (Organic Materials and Combinatorial Chemistry), University of Ulm, Albert-Einstein-Allee 11, 89081, Ulm, Germany
cMax-Planck-Institute for Polymer Research, Ackermannstrasse 10, 55128, Mainz, Germany
First published on 9th November 2004
Abstract
Based on femtosecond time-resolved spectroscopy and single photon timing experiments, intramolecular photoinduced charge transfer has been investigated in two systems containing a peryleneimide chromophore (P) and thiophene (T) groups. The first compound bearing a single thiophene ring (PT1) is used as model and shows a behavior similar to P, studied previously, while in the compound with two thiophene rings attached (PT2) electron transfer from the thiophene donor to the peryleneimide acceptor is observed in benzonitrile. Femtosecond fluorescence upconversion and femtosecond transient absorption experiments in benzonitrile indicate that this ion-pair state formation occurs in 19 ps. This ion-pair state then decays with two time constants of 1400 and 820 ps, probably corresponding to different conformations of the thiophene rings.
Introduction
Oligothiophenes are currently of interest for their use in molecular electronics and charge storage.1,2 They were first used to build thin film field effect transistors in 1990 and later reached high performances in terms of carrier mobility and on/off ratio.3 They are also of interest in photovoltaic elements.4,5 Their tunable optical properties6 are motivating the development of a novel class of optoelectronic materials. An important progress in realizing high efficiencies of photo-induced charge separation has been achieved by synthesizing compounds which combine an electron donor system with a suitable electron acceptor chromophore.4 The peryleneimide chromophore, which is an extremely (photo)stable electron acceptor moiety has been studied in detail in our research group.7–16 In contrast to other linked peryleneimide systems investigated earlier12,15,16 the ion pair state found in this study is fluorescent. In this way they resemble other electron donor–acceptor systems with emitting ion pair states reported as symmetric or asymmetric biaryl derivatives.17–23 Contrary to the latter compounds they absorb at much longer wavelengths and they provide the possibility to tune the occurrence of charge transfer by modification of the number of covalently linked thiophene moieties. In this study we report on the excited state dynamics and ion-pair state formation in two peryleneimide (P) substituted thiophene compounds. The intramolecular photoinduced charge transfer process in two different peryleneimide substituted oligothiophenes (PT1, PT2 molecular structures depicted in Scheme 1) was investigated by steady-state and time resolved fluorescence and transient absorption spectroscopy in solvents with different polarities. The results obtained for PT2 are compared with the ones obtained for the compound PT1 which serves as a model compound. The electron transfer process was characterised by comparison with results obtained previously in polyphenylene dendrimers with a triphenylamine core and substituted with the same peryleneimide moiety at the periphery.12,15 All measurements were performed on deoxygenated PT1 and PT2 solutions.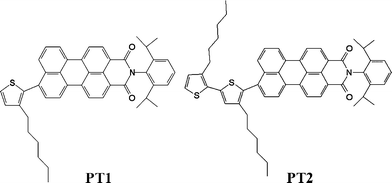 | ||
| Scheme 1 Molecular structure of the mono-thiophenylperyleneimide PT1 (left) and bis-thiophenylperyleneimide PT2 (right). | ||
Experimental
Synthesis and steady state measurements
The synthesis of the mono-thiophene and bis-thiophene substituted with one peryleneimide (P) chromophore has been reported previously.24 As a model for the locally excited state of the compound showing intramolecular charge separation we investigated a mono-thiophene substituted with a peryleneimide chromophore (Scheme 1). All stationary measurements have been recorded using a fluorimeter (Lambda 40, Perkin Elmer) and a spectrophotometer (Fluorolog, Perkin Elmer). To investigate the dependence of all fast kinetic processes (especially electron transfer) on the solvent polarity both compounds were investigated in five different solvents ranging from low to high polarity (toluene, ethyl acetate, tetrahydrofuran (THF), benzonitrile and acetonitrile). The optical density at the absorption maximum of all solutions was kept below 0.1 in a 1 cm cuvette. The excitation wavelength was set to 495 nm. As reference, a polyphenylene dendrimer substituted with one peryleneimide (m-C1P1)7 was used, which is characterized by a quantum yield of fluorescence of 1 at an excitation wavelength of 495 nm in toluene.Picosecond and femtosecond fluorescence time-resolved experiments
The picosecond time-resolved measurements of PT1 and PT2 have been performed in two different solvents of low (toluene) and high polarity (benzonitrile). All measurements were carried out in 1 cm optical path length cuvettes at an optical density of ca. 0.1 at the excitation wavelength of 488 nm, which is very close to the absorption maximum. The fluorescence decay times of PT1 and PT2 have been determined by single-photon-timing measurements (SPT) described in detail previously.25 In brief, the second harmonic output of a Ti:sapphire laser (Tsunami, Spectra Physics) has been used to excite the samples at 488 nm at a repetition rate of 4 MHz. As detector the system uses a microchannel plate photomultiplier (R3809U, Hamamatsu) mounted at the exit of a subtractive double monochromator (9030DS, Sciencetech). A time-correlated Single Photon Timing PC module (SPC 630, Picoquant) was used to obtain the fluorescence decay histogram in 4096 channels. The decays were recorded with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts in the peak channel in time windows of 25 and 15 ns corresponding to 6 and 3.7 ps channel−1 and analyzed globally with a time resolved fluorescence analysis (TRFA) software26 based on iterative reconvolution with the instrumental response function (IRF) obtained using light scattered by an aqueous suspension of ludox. The full width at half-maximum (FWHM) of the IRF was typically in the order of 40 ps. The quality of the fits has been judged by the fit parameters χ2
(<1.2), Zχ2
(<3) and the Durbin–Watson parameter (1.8 < DW < 2.2) as well as by the visual inspection of the residuals and autocorrelation function.27 To obtain a better insight into the fast kinetic processes that may occur, additional femtosecond fluorescence upconversion experiments were performed on PT1 and PT2 in benzonitrile. The fluorescence upconversion detection setup has also been described in detail in a previous publication.28 In short, the fluorescence light emitted from the sample is collected, directed and overlapped with a gate pulse (800 nm, ca. 100 µJ) derived from the RGA onto an LBO crystal. By tuning the incident angle of these two beams relative to the crystal plane the sum frequency from this light and the gate pulse is generated. The time resolved traces are then collected by detecting this sum frequency light while changing the relative delay of the gate pulse versus the sample excitation time. As detector a photomultiplier tube (R1527p, Hamamatsu) placed at the exit of a 30 cm monochromator was used. The electrical signal from the multiplier tube was gated by a boxcar averager (SR 520, Stanford Research Systems) and detected by a lock-in amplifier (SR830, Stanford Research Systems).The prompt response of this arrangement (including laser sources) was determined by detection of scattered light under otherwise identical conditions and found to be approximately 250 fs (FWHM). This value was used in the analysis of all measurements for deconvolution of the data sets. The measurements were performed in 1 mm optical path length cuvettes and the optical density of the solutions was ca. 0.4 per mm at the excitation wavelength of 495 nm.
000 counts in the peak channel in time windows of 25 and 15 ns corresponding to 6 and 3.7 ps channel−1 and analyzed globally with a time resolved fluorescence analysis (TRFA) software26 based on iterative reconvolution with the instrumental response function (IRF) obtained using light scattered by an aqueous suspension of ludox. The full width at half-maximum (FWHM) of the IRF was typically in the order of 40 ps. The quality of the fits has been judged by the fit parameters χ2
(<1.2), Zχ2
(<3) and the Durbin–Watson parameter (1.8 < DW < 2.2) as well as by the visual inspection of the residuals and autocorrelation function.27 To obtain a better insight into the fast kinetic processes that may occur, additional femtosecond fluorescence upconversion experiments were performed on PT1 and PT2 in benzonitrile. The fluorescence upconversion detection setup has also been described in detail in a previous publication.28 In short, the fluorescence light emitted from the sample is collected, directed and overlapped with a gate pulse (800 nm, ca. 100 µJ) derived from the RGA onto an LBO crystal. By tuning the incident angle of these two beams relative to the crystal plane the sum frequency from this light and the gate pulse is generated. The time resolved traces are then collected by detecting this sum frequency light while changing the relative delay of the gate pulse versus the sample excitation time. As detector a photomultiplier tube (R1527p, Hamamatsu) placed at the exit of a 30 cm monochromator was used. The electrical signal from the multiplier tube was gated by a boxcar averager (SR 520, Stanford Research Systems) and detected by a lock-in amplifier (SR830, Stanford Research Systems).The prompt response of this arrangement (including laser sources) was determined by detection of scattered light under otherwise identical conditions and found to be approximately 250 fs (FWHM). This value was used in the analysis of all measurements for deconvolution of the data sets. The measurements were performed in 1 mm optical path length cuvettes and the optical density of the solutions was ca. 0.4 per mm at the excitation wavelength of 495 nm.
Femtosecond transient absorption measurements
The amplified femtosecond double OPA laser system has also been described previously.29 The laser source for femtosecond pulses was a regeneratively amplified (RGA) Ti:sapphire system (Spitfire, Positive Light). The output of the RGA was used to pump an optical parametric amplifier (OPA 800-II, Spectra Physics), which was tuned to give the sample excitation pulses at 495 nm. To avoid photodegradation, the femtosecond transient absorption experiments were performed at 90 nJ excitation energy. A small percentage of the RGA light was used to generate a white light continuum in a sapphire plate which then served as probe light. The actual detection was done by a CCD Camera (EEV 30, Princeton Instruments) mounted at the exit of a 30 cm focal length spectrograph (SP300i, Acton Research). The entire system provides pulses with duration of 300 fs (FWHM) and covers a wavelength range between 350 and 700 nm.The transient signal of the samples was derived from a sequence of measurements. At each delay position, four spectra were recorded with the CCD-camera. By selectively blocking the pump and/or white light beam, the absorbance was determined as:
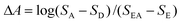 | (1) |
Results and discussion
Stationary measurements
The normalized absorption and emission spectra of PT1 and PT2 dissolved in toluene and benzonitrile are displayed in Fig. 1. The absorption spectra of PT1 in toluene and benzonitrile and PT2 in toluene are almost identical to the ones recorded for a series of polyphenylene dendrimers substituted with a varying number of peryleneimide chromophores investigated earlier.7,11,14 This suggests that under those conditions the Franck–Condon excited state resembles strongly that of P. | ||
| Fig. 1 Normalized steady-state absorption and emission spectra (495 nm excitation wavelength) for PT1 (top) and PT2 (bottom) in toluene (solid lines) and benzonitrile (dotted lines). | ||
The maximum (Table 1) and features (Fig. 1) of the emission spectrum of PT1 in toluene are nearly identical with those of m-C1P1 and shifted 600 cm−1 to lower energy compared to P.30 In benzonitrile PT1 shows a bathochromic shift of 1400 cm−1 compared to the same spectrum recorded in toluene, while for P or m-C1P1 the shift is only 700 to 800 cm−1.30 Contrary to the peryleneimide model compound16 in benzonitrile the vibrational structure has disappeared in the emission spectrum of PT1 in benzonitrile. This suggests that in benzonitrile PT1 undergoes a conformational and/or solvent relaxation which does not occur for peryleneimide model compounds. This could be either a rotation of the thiophene moiety into the plane of the ring of P or solvent relaxation related to the higher excited state dipole moment of PT1 compared to P and related model compounds. Both suggest excited state relaxation of PT1 leading to increased conjugation and/or an increased excited state dipole moment. Contrary to PT1 in toluene, PT2 emits at 1300 cm−1 lower energy than polyphenylene model compounds as m-C1P1, while showing no fine structure. This indicates that for PT2 the excited state relaxation already occurs in toluene. Furthermore for PT2 the red shift between toluene and benzonitrile is 2300 cm−1 with a fluorescence maximum at 697 nm. Moreover, in benzonitrile for PT2 a larger Stokes shift compared to PT1 is observed and the vibrational fine structure is lost in both the absorption and emission spectrum. As a preliminary indication the stationary results show the possibility of charge transfer upon excitation of the donor–acceptor system PT2 in more polar solvents.
=495 nm)
| Compound | P | m-C1P1 | PT1 | PT2 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent | ε a | λ em-max/nm | Φ F b | λ em-max/nm | Φ F b | λ em-max/nm | Φ F b | λ em-max/nm | Φ F b |
| a Dielectric constant, ε, is taken from ref. 39. b Fluorescence quantum yields have been determined using Rhodamine 101 and Cresyl violet perchlorate as ref. 40. | |||||||||
| Toluene | 2.43 | 537 | 1 | 555 | 1 | 557 | 1 | 600 | 0.47 |
| Ethyl acetate | 6.02 | 537 | 1 | — | — | 576 | 0.95 | 642 | 0.44 |
| THF | 7.52 | 537 | 1 | 565 | 1 | 584 | 0.96 | 647 | 0.44 |
| Benzonitrile | 26 | 553 | 0.9 | 580 | 0.9 | 605 | 0.90 | 697 | 0.18 |
An overview of the values of quantum yields of fluorescence (ΦF) of PT1 and PT2 in four different solvents of increasing polarity is given in Table 1. For m-C1P1, P and PT1, only a small decrease from 1 (toluene) to 0.9 (benzonitrile) is observed. A significant decrease in quantum yield of fluorescence is observed for PT2 with increasing polarity of the solvent from 0.47 (toluene) to 0.18 (benzonitrile).
While P shows a small shift of the emission maximum from 18600 cm−1 in toluene to 18000 cm−1 in benzonitrile, the corresponding change in PT2 is much larger (see Fig. A in the ESI†).
Single photon timing experiments
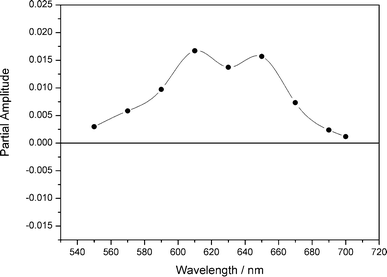
The fluorescence decay times of PT1 and PT2 were determined by SPT experiments in two solvents of low (toluene) and high (benzonitrile) polarity. For each compound the measurements were performed at different fluorescence detection wavelengths throughout the entire emission spectra. A typical plot of the fluorescence decays of PT1 and PT2 in benzonitrile detected at 600 nm is displayed in Fig. B in the ESI.† The fluorescence decay times obtained from global analysis are summarized in Table 2 while for PT2 the respective amplitudes are given in Fig. 2 and 3. | ||
| Fig. 2 Wavelength dependence of the partial amplitudes of the decay times of the PT2 in benzonitrile determined by SPT. <30 ps component (▲), 820 ps component (○), 1200 ps component (●). | ||
 | ||
| Fig. 3 Wavelength dependence of the 19 ps (●) component for PT2 in benzonitrile determined by femtosecond fluorescence upconversion. | ||
Global analysis of decays recorded at different emission wavelengths revealed a mono-exponential decay for PT1 in both solvents with a time constant of 4.28 ns (toluene) and 4.04 ns (benzonitrile) corresponding to a rate constant of 2.3 × 108 s−1 and 2.5 × 108 s−1, respectively. The same decay time constants have been found earlier for a series of polyphenylene dendrimers substituted with varying number of peryleneimide chromophores, in which this component is attributed to the fluorescence of the P.7,8 Combining the fluorescence decay time and fluorescence quantum yield of PT1 in toluene and benzonitrile yielded a fluorescence rate constant of 2.3 × 108 and 2.25 × 108 s−1, respectively. This is close to the fluorescence rate constant of P (2.3 × 108 s−1) indicating that both molecules have a similar transition dipole and hence also a similar HOMO and LUMO both in the Franck–Condon (FC) excited state (as revealed from the absorption spectra) and the relaxed excited state as revealed from the fluorescence data. In contrast, the global analysis of the fluorescence decays of PT2 (see Fig. B in the ESI†) required a triple-exponential decay in toluene and a quadruple-exponential decay in benzonitrile to obtain a good fit of the traces. The fluorescence decay parameters are listed in Table 2. A 4 ns component with an amplitude of less than 5% is due to an impurity in PT2. The relative amplitudes of the two relevant kinetic components of PT2 in toluene in function of detection wavelength are reported in Fig. 4. The fastest component (<30 ps) with positive amplitude at shorter wavelengths and negative amplitude at long detection wavelengths can be attributed to a relaxation (vide infra) of the electronically excited state of the PT2. The stationary emission spectrum must be attributed mainly to the state formed with this sub-30 ps time constant. The bathochromic shift of the steady-state emission spectra with increasing polarity suggests that this state is due to more extensive delocalization of the excited state possibly mixed in with charge separation. The <30 ps component can be attributed to the formation of a more planar excited state with more extensive conjugation between bis-thiophene and P. The 1.4 ns component can be attributed to fluorescence from the latter state. The fluorescence rate constant obtained for this relaxed conjugated state can be estimated to be 3.3 × 108 s−1, which is 30% larger than the fluorescent rate constant of P in spite of the red shift. The increase of the oscillator strength clearly excludes the formation of a twisted intramolecular charge transfer (TICT) state. After excitation the torsion angle between the thiophene moieties and/or the thiophene and P will decrease, leading to a more extensive delocalization of the HOMO and/or LUMO and the occurrence of a net charge transfer from the bisthiophene to the P moiety. This more extensive relaxation along a torsional and possibly a solvent coordinate explains the larger Stokes shift of PT2 compared with PT1 in toluene and the loss of fine structure in the emission spectrum compared to that of PT1 in toluene where the orbitals of the single thiophene moiety are energetically further away from those of P leading to less extensive electronic interaction.
 | ||
| Fig. 4 Wavelength dependence of the partial amplitudes of the decay times of PT2 in toluene determined by SPT. <30 ps component (▲), 1410 ps component (●). | ||
The amplitudes of the three relevant kinetic components of PT2 in benzonitrile are reported in Fig. 2. The fastest sub-30 ps kinetic component with a large positive amplitude at short wavelengths which becomes negative at longer wavelengths can be attributed to the formation of a charge transfer state characterized by the absorption spectra of the P-radical anion and the thiophene radical cation (vide infra) from a less polar precursor excited state formed upon excitation and emitting between 600 and 700 nm. Due to the increased polarity of benzonitrile, this delocalized state will be characterized by a more pronounced charge separation compared to toluene. The amplitude of the two components of 1.2 ns and 820 ps increases with the wavelength (Fig. 2). The amplitudes of the 1.2 and 0.82 ns components have a similar trend which is opposite to that of the fast decaying component. The observed emission with maximum at 697 nm is mainly due to those two components of the decay and hence to the charge separated state. The two different components, (820 and 1200 ps) are attributed to subpopulations in the sample having different torsional angles (CTA and CTB) between the thiophene rings or between the bis-thiophene moiety and P. The possible occurrence of different conformations and/or orientation based on theoretical calculations, especially regarding the torsional (inter-ring) barriers and the enthalpy difference between trans- and cis-conformers and the energy of the excited states has been discussed.31,32
As the emission of PT2 in benzonitrile occurs mainly from the charge transfer state, its fluorescence rate constant can be estimated to be of the order of 2 × 108 s−1 from the observed fluorescence quantum yield and the average decay rate constant, assuming that this species is formed with nearly 100% efficiency. Although this value is 25% smaller than the fluorescent rate constant of P, it will be characterized by an oscillator strength which is 40% larger than that of P if one considers the red shift of the emission maximum.
Femtosecond fluorescence upconversion
To be able to elucidate fast kinetic components which occur on a time scale less than 30 ps and to validate the presence of the short picosecond (<30 ps) decay component suggested by the SPT experiments, femtosecond fluorescence upconversion measurements were performed. The fluorescence decays have been recorded in two time windows of 30 and 300 ps for PT1 and PT2 in benzonitrile. Representative fluorescence decay traces of PT2 in benzonitrile measured in a 30 ps time window are depicted in Fig. C in the ESI.† By global analysis of PT1 and PT2 two short kinetic components with time constants of (0.5–1.5) and 4.2 ps were found. Besides these ps components also ns components were observed which could not be resolved with the femtosecond fluorescence upconversion setup. In PT2 an extra component not observed in PT1, with a time constant of 19 ps, was found. The decay time of the long nanosecond component(s) obtained from SPT experiments was kept fixed during the global analysis.Two fast components of (0.5–1.5) and 4.2 ps have been observed both in PT1 and PT2. These kinetic components occurring on those short time scales can be attributed to intramolecular vibrational redistribution (IVR)33 and vibrational/solvent relaxation (VR)34 processes. Kinetic components occurring on similar time scales were observed for other P substituted compounds.10
An additional (19 ps) kinetic component (Fig. 3) is observed in PT2 compared to PT1 which suggests that it can be attributed to the formation of the charge transfer state. The 19 ps component was also recovered in femtosecond transient absorption experiments (vide infra).
Femtosecond monochromatic and polychromatic transient absorption experiments
To further identify the properties of the states of PT2 and PT1, additional independent transient absorption experiments were performed. The time-resolved transient absorption spectra were recorded for PT1 and PT2 in three time windows of 50, 420 and 1400 ps at 512 delay positions.To obtain the different kinetic components, all monochromatic transient absorption traces were globally analyzed over the three time windows (Fig. D in the ESI†).
The three-dimensional plot of the transient absorption data for PT1 in benzonitrile in a 1400 ps time window is shown in Fig. 5A. Two features are present immediately after excitation and decay on nanosecond time scale. While the negative signal around 520 nm appears due to a ground-state bleaching and stimulated emission, the positive signal situated at 650 nm can be attributed to the S1–Sn absorption of the peryleneimide chromophore.14
 | ||
| Fig. 5 Three-dimensional plot of the transient absorption spectra of the model compound PT1 in benzonitrile recorded in 1400 ps (A) and 50 ps time windows (B). | ||
In the transient absorption spectrum of PT1 in benzonitrile, a small hypsochromic shift from 655 to 650 nm is observed in the S1–Sn absorption band in the first few picoseconds as depicted in Fig. 5B. This small shift in the S1–Sn absorption band of the model compound PT1, where the stationary data do not indicate the formation of a polar state, is a consequence of a vibrational and/or solvent relaxation process in the electronically excited state of the peryleneimide moiety as also observed previously in other peryleneimide substituted compounds.14,15
Global analysis of the transient absorption decays recorded for the model compound PT1 in benzonitrile revealed three kinetic components with time constants of (0.5–1.5) ps, 4.2 ps and 4 ns. The two fast components have also been found by femtosecond fluorescence upconversion (vide supra). The 4 ns component can again be attributed to the excited state decay related to peryleneimide fluorescence. The wavelength dependence of the 4.2 ps and 4 ns components are shown in Fig. 6. The 4 ns bands resemble the combination of ground state depletion, stimulated emission and S1–Sn absorption found for other peryleneimide substituted compounds.16
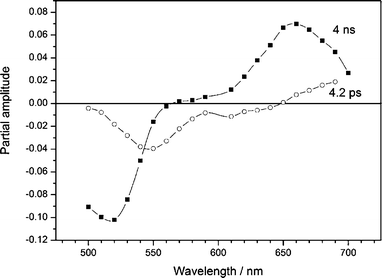 | ||
| Fig. 6 Wavelength dependence of the partial amplitudes of the decay times for PT1 in benzonitrile determined by femtosecond transient absorption. 4.2 ps component (○), 4 ns component (■). | ||
For PT2 in benzonitrile a different behaviour is observed as shown in Fig. 7A. The negative band with maximum at 530 nm can also here be attributed to ground state bleaching and stimulated emission from the unrelaxed charge transfer state. The positive absorption band with maxima around 700 nm can be attributed to the S1–Sn absorption band of the conjugated thiophene–P system. The red shift is probably due to a more extended conjugation compared to PT1. Furthermore, an additional shoulder between 570 and 640 nm which is not present in PT2 in toluene (vide infra) or in the model compound PT1 in benzonitrile is observed. This shoulder is formed on a 19 ps time scale as indicated by both transient absorption and fluorescence upconversion experiments and starts to grow immediately after appearance of the conjugated thiophene–P S1–Sn absorption band. This is shown more clearly in Fig. 7B which displays data from the same sample, however recorded in a shorter time window of 50 ps. This shoulder between 570 and 640 nm consists of the peryleneimide radical anion absorption and bis-thiophene radical cation absorption for which maxima at 620 and 580 nm, respectively, were reported previously.12,15,16,35,36 Both bands are however shifted to the red due to the more extended conjugation in PT2 compared to P and related compounds. These bands overlap partially with the S1–Sn absorption band of the conjugated thiophene–P system. This overlap can explain why the absorption with maxima around 700 nm does not decay with the same rate as the formation of the radical cation and anion absorption bands mentioned earlier.
 | ||
| Fig. 7 Three-dimensional plot of the transient absorption spectra of PT2 in benzonitrile recorded in 1400 ps (A) and 50 ps time windows (B). | ||
For PT2 in benzonitrile four decay time constants were obtained from the global analysis. In the largest time window of 1400 ps used for recording the transient absorption traces it is not possible to discriminate between the 820 and 1200 ps components found in the SPT measurements. As a consequence instead of these two kinetic components found for PT2 by SPT, an average component of 1150 ps has been found by global analysis of the femtosecond transient absorption traces (Fig. 8, ultrafast component (0.5–1.5) ps not shown). One can observe that while the 1150 ps component shows a negative/positive amplitude behaviour corresponding to ground state depletion and transient absorption of the radical ions, the amplitude of the 19 ps component has a negative amplitude over the entire detection wavelength region. The negative amplitude of the 19 ps component at wavelengths where no (induced) emission of originally populated less polar excited state occurs (see Fig. 3) indicates that this component must be attributed to transient absorption of a species formed with a 19 ps time constant from a precursor state and to stimulated emission of the precursor state. The 19 ps component is not observed in PT1 and can therefore be attributed to the formation of the charge transfer state. In the wavelength region where the 19 ps component displays a negative amplitude, the 1150 ps component has a positive amplitude. This indicates that in this wavelength region the 1150 ps component can be attributed to transient absorption of a species decaying with a 1150 ps time constant and of which the transient absorption is more important than the induced emission. This component can therefore be attributed to the decay of the polar charge transfer state, which is formed with a 19 ps time constant. The absorption of this species between 550 and 600 nm, slightly bathochromic to the P-radical anion and the bithiophene radical cation indicates that this species resembles a radical ion-pair of P and bis-thiophene, but with more extended conjugation.
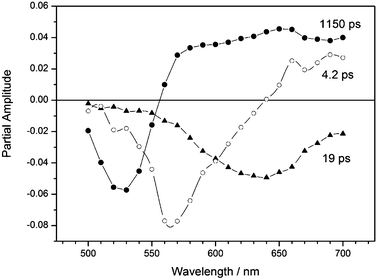 | ||
| Fig. 8 Wavelength dependence of the partial amplitudes of the decay times for PT2 in benzonitrile determined by femtosecond transient absorption. 4.2 ps component (○), 19 ps component (▲) and 1150 ps component (●). | ||
The short component of 4.2 ps found for PT2 and PT1 in benzonitrile has the same amplitude behaviour as shown in Fig. 8 and 6, respectively. For both compounds the 4.2 ps component has a negative amplitude at shorter wavelengths and a positive amplitude at longer wavelengths. The same features are observed in the fluorescence upconversion experiments (data not shown), however, the signs are of course reversed. This shorter component can be attributed to vibrational/solvent relaxation processes as mentioned earlier. The same amplitude behaviour is observed for the 9 ps component found for PT2 in toluene as shown in Fig. 9.
 | ||
| Fig. 9 Three-dimensional plot of the transient absorption spectra of PT2 in toluene recorded in a 50 ps time window (A). Wavelength dependence of the partial amplitudes of the vibrational/solvent relaxation component with a decay time of 9 ps of PT2 in toluene as determined by fs-TA (B). | ||
The three-dimensional plot of the transient absorption results obtained for PT2 in the less polar solvent toluene (Fig. 9) is characterized by a negative band with a maximum at 530 nm. This band which appears immediately after excitation can also be attributed to ground state bleaching and stimulated emission from the vibrationally relaxed state. The excited state absorption with a maximum around 700 nm which appears immediately after excitation, can be attributed to the S1–Sn absorption band of the conjugated state of bis-thiophene-P. The S1–Sn absorption band of PT2 is shifted to the red compared to PT1 because of the more extended conjugation in PT2 which was also suggested by the red shift of the emission spectrum in toluene. In the 550–640 nm wavelength region, the negative band is due to stimulated emission.
Global analysis of the transients reveals three decay components: a component which decays on a ns time scale, a component with a decay time of 9 ps and a component with a much shorter decay time which is wavelength dependent. The amplitude of the 9 ps component is negative (Fig. 9) at detection wavelengths up to 640 nm and becomes positive at longer wavelengths. This corresponds to the features of the amplitude and to the time scale of the vibrational/solvent relaxation component observed for other P-substituted compounds in toluene.9 The negative amplitudes between 520 and 640 nm indicate that at those wavelengths the emission of the unrelaxed excited state prevails while at wavelengths above 640 nm the emission of vibrationally relaxed state that is populated with a 9 ps time constant process prevails.
The free energy change of charge separation (ΔGcs), to form the polar charge transfer state, was calculated for PT2 in toluene and benzonitrile using the Rehm–Weller equation.37 With the values of oxidation potential of the bi-thiophene (0.94 eV)24 and the redox potential of the P chromophore (−1.42 eV)24 at an average distance of 8 Å between the two moieties the resulting ΔGcss were −0.31 kcal mol−1 in toluene and −9.7 kcal mol−1 in benzonitrile.
The resulting kinetic scheme of the physical processes which occur in benzonitrile and toluene upon photoexcitation of PT2 is shown in Scheme 2. The more extensive charge transfer observed for PT2 compared to PT1 can be related to the lower ionization potential of bis-thiophene compared to mono-thiophene.
 | ||
| Scheme 2 The kinetic scheme of the photophysical processes with the corresponding time constants occurring in PT2 in toluene and benzonitrile. FC=vibrationally unrelaxed state; RES=vibrationally relaxed state; CTA,B=charge transfer state with different conformations (torsion angle). | ||
Conclusions
We have presented data indicating that for a peryleneimide substituted bis-thiophene electron transfer occurs from the bis-thiophene (electron donor) to the peryleneimide (electron acceptor). Performing femtosecond transient absorption experiments, a broad new absorption band—consisting of the radical cation of the bis-thiophene and radical anion of the peryleneimide—was revealed in PT2 in benzonitrile which was not present in the model compound PT1 or in PT2 in toluene. These bands are formed within 19 ps and decay on a nanosecond time scale as demonstrated by transient absorption and time resolved fluorescence experiments. The radical anion and cation absorption bands mentioned above are not observed in the less polar solvent toluene indicating that no charge transfer state is formed in toluene. In benzonitrile, the observation of two CT states can probably be related to two conformations. The interconversion between these two conformations will require rotation of a thiophene moiety around the bond with a partial double bound character and will hence not occur on a time scale of 1 ns.Acknowledgements
Marc Lor thanks the Vlaams instituut voor de bevordering van het wetenschappelijk en technologisch onderzoek (IWT). The authors gratefully acknowledge the FWO, de ‘Nationale Loterij’, the Flemish Ministry of Education through GOA 1/96 and GOA 2001/2, the Bundesministerium for Education and Research of the Federal Republic of Germany, a Max-Planck Research Award and the support of Federal Science Policy (Belgium) through IAP-V-03.References
- R. S. Becker, J. S. De Melo, A. L. Macanita and F. Elisei, Comprehensive investigation of the solution photophysics and theoretical aspects of oligothiophenes of 1-7 rings, Pure Appl. Chem., 1995, 67, 9–16 CrossRef CAS.
- H. S. Majumdar, A. Bolognesi and A. J. Pal, Memory applications of a thiophene-based conjugated polymer: capacitance measurements, J. Phys. D: Appl. Phys., 2003, 36, 211–215 CrossRef CAS.
- A. Dodabalapur, L. Torsi and H. E. Katz, Organic transistors: two-dimensional transport and improved electrical characteristics, Science, 1995, 268, 270–271 CrossRef CAS.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Plastic Solar Cells, Adv. Funct. Mater., 2001, 11, 15–26 CrossRef CAS.
- M. Granström, K. Petrisch, A. C. Arias, A. Lux, M. R. Andersson and R. H. Friend, Laminated fabrication of polymeric photovoltaic diodes, Nature, 1998, 395, 257–260 CrossRef CAS.
- A. Andronov, P. R. L. Malenfant and J. M. F. Fréchet, Synthesis and steady-state photophysical properties of dye-labeled dendrimers having novel olgothiophene cores: a comparative study, Chem. Mater., 2000, 12, 1463–1472 CrossRef CAS.
- M. Maus, S. Mitra, M. Lor, J. Hofkens, T. Weil, A. Herrmann, K. Müllen and F. C. De Schryver, Intramolecular energy hopping in polyphenylene dendrimers with an increasing number of peryleneimide chromophores, J. Phys. Chem. A, 2001, 105, 3961–3966 CAS.
- M. Maus, R. De, M. Lor, T. Weil, S. Mitra, U.-M. Wiesler, A. Herrmann, J. Hofkens, T. Vosh, K. Müllen and F. C. De Schryver, Intramolecular energy hopping and energy trapping in polyphenylene dendrimers with multiple peryleneimide donor chromophores and a terryleneimide acceptor trap chromophore, J. Am. Chem. Soc., 2001, 123, 7668–7676 CrossRef CAS.
- G. De Belder, G. Schweitzer, S. Jordens, M. Lor, M. Sivaprasad, J. Hofkens, S. De Feyter, M. Van der Auweraer, A. Herrmann, T. Weil, K. Müllen and F. C. De Schryver, Singlet–Singlet annihilation in multichromophoric peryleneimide dendrimers, determined by fluorescence upconversion, Chem. Phys. Chem., 2001, 1, 49–55 CrossRef.
- G. De Belder, S. Jordens, M. Lor, G. Schweitzer, R. De, T. Weil, A. Herrmann, U. K. Wiesler, K. Müllen and F. C. De Schryver, Femtosecond fluorescence upconversion study of rigid dendrimers containing peryleneimide chromophores at the rim, J. Photochem. Photobiol. A: Chem., 2001, 145, 61–70 CrossRef CAS.
- M. Lor, R. De, S. Jordens, G. De Belder, G. Schweitzer, M. Cotlet, J. Hofkens, T. Weil, A. Herrmann, K. Müllen, M. Van der Auweraer and F. C. De Schryver, Generation-Dependent energy dissipation in rigid dendrimers studied by femtosecond to nanosecond time-resolved fluorescence spectroscopy, J. Phys. Chem., 2002, 106, 2083–2090 Search PubMed.
- M. Lor, J. Thielemans, L. Viaene, M. Cotlet, J. Hofkens, T. Weil, C. Hampel, K. Müllen, J. W. Verhoeven, M. Van der Auweraer and F. C. De Schryver, Photoinduced electron transfer in a rigid first generation triphenylamine core dendrimer substituted with a peryleneimide acceptor, J. Am. Chem. Soc., 2002, 124, 9918–9925 CrossRef CAS.
- G. Schweitzer, R. Gronheid, S. Jordens, M. Lor, G. De Belder, T. Weil, E. Reuter, K. Müllen and F. C. De Schryver, Intramolecular directional energy transfer processes in dendrimers containing perylene and terrylene chromophores, J. Phys. Chem. A, 2003, 107, 3199–3207 CrossRef CAS.
- S. Jordens, G. De Belder, M. Lor, M. Van der Auweraer, T. Weil, A. Herrmann, U.-W. Wiesler, K. Müllen and F. C. De Schryver, Generation dependent singlet-singlet annihilation within multichromophoric dendrimers studied by polychromatic transient absorption, Photochem. Photobiol. Sci., 2003, 2, 1118–1124 RSC.
- M. Lor, S. Jordens, G. De Belder, G. Schweitzer, E. Fron, L. Viaene, M. Cotlet, T. Weil, K. Müllen, J. W. Verhoeven, M. Van der Auweraer and F. C. De Schryver, Direct proof of electron transfer in a rigid first generation triphenyl amine core dendrimer substituted with a peryleneimide acceptor, Photochem. Photobiol. Sci., 2003, 2, 501–510 RSC.
- M. Lor, L. Viaene, R. Pilot, E. Fron, S. Jordens, G. Schweitzer, T. Weil, K. Müllen, J. W. Verhoeven, M. Van der Auweraer and F. C. De Schryver, Photophysical study of electron transfer and energy hopping processes in first generation mono- and multichromophoric triphenylamine core dendrimers, J. Phys. Chem. B, 2004, 108, 10721–10731 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures, Chem. Rev., 2003, 103, 3899–4031 CrossRef.
- M. Jurczok, P. Plaza, W. Rettig and M. M. Martin, Ultrafast electron transfer in acceptor substituted bianthryl derivatives, Chem. Phys., 2000, 256, 137–149 CrossRef CAS.
- M. Mac, P. Kwiatkowski and U. Pischel, Temperature dependence of bianthryl dual fluorescence, Chem. Phys. Lett., 2002, 357, 440–449 CrossRef CAS.
- A. Onkelinx, F. C. De Schryver, L. Viaene, M. Van der Auweraer, K. Iwai, M. Yamamoto, M. Ichikawa, H. Masuhara, M. Maus and W. Rettig, Radiative depopulation of the excited intramolecular charge-transfer state of 9-(4-(N,N-Dimethylamino)phenyl)phenanthrene, J. Am. Chem. Soc., 1996, 118, 2892–2902 CrossRef CAS.
- J. Herbich and A. Kapturkiewicz, Radiative and radiationless depopulation of the excited intramolecular charge transfer states: aryl derivatives of aromatic amines, Chem. Phys., 1991, 158, 143–153 CrossRef CAS.
- K. Tominaga, G. C. Walker, T. J. Kang, P. F. Barbara and T. Fonseca, Reaction rates in the phenomenological adiabatic excited-state electron-transfer theory, J. Phys. Chem., 1991, 95, 10485–10492 CrossRef CAS.
- W. Verbouwe, L. Viaene, M. Van der Auweraer, F. C. De Schryver, H. Masuhara, R. Pansu and J. Faure, Photoinduced intramolecular charge transfer in diphenylamino-substituted triphenylbenzene, biphenyl and fluorene, J. Phys. Chem. A, 1997, 101, 8157–8165 CrossRef CAS.
- J. Cremer, N. Pschierer, K. Mullen and P. Bauerle, in preparation.
- M. Maus, E. Rousseau, M. Cotlet, G. Schweitzer, J. Hofkens, M. Van der Auweraer and F. C. De Schryver, New picosecond laser system for easy tunability over the whole ultraviolet/visible/near infrared wavelength range based on flexible harmonic generation and optical parametric oscillation, Rev. Sci. Instrum., 2001, 72, 36–40 CrossRef CAS.
- TRFA Global Analysis Program, a program developed in cooperation between the Management of Technology Institute, Belarussian State University, Minsk, and the Division of Photochemistry and Spectroscopy, University of Leuven, ver. 1.0, 2000–2001 Search PubMed.
- D. V. O. Connor and D. Philips, Time-Correlated Single Photon Counting, Academic Press, London, 1984 Search PubMed.
- Y. Karni, S. Jordens, G. De Belder, G. Schweitzer, J. Hofkens, T. Gensch, M. Maus, F. C. De Schryver, A. Herrmann and K. Mullen, Intramolecular evolution from a locally excited state to an excimer-like state in a multichromophoric dendrimer evidenced by a femtosecond fluorescence upconversion study, Chem. Phys. Lett., 1999, 310, 73–78 CrossRef CAS.
- G. Schweitzer, L. Xu, B. Craig and F. C. De Schryver, A double OPA femtosecond laser system for transient absorption spectroscopy, Opt. Commun., 1997, 142, 283–288 CrossRef CAS.
- G. De Belder, PhD Thesis, Katholieke Universiteit Leuven, Belgium, 2001.
- R. S. Becker, J. S. De Melo, A. L. Macanita and F. Elisei, Comprehensive evaluation of the absorption, photophysical, energy transfer, structural and theoretical properties of α-oligothiophenes with one to seven rings, J. Phys. Chem., 1996, 100, 18683–18695 CrossRef CAS.
- G. Lanzani, G. Cerullo, S. Stagira and S. De Silvestri, Excited state dynamics of oligothiophenes studied by transient pump–probe spectroscopy, J. Photochem. Photobiol. A: Chem., 2001, 144, 16–19 CrossRef.
- T. Nakabayashi, H. Okamoto and M. Tasumi, Probe-wavelength dependence of picosecond time-resolved anti-stokes Raman spectrum of canthaxanthin: determination of energy states of vibrationally excited molecules generated via internal conversion from the lowest excited singlet state, J. Phys. Chem. A, 1997, 101, 3494–3500 CrossRef CAS.
- P. K. McCarty and G. J. Blanchard, An experimental examination of the competition between polar coupling and local organization in determining vibrational population relaxation, J. Phys. Chem., 1996, 100, 14592–14597 CrossRef.
- C. H. Evans and J. C. Scaiano, Photochemical generation of radical cations from α-terthienyl and related thiophenes: kinetic behavior and magnetic field effects on radical-ion pairs in micellar solution, J. Am. Chem. Soc., 1990, 112, 2694–2701 CrossRef CAS.
- W. Paa, J.-P. Yang and S. Rentsch, Ultrafast intersystem crossing in thiophene oligomers investigated by fs-pump–probe spectroscopy, Synth. Met., 2001, 119, 525–526 CrossRef CAS.
- D. Rehm and A. Weller, Kinetics of fluorescence quenching by electron and H-atom transfer, Isr. J. Chem., 1970, 8, 259–271 CAS.
- J. Hofkens, L. Latterini, G. De Belder, T. Gensch, M. Maus, T. Vosch, Y. Karni, G. Schweitzer, F. C. De Schryver, A. Hermann and K. Müllen, Photophysical study of a multi-chromophoric dendrimer by time-resolved fluorescence and femtosecond transient absorption spectroscopy, Chem. Phys. Lett., 1999, 304, 1–9 CrossRef CAS.
- CRC Handbook of Physics and Chemistry, ed. D. R. Lide, CRC Press, Boca Raton, FL, 73rd edn., 1992 Search PubMed.
- D. F. Eaton, Handbook of Organic Photochemistry, ed. J. C. Scaiano, CRC Press, Boca Raton, FL, 1989, vol. 1, p. 231 Search PubMed.
Footnotes |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Fig. A–D. See http://www.rsc.org/suppdata/pp/b4/b409346c/ |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |