Intramolecular electron transfer in diastereomeric naphthalene–amine dyads: a fluorescence and laser flash photolysis study†‡
Sergio
Abad
a,
Uwe
Pischel
*b and
Miguel A.
Miranda
*a
aInstituto de Tecnología Química, UPV-CSIC, Universidad Politécnica de Valencia, Av. de los Naranjos s/n, E-46022, Valencia, Spain. E-mail: mmiranda@qim.upv.es; Fax: +34 96 387 78 09; Tel: +34 96 387 78 07
bCEQUP/Departamento de Química, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre, 4169-007, Porto, Portugal. E-mail: upischel@fc.up.pt; Fax: +351 22 608 29 59; Tel: +351 22 608 28 85
First published on 4th November 2004
Abstract
Two dyads containing a naphthalene-like chromophore linked to a pyrrolidine-derived moiety, namely (S,S)- and (R,S)-NPX–PYR, have been synthesised by esterification of (S)- or (R)-naproxen (NPX) with (S)-N-methyl-2-pyrrolidinemethanol (PYR) and submitted to photophysical studies (steady-state and time-resolved fluorescence, as well as laser flash photolysis). The emission spectra of the dyads in acetonitrile were characterised by a typical band centred at 350 nm, identical to that of the reference compound (S)-NPX. However the intensities were clearly different, revealing a significant intramolecular quenching in the dyads, as well as a remarkable stereodifferentiation (factor of 1.6). Accordingly, the fluorescence lifetimes of the two dyads were different from each other and markedly shorter than that of (S)-NPX. The quenching mechanism is intramolecular electron transfer, that is thermodynamically favoured. Exciplex formation, that is nearly thermoneutral, does not compete efficiently. The electron transfer rate constants for (S,S)- and (R,S)-(NPX–PYR) were 1.8 × 108 and 2.8 × 108 s−1, respectively. By contrast, no significant intramolecular quenching was observed for the excited triplet states (λmax = 440 nm), generated by laser flash photolysis; this is in agreement with the fact that intramolecular electron transfer is thermodynamically disfavoured, due to the lower energy of excited triplets.
Introduction
Excited state interaction between aromatic chromophores and electron-donating amines provides a classical example of photoinduced electron transfer reactions.1–3 Several of its aspects, including the influence of factors like solvent polarity,4 electronic structure of the electron donor5 and excited state energy,6 are connected with the driving force of electron transfer by the well-known Rehm–Weller equation.1,7 Less straightforward predictable is the influence of geometrical parameters, like steric hindrance and chiral discrimination.8–12The investigation of stereoselective photochemical processes has become an attractive topic in recent years.11–13 As regards the interaction of excited aromatics with electron donors, Irie and co-workers have investigated the intermolecular quenching of (R)-(−)-1,1’-binaphthyl by chiral benzylamines.14 In non-polar solvents like n-hexane, the enantiodifferentiations observed for photophysical behaviour of these systems (factors of up to 7.9) are among the highest ones reported for excited state quenching. However, in polar acetonitrile no stereodifferentiation was noted. The authors argued that in polar media electron transfer becomes dominant, while in non-polar solvents exciplex formation is the major quenching pathway. Intuitively, exciplex formation should be subjected to geometrical influences, due to closer interaction between donor and acceptor via orbital overlap. On the other hand, it is known that electron transfer can happen over longer distances, without the need to approach donor and acceptor within van der Waals distance. Other related work included binaphthol and its derivatives, whose fluorescence was quenched by chiral amines, with enantiodifferentiation factors of maximal 1.16.15 Also 2,2’-dimethyl-1,1’-bianthryl has been used as chiral aromatic chromophore for exciplex-induced quenching by chiral N,N-dimethylaniline derivatives.16 Again, the largest enantiodifferentiation factors were around 1.15. Related enantiomeric binaphthyl derivatives were successfully used for the development of chiral chemosensors based on fluorescence quenching by chiral amines.17
In our recent research we became interested in the behaviour of classical chromophores like naphthalene or benzophenone in diastereomeric dyads, modified with potential quenchers acting via electron, hydrogen or energy transfer. These quenchers were selected to enable the investigation of the impact of chiral information on the dynamics of basic photochemical mechanisms.18–26 In this context, we have now synthesised two novel asymmetric dyads [(S,S)- and (R,S)-NPX–PYR] composed of a naphthalene derivative 2-(S)-(+)- and 2-(R)-(−)-6-methoxy-2-naphthylpropionic acid, naproxen, NPX], and (S)-N-methyl-2-pyrrolidinemethanol (PYR) (cf.Chart 1).
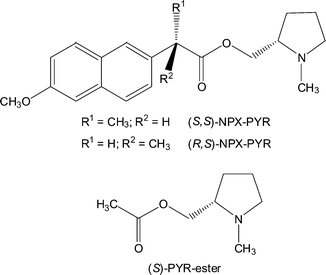 | ||
| Chart 1 | ||
The pyrrolidine moiety is a potential electron donor, which has allowed us to investigate chiral discrimination in electron-transfer induced intramolecular quenching. A remarkable diastereodifferentiation of the intramolecular electron transfer rate constant for the excited singlet state of the naphthalene chromophore has been observed. Strikingly, it has been suggested by several authors that exciplex formation is a precondition for the observation of stereoselectivity in electron donor–acceptor interactions.12 As will be shown below, electron transfer is the main quenching pathway in our dyads; nevertheless, diastereodifferentiation is observed. Part of these observations were the subject of a preliminary communication;23 now we wish to report our results in full, including a complete time-resolved and steady-state fluorescence study. Besides, the compounds have been submitted to laser flash photolysis, which is directly related to the observation of the long-lived excited triplet state of naproxen and its radical cation, formed by photoionisation.27,28 The triplet state has been shown to be unreactive towards electron transfer from the pyrrolidine unit, owing to a highly unfavourable thermodynamics. However, the radical cation of naproxen can be reduced by a ground-state electron transfer from the pyrrolidine unit.
Experimental
Materials
All chemicals for the synthesis of the compounds were purchased from Aldrich with the exception of (R)-(−)-6-methoxy-2-naphthylpropionic acid [(R)-naproxen, NPX], which was from ChiroTecnology, and were used as received. Acetonitrile, used as solvent for photophysical experiments and chromatography, was of HPLC quality from Merck. LiChrosorb RP-18 reverse phase (7 µm, 250 × 25 mm) from Merck was used as column for purification of the dyads.Spectroscopic measurements
UV/Vis absorption measurements were performed on a Shimadzu UV-2101PC spectrometer. Fluorescence spectra were recorded on a FS900 fluorimeter, and lifetimes were measured with a FL900 setup, both from Edinburgh Instruments. Lifetime measurements are based on single-photon-counting using a hydrogen flashlamp (1.5 ns pulse width) as excitation source. The kinetic traces were fitted by monoexponential decay functions using a re-convolution procedure to separate from the lamp pulse profile.Laser flash photolysis studies in the kinetic mode were carried out with a pulsed XeCl excimer laser (λexc = 308 nm, ca. 17 ns pulse width, ≤ 100 mJ per pulse). As detecting light source a pulsed Lo255 Oriel xenon lamp was used. The observation wavelength was selected with a 77200 Oriel monochromator and the signal amplified by an Oriel photomultiplier tube (PMT) system made up of a 77348 side-on PMT tube, 70680 PMT housing and a 70705 PMT power supply. The signal was registered with a TDS-640A Tektronix oscilloscope and subsequently transferred to a personal computer.
All measurements were either performed at room temperature (23 °C) with air-equillibrated acetonitrile solutions (fluorescence) or in nitrogen-purged solutions for the removal of oxygen (laser flash photolysis). Cuvettes were of 1 cm optical path length, and the absorbance at excitation wavelength was kept at ca. 0.2, in order to avoid non-linear effects like self-absorption or inhomogeneous transient distribution.
Synthesis of the dyads and the acetate of (S)-N-methyl-2-pyrrolidinemethanol
The synthesis of the dyads was performed by converting 0.9 mmol (S)-(+)- and (R)-(−)-6-methoxy-2-naphthylpropionic acid with 6.9 mmol thionyl chloride (in 10 mL dry dichloromethane) into the activated acyl chloride. The obtained crude acyl chloride was used without further purification and reacted with 1.0 mmol (S)-N-methyl-2-pyrrolidinemethanol in the presence of 1.6 mmol triethylamine as acid scavenger in 10 mL dichloromethane. After standard work-up, the compounds were purified by preparative reverse-phase HPLC using acetonitrile as eluent.The acetate of (S)-N-methyl-2-pyrrolidinemethanol was synthesised by reaction of 1.1 mmol (S)-N-methyl-2-pyrrolidinemethanol with a slight excess of acetyl chloride (1.3 mmol) in 10 mL dry dichloromethane. Triethylamine (1.5 mmol) was used as acid scavenger. After standard work-up, the product was purified by vacuum distillation. All compounds were analytically characterised by 1H and 13C NMR spectroscopy, as well as elemental analysis and/or HRMS.
Results and discussion
Absorption spectra
The absorption spectra of both diastereomeric compounds (S,S)-NPX–PYR and (R,S)-NPX–PYR in acetonitrile are shown in Fig. 1. | ||
| Fig. 1 Absorption spectra of (S,S)-NPX–PYR (solid line) and (R,S)-NPX–PYR (dotted line) in acetonitrile. The latter has been slightly shifted upwards to avoid complete overlap. | ||
The dyads display fine-structured bands, akin to the parent naproxen (NPX).27,28 These absorptions are ascribed to π,π*-type transitions and are not altered compared to naproxen, neither in oscillator strength nor in spectral position. This indicates the absence of important ground-state interactions between naphthalene chromophore and amine. Furthermore, no significant differences in the spectra of both diastereomeric dyads were noted, i.e., chiral information has neither significant impact on the spectral distribution nor oscillator strength of the π,π*-transitions.
Fluorescence measurements
The fluorescence spectra of (S,S)-NPX–PYR, (R,S)-NPX–PYR, and (S)-NPX in acetonitrile are characterised by a typical emission band centred at 350 nm.27,28 The excitation spectra (not shown) present the same bands as the absorption spectra, which unambiguously confirms that the emission originates from the naphthalene chromophore. Steady-state fluorescence spectra of the dyads obtained upon excitation at 280 nm, with optically matched solutions in acetonitrile, revealed an important quenching compared to parent (S)-NPX (cf.Fig. 2). | ||
Fig. 2 Fluorescence emission spectra (λexc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =280 nm) of optically matched solutions of (a) (S)-NPX, (b) (S,S)-NPX–PYR and (c) (R,S)-NPX–PYR in aerated acetonitrile. The inset shows the exciplex emission bands of (b) (S,S)-NPX–PYR and (c) (R,S)-NPX–PYR in acetonitrile. =280 nm) of optically matched solutions of (a) (S)-NPX, (b) (S,S)-NPX–PYR and (c) (R,S)-NPX–PYR in aerated acetonitrile. The inset shows the exciplex emission bands of (b) (S,S)-NPX–PYR and (c) (R,S)-NPX–PYR in acetonitrile. | ||
This quenching can be explained by exciplex formation or photoinduced electron transfer, which are typical photoinduced reactions between excited naphthalene chromophores and electron donors like amines.14,29–35 The two pathways should be considered as main quenching mechanisms. Other possibilities like hydrogen transfer or energy transfer can be excluded. It is generally accepted that π,π*-excited states, like the present one, are not very efficient hydrogen atom acceptors. Further, energy transfer should be an up-hill process, since the excited state energy of pyrrolidine can be estimated to lie above that of singlet-excited naproxen, based on the generally strongly blue-shifted absorption spectra of saturated amines. In general, exciplex formation is favoured in non-polar solvents, while in polar media photoinduced electron transfer dominates.3 Hence, in acetonitrile as reaction medium, electron transfer would be expected, if thermodynamically allowed. The energetics of radical ion pair formation resulting from photoinduced electron transfer can be calculated with eqn. (1).7
 | (1) |
The amount of quenching, i.e., the quantum yield of electron transfer Φet, is significantly different for the two diastereomers, which must be related to a stereodifferentiation in the electron transfer process (cf.Fig. 2). The (R,S) combination shows a higher efficiency than the (S,S) dyad, Φet = 0.66 and 0.52, respectively. The same trend is observed in time-resolved measurements shown in Fig. 3, where (S)-NPX has a lifetime of τ0 = 7.4 ns in aerated acetonitrile, while the singlet lifetimes of the dyads are significantly shorter.
 | ||
| Fig. 3 Fluorescence decay traces (λexc=280 nm, λobs=350 nm) of (a) (S)-NPX, (b) (S,S)-NPX–PYR and (c) (R,S)-NPX–PYR in aerated acetonitrile. | ||
As the τf value of (R,S)-NPX–PYR is smaller than that obtained for (S,S)-NPX–PYR (2.3 vs. 3.0 ns); this leads to the same conclusion of stereodifferentiation in the observed electron transfer, with the (R,S) dyad reacting more efficiently. Quantum yields for electron transfer as obtained from the lifetime data are virtually the same as from the steady-state experiment, i.e., 0.68 and 0.59 for (R,S)- and (S,S)-NPX–PYR, respectively. The unimolecular rate constant for photoinduced intramolecular electron transfer38 in the dyads can be calculated with eqn. (2), resulting in ket(R,S) = 2.8 × 108 s−1 and ket(S,S) = 1.8 × 108 s−1. These data indicate a factor of ca. 1.6 for the diastereodifferentiation [ket(R,S)/ ket(S,S)] between both asymmetric dyads.
 | (2) |
 | (3) |
The reason for the sensitivity of the electron transfer rate constant to the chiral information in the present system must be sought in the steric hindrance associated with the necessary approach of the donor and acceptor moiety.8–10 As the reaction is only moderately exergonic, such a steric effect could have a strong impact on the actual height of the activation barrier, as can be predicted from the reactivity–selectivity principle.
Finally, an intermolecular control experiment was performed. The fluorescence of (S)- and (R)-NPX was quenched by the acetate of (S)-N-methyl-2-pyrrolidinemethanol [(S)-PYR ester] (cf.Chart 1) in acetonitrile (see Fig. 4).
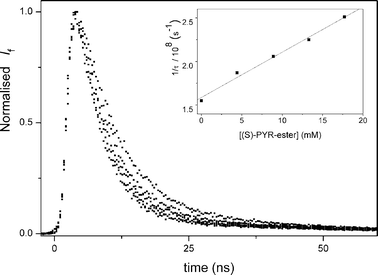 | ||
| Fig. 4 Fluorescence decay traces (λexc=280 nm, λobs=350 nm) of (a) (S)-NPX in the presence of increasing concentrations of (S)-PYR ester. The inset shows the corresponding plot of the reciprocal singlet lifetime versus (S)-PYR ester concentration. | ||
The bimolecular quenching rate constants can be simply determined by application of the Stern–Volmer equation: I0/I = 1 + kqτ0[(S)-PYR ester]. Bimolecular quenching rate constants obtained from steady-state fluorescence measurements were 1.2 × 1010 M−1 s−1 and 1.0 × 1010 M−1 s−1 for (S)/(S) and (R)/(S) combination, respectively. However, the values derived from time-resolved measurements were the same for both combinations and somewhat smaller, 5.2 × 109 M−1 s−1. The absence of stereodifferentiation in the intermolecular case emphasises the decisive role of the spacer in the intramolecular dyads, which reduces the degree of freedom of the chromophore and the quencher, compared to the intermolecular case.
Laser flash photolysis studies
Laser flash photolysis studies (nanosecond timescale) were performed for (S)-NPX and the two diastereomeric dyads NPX–PYR in nitrogen-purged acetonitrile solution. After laser flash excitation of (S)-NPX at 308 nm two transient species were observed (cf.Fig. 5) that were assigned according to the literature data.27,28 The absorption band with its maximum at 440 nm is identified as a triplet–triplet absorption and belongs to the excited triplet state of naproxen. The second transient species was identified as the naphthalene-like radical cation with λmax of 620 nm, formed in a photoionisation mechanism.27,28 | ||
| Fig. 5 Transient absorption spectra of (S)-NPX (solid line), (S,S)-NPX–PYR (dash line) and (R,S)-NPX–PYR (dotted line) 1 µs after the laser flash. The inset shows the corresponding transient decays at 440 nm. | ||
For (S,S)- and (R,S)-NPX–PYR, only the triplet–triplet absorption band with a maximum at 440 nm was detected after the laser pulse (cf.Fig. 5). The kinetic behaviour of the triplet is virtually the same as found for naproxen itself (see inset Fig. 5), indicating the lack of triplet quenching in the dyads. Thermodynamics for naproxen triplet state quenching by electron transfer clearly disfavours such a process. An endergonic driving force of ca. 0.8 eV can be calculated with eqn. (1) using a naproxen excited triplet state energy of ca. 2.70 eV27,28 and the known electrochemical potentials (see above). The absence of the signal at 620 nm in the transient spectra of the dyads can be explained by a fast (<20 ns) ground state electron transfer from the pyrrolidine moiety to the electron-deficient radical cation of naproxen.
An interesting observation has been made by comparing the signal amplitudes of the triplet–triplet absorption at 440 nm, measured under the same conditions, for (S)-NPX and both dyads NPX–PYR (cf.Fig. 5). Clearly, the largest signal is obtained for (S)-NPX. Obviously, if the naproxen excited singlet state in the dyads is quenched by electron transfer (see above), the amount of triplet state populated should be minor compared with (S)-NPX, where such a deactivation pathway does not contribute. However, based on the electron transfer quantum yield of ca. 60% for excited singlet state quenching, the triplet signal is too strong by far [only ca. 30% less than the signal for (S)-NPX]. This must be explained by an additional pathway leading to triplet state population, different from intersystem crossing. We assume that the initially formed singlet radical ion pair undergoes an intersystem crossing to the triplet radical ion pair, which subsequently converts to the lower lying excited triplet state via back electron transfer.3
The corresponding intermolecular control experiment was performed in order to investigate the triplet state interaction of (S)- and (R)-NPX with (S)-PYR ester. In agreement with the observation for the intramolecular case (see above), the excited triplet state remained unquenched upon addition of increasing amounts of (S)-PYR ester, due to the unfavourable thermodynamics for both electron transfer and exciplex formation. Although at the highest (S)-PYR ester concentration (ca. 0.5 M) employed in this experiment all excited singlet state of naproxen should be practically quenched, a considerable triplet signal still remained observable. This is in agreement with our rationalisation that triplet state can be populated via the triplet radical ion pair (formed by intersystem crossing from the singlet radical ion pair). Further, at these high amine concentrations a new transient with maximum at ca. 340 nm was noted (cf.Fig. 6).
 | ||
| Fig. 6 Transient absorption spectra of (S)-NPX in the presence of 0.5 M (S)-PYR ester 1 µs after the laser flash. The inset shows the corresponding transient decay at 340 nm. | ||
This species can be attributed, on the basis of its absorption spectrum and its quenching by oxygen, to the naproxen radical formed by protonation of the radical anion,29,39 thus supporting the involvement of electron transfer in the excited singlet state quenching.
Finally, the decay of the radical cation of (S)- and (R)-NPX at λobs = 610 nm in the presence of (S)-PYR ester was investigated as well. The lifetime of this species was determined in the presence of increasing amine concentrations.
The decays are shown in Fig. 7, while the plot of reciprocal lifetimes vs. quencher concentration can be found in the inset. From these data, bimolecular quenching rate constants of 3.6 × 109 and 3.7 × 109 M−1 s−1 for (S)- and (R)-naproxen, respectively, were estimated. Obviously, the process seems too fast to allow for the observation of significant enantiodifferentiation.
 | ||
| Fig. 7 Transient decay traces of (S)-NPX at 610 nm in the presence of increasing concentrations of (S)-PYR ester. The inset shows the corresponding plot of the reciprocal radical-cation lifetime versus (S)-PYR ester concentration. | ||
Conclusions
A remarkable stereodifferentiation has been observed in the intramolecular interaction of naphthalene-like excited singlet state with a pyrrolidine-derived quencher. This is very interesting, as it involves an electron transfer process, thermodynamically favoured in acetonitrile, rather the exciplex formation. By contrast, no significant quenching is observed for the corresponding triplet excited states.Acknowledgements
We thank the Generalitat Valenciana (Grupos 03/082) and the Spanish Ministry of Science and Technology (MCYT; Grant BQU 2001-2725 and fellowship to S. A.) for financial support. The Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged for a research fellowship for U. P.References
- D. Rehm and A. Weller, Kinetics of fluorescence quenching by electron and H- atom transfer, Isr. J. Chem., 1970, 8, 259–271 CAS.
- N. Mataga and H. Miyasaka, Electron transfer and exciplex chemistry, Adv. Chem. Phys., 1999, 107, 431–496 CAS.
- G. J. Kavarnos and N. J. Turro, Photosensitization by reversible electron transfer: theories, experimental evidence and examples, Chem. Rev., 1986, 86, 401–449 CrossRef CAS.
- H. Knibbe, D. Rehm and A. Weller, Intermediates and kinetics of fluorescence quenching by electron transfer, Ber. Bunsen-Ges. Phys. Chem., 1968, 72, 257–263 CAS.
- X. Allonas and P. Jacques, Factors affecting adiabaticity in bimolecular photoinduced electron transfer reaction between anthracene derivatives and organic donors, Chem. Phys., 1997, 215, 371–378 CrossRef CAS.
- U. Pischel and W. M. Nau, Switch-over in photochemical reaction mechanism from hydrogen abstraction to exciplex-induced quenching: interaction of triplet-excited versus singlet-excited acetone versus cumyloxyl radicals with amines, J. Am. Chem. Soc., 2001, 123, 9727–9737 CrossRef CAS.
- A. Weller, Photoinduced electron transfer in solution: exciplex and radical ion pair formation free enthalpies and their solvent dependence, Z. Phys. Chem. (Munich), 1982, 133, 93–98 CAS.
- I. R. Gould and S. Farid, Steric effects in photoinduced electron-transfer reactions, J. Phys. Chem., 1993, 97, 13067–13072 CrossRef CAS.
- P. Jacques, X. Allonas, P. Suppan and M. von Raumer, The interplay between steric hindrance and exergonicity in the rates of excited state quenching, J. Photochem. Photobiol. A: Chem., 1996, 101, 183–184 CrossRef CAS.
- U. Pischel, X. Zhang, B. Hellrung, E. Haselbach, P.-A. Muller and W. M. Nau, Fluorescence quenching of n,p*-excited azoalkanes by amines: what is a sterically hindered amine?, J. Am. Chem. Soc., 2000, 122, 2027–2034 CrossRef CAS.
- H. Rau, Asymmetric photochemistry in solution, Chem. Rev., 1983, 83, 535–547 CrossRef CAS.
- Y. Inoue, Asymmetric photochemical reactions in solution, Chem. Rev., 1992, 92, 741–770 CrossRef CAS.
- A. G. Griesbeck and U. J. Meierhenrich, Asymmetric photochemistry and photochirogenesis, Angew. Chem., Int. Ed., 2002, 41, 3147–3154 CrossRef.
- T. Yorozu, K. Hayashi and M. Irie, Chiral discrimination in fluorescence quenching, J. Am. Chem. Soc., 1981, 103, 5480–5484 CrossRef CAS.
- W. Iwanek and J. Mattay, Ground state and excited state association: chiral recognition between 2,2′-dihydroxy-1,1′-binaphthyl and amines, J. Photochem. Photobiol. A: Chem., 1992, 67, 209–226 CrossRef CAS.
- T. Nishiyama, K. Mizuno, Y. Otsuji and H. Inoue, Chiral discrimination in exciplex quenching of 2,2′-dimethyl-1,1′-bianthryl-N,N-dimethylaniline system, Chem. Lett., 1994, 2227–2228 CAS.
- D. Wang, T.-J. Liu, W.-C. Zhang, W. T. Slaven IV and C.-J. Li, Enantiomeric discrimination of chiral amines with new fluorescent chemosensors, Chem. Commun., 1998, 1747–1748 RSC.
- M. A. Miranda, A. Lahoz, R. Martínez-Mánez, F. Boscá, J. V. Castell and J. Pérez-Prieto, Enantioselective discrimination in the intramolecular quenching of an excited aromatic ketone by a ground-state phenol, J. Am. Chem. Soc., 1999, 121, 11569–11570 CrossRef CAS.
- M. A. Miranda, A. Lahoz, F. Boscá, M. R. Metni, F. B. Abdelouahab, J. V. Castell and J. Pérez-Prieto, Regio- and stereo-selectivity in the intramolecular quenching of the excited benzoylthiophene chromophore by tryptophan, Chem. Commun., 2000, 2257–2258 RSC.
- M. A. Miranda, L. A. Martínez, A. Samadi, F. Boscá and I. M. Morera, Stereoselective intramolecular hydrogen abstraction by a chiral benzophenone derivative, Chem. Commun., 2002, 280–281 RSC.
- F. Boscá, I. Andreu, I. M. Morera, A. Samadi and M. A. Miranda, Chiral discrimination in the intramolecular abstraction of allylic hydrogens by benzophenone triplets, Chem. Commun., 2003, 1592–1593 RSC.
- U. Pischel, S. Abad, L. R. Domingo, F. Boscá and M. A. Miranda, Diastereomeric differentiation in the quenching of excited states by hydrogen donors, Angew. Chem., Int. Ed., 2003, 42, 2531–2534 CrossRef CAS.
- U. Pischel, S. Abad and M. A. Miranda, Stereoselective fluorescence quenching by photoinduced electron transfer in naphthalene–amine dyads, Chem. Commun., 2003, 1088–1089 RSC.
- S. Abad, U. Pischel and M. A. Miranda, Wavelength-dependent stereodifferentiation in the fluorescence quenching of asymmetric naphthalene-based dyads by amines, submitted for publication, J. Phys. Chem. A Search PubMed , in press.
- M. C. Jiménez, S.-E. Stiriba, R. Tormos, J. Pérez-Prieto and M. A. Miranda, Stereoselectivity in the triplet decay of chiral benzophenone–naphthalene bichromophoric systems, Photochem. Photobiol. Sci., 2004, 3, 36–38 RSC.
- J. Pérez-Prieto, A. Lahoz, F. Boscá, R. Martínez-Mánez and M. A. Miranda, Stereodifferentiation in the decay of triplets and biradicals involved in intramolecular hydrogen transfer from phenols or indoles to p,p* aromatic ketones, J. Org. Chem., 2004, 69, 374–381 CrossRef CAS.
- L. J. Martinez and J. C. Scaiano, Characterization of the transient intermediates generated from the photoexcitation of nabumetone: a comparison with naproxen, Photochem. Photobiol., 1998, 68, 646–651 CrossRef CAS.
- F. Boscá, M. L. Marin and M. A. Miranda, Photoreactivity of the nonsteroidal anti-inflammatory 2-arylpropionic acids with photosensitzing side effects, Photochem. Photobiol., 2001, 74, 637–655 CAS.
- H. D. Burrows, Flash photolytic study of the photoreduction of naphthalene by triethylamine, Photochem. Photobiol., 1974, 19, 241–243 CAS.
- J. A. Ibemesi, M. A. El-Bayoumi and J. B. Kinsinger, Solvent-induced intramolecular exciplex fluorescence of b-naphthylmethyl amine, Chem. Phys. Lett., 1978, 53, 270–272 CrossRef CAS.
- S.-P. Van and G. S. Hammond, Amine quenching of aromatic fluorescence and fluorescent exciplexes, J. Am. Chem. Soc., 1978, 100, 3895–3902 CrossRef CAS.
- F. Meeus, M. V. d. Auweraer and F. C. D. Schryver, Thermodynamic and kinetic aspects of the intermolecular exciplex formation between 2-methylnapthalene and aliphatic amines, J. Am. Chem. Soc., 1980, 102, 4017–4024 CrossRef CAS.
- D. Avnir, E. Wellner and M. Ottolenghi, Photophysical recognition of chiral surfaces, J. Am. Chem. Soc., 1989, 111, 2001–2003 CrossRef CAS.
- T. Sakurai, K. Miyoshi, M. Obitsu and H. Inoue, Solvent and temperature effects on the intramolecular fluorescence quenching in dipeptide model compounds. 1, Ber. Bunsen-Ges. Phys. Chem., 1996, 100, 46–54 CAS.
- F. Pina, J. C. Lima, C. Lodeiro, J. Seixas de Melo, P. Díaz, M. T. Albelda and E. García-España, Long range electron transfer quenching in polyamine chains bearing a terminal naphthalene unit, J. Phys. Chem. A, 2002, 106, 8207–8212 CrossRef CAS.
- J. W. Arbogast, C. S. Foote and M. Kao, Electron transfer to triplet C60, J. Am. Chem. Soc., 1992, 114, 2277–2279 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry; 2nd edn., Marcel Dekker, Inc., New York, 1993 Search PubMed.
- Notably, at the chosen dyad concentration of ca. 5 × 10−5 M the contribution of intermolecular quenching is less than 1%, calculated with kq = 5.1 × 109 M−1 s−1 for the naphthalene–triethylamine system in acetonitrile (cf.ref. 32) and t0 = 7.44 ns for (S)-naproxen in aerated acetonitrile (this work).
- F. S. Dainton, T. J. Kemp, G. A. Salmon and J. P. Keene, Formation of triplet states of solutes in radiolysis of organic liquids, Nature, 1964, 203, 1050–1052 CAS.
Footnotes |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Optimised MOPAC (AM1) extended and folded geometries. See http://www.rsc.org/suppdata/pp/b4/b409729g/ |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |