A study of 2-azido-3,5-dichlorobiphenyl by nano- and picosecond laser flash photolysis and computational methods†
Nina P.
Gritsan
*ab,
Dmitrii A.
Polshakov
a,
Meng-Lin
Tsao
a and
Matthew S.
Platz
*a
aDepartment of Chemistry, The Ohio State University, Columbus, Ohio 43210. E-mail: platz.1@osu.edu; Fax: 1 614 292 5151; Tel: 1 614 292 0401
bInstitute of Chemical Kinetics and Combustion and Novosibirsk State University, 630090 Novosibirsk, Russia. E-mail: gritsan@kinetics.nsc.ru; Fax: 7 3832 342350; Tel: 7 3832 333053
First published on 13th May 2004
Abstract
The dynamics of singlet 3,5-dichloro-2-biphenylnitrene and the products of its rearrangement were monitored by pico- and nanosecond laser flash photolysis and the results were consistent with the predictions of DFT and ab initio molecular orbital calculations.
Introduction
We have recently reported a study of the photochemistry of ortho-biphenyl azide (1a) using laser flash photolysis (LFP) methods.1 It is well known2 that photolysis and pyrolysis of 1a produces carbazole (2a) (Scheme 1). It is one of the most synthetically useful photochemical reactions of aryl azides. | ||
| Scheme 1 | ||
The system is complicated by the fact that the key intermediate of this reaction, singlet ortho-biphenyl nitrene (X = H, 3Sa), undergoes two processes, (1) and (2), at competitive rates, and that azirine formation is reversible (Scheme 2).1
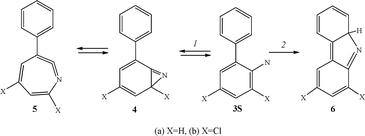 | ||
| Scheme 2 | ||
LFP of 1a in fluid solution does not produce singlet nitrene 3Sa as a detectable species on the nanosecond time scale. Instead, benzazirine 4a and isocarbazole 6a were observed immediately (<10 ns) after the laser pulse.1
The ortho-phenyl group directs azirine formation away from the substituent,3 ortho-alkyl and fluorine substituents behave similarly.4 Two ortho-methyl or halogen substituents can extend the lifetime of a singlet aryl nitrene by one or two orders of magnitude by retardation of azirine formation (process 1).4,5 Substituents are not expected to influence the rate of formation of isocarbazole 6 (process 2). Thus, it seems reasonable to predict that replacement of the ortho-hydrogen of 2-biphenylnitrene with halogen (X = Cl) might simplify the chemistry of the singlet 2-biphenylnitrene and allow straightforward study of process 2, the formation of isocarbazole 6. In addition, we have applied ultrafast transient absorption techniques to monitor the dynamics of singlet nitrene 3Sb (X = Cl) on the picosecond time scale at ambient temperature. Herein, we are pleased to report the results of our study.
Experimental
Laser flash photolysis: nanosecond time scale
A Nd:YAG laser (Spectra Physics LAB-150-10, 266 nm, 5 ns, 50 mJ) or XeCl excimer laser (Lambda Physik, 20 ns, 50 mJ, 308 nm) was used as the excitation light source. The spectrometer has been described previously.1,6 Typically, solutions were prepared in dry spectroscopic grade solvents to an optical density (OD) of about 1.5–2.0 at the excitation wavelength (266 or 308 nm). The temperature was maintained at 77 K by means of boiling liquid nitrogen or varied over the range of 150–300 K by passing a thermo-stabilized nitrogen stream over the sample and held to within ±1 K. In these experiments, a quartz cuvette was placed in a quartz cryostat. The sample solutions were changed after every laser shot, unless otherwise indicated.Picosecond pump–probe experiments
For detection of kinetics on the picosecond time scale, we used a pump–probe spectrometer.7 A regenerative amplified, kHz repetition rate Ti:S laser system produces up to 20 µJ pump pulses with center wavelength 265 nm, using two Type I BBO crystals. Typical probe pulse energies used were 3–5 µJ at the sample. The pump beam was focused to a 0.6 mm diameter spot. Probe pulses between 370–750 nm were generated by focusing the laser fundamental in a water cell. Polarization of pump and probe were set to the magic angle (54.7°). After traversing the sample, the probe beam was passed through an f = 0.25 m monochromator to isolate an approximately 10 nm portion of the continuum for single wavelength experiments.Transient absorption spectra were recorded by scanning the monochromator. Since the total group delay between the shortest and the longest wavelength of the continuum is about 3 ps, it is not necessary to apply group delay (’chirp’) correction to transient spectra recorded after 10 ps. The instrument response was approximately 220 fs (Gaussian full width at half-maximum, fwhm), which was determined by difference frequency mixing between the third harmonic and 10 nm portion of the continuum in a thing BBO crystal. To minimize the effects of photoproducts we prepared the sample in a large reservoir (1 L) with a concentration of 0.003 M. The sample solution was continuously circulating through the jet system as described.7
Quantum chemical calculations
The geometries of the singlet and triplet states of 3,5-dichloro-2-biphenylnitrene (3b) were fully optimized at the CASSCF/6-31G* (5D) level.8,9 An (8,8) active space was employed for the calculation; the selected molecular orbitals were similar to those used in the calculation of unsubstituted phenylnitrene.10 The dynamic electron correlation was included using CASPT2/6-31G* (5D) calculations11 with the CASSCF optimized geometries. During the CASPT2 calculations of 3Sb, an intruder problem12 was present as a low reference weight was obtained. This problem was solved by applying an initial 0.1 h level shift12 prior to the CASPT2 calculations of 3Sb and 3Tb. Both CASSCF and CASPT2 calculations were carried out with a MOLCAS program (version 5.0).13To compute the possible reaction pathways of the singlet nitrene, geometries of proposed reactive intermediates and transition states were fully optimized using the B3LYP14 method with the 6-31G* (6D) basis set. Vibrational frequencies were calculated to analyze the nature of the stationary points (minimum or transition state) and were used to account for ZPE differences. Single-point energies were then calculated using the B3LYP/6-311+G(2d,p) method15 for B3LYP/6-31G* optimized geometries. For the transition states connecting the open-shell singlet nitrene and a closed-shell intermediate, a standard DFT calculation fails to predict the chemistry properly. Thus, the broken symmetry DFT calculations (UB3LYP method with mixed HOMO and LUMO to destroy the spatial symmetry) were used for the calculations of these transition states. The calculated <S2> values of these transition states were found to be small and to fall in the range of 0.04–0.28, except TS3Sb-6b in which the <S2> value was very high (≈0.6). Time dependent DFT (TD-DFT) calculations16 were performed at the B3LYP/6-31G* level. All DFT calculations were carried out with GAUSSIAN 98.17
Materials
All solvents (Aldrich) are spectroscopic grade and were used as received. THF was refluxed over sodium and distilled under argon prior to use.2-azido-3,5-dichlorobiphenyl
A solution of 2.2 g (13 mmol) of ortho-amino-biphenyl in 40 mL of CCl4 was stirred in an ice bath and continuously bubbled with chlorine gas. The reaction flask was connected to a chlorine gas trap. The white precipitate, which separated was collected after one hour, washed with CCl4, and dried by exposure to the air. The crude product of 3.33 g was passed through a silica gel column with 10% ethyl acetate and 90% hexane to separate the dichlorinated compound (first component to run through the column) from the monochlorinated product to give 1.47 g (41% yield) of 3,5-dichloro-2-aminobiphenyl hydrochloride, as a white solid. GC/MS analysis found M+ at 237 m/z and M+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶(M + 2)+∶(M + 4)+ was 9∶6∶1. To a solution of 1.47 g (5.4 mmol) of 3,5-dichloro-2-amino-biphenyl hydrochloride in 10 mL of THF was added a mixture of concentrated hydrochloric acid (2.5 mL) and water (20 mL). The reaction mixture was stirred for 15 min in an ice bath, and a solution of 700 mg (10 mmol) of NaNO2 in 10 mL was added drop-wise to the solution, keeping the temperature of the reaction mixture below 5 °C. The reaction mixture was stirred for 15 min at 0 °C, and excess nitrous acid was removed by the addition of urea. To the cooled reaction mixture was added drop-wise a solution of 900 mg (13.8 mmol) of NaN3 in 10 mL of water. After the addition was complete, the reaction mixture was stirred for 1.5 h at room temperature. The organic material was extracted with diethyl ether, and the extract was washed with dilute hydrochloric acid (0.1 M) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was developed on a silica gel column with n-pentane and was then recrystallized in methanol/pentane under dry ice to give 470 mg (1.8 mmol, 33% yield) of 3,5-dichloro-2-biphenylazide as white crystals (needles). Mp 53.5–54.5 °C; IR (KBr) 2114 cm−1
(N3); HRMS calcd. for C12H7Cl2N3 263.00115, found 262.9994 m/z.
∶(M + 2)+∶(M + 4)+ was 9∶6∶1. To a solution of 1.47 g (5.4 mmol) of 3,5-dichloro-2-amino-biphenyl hydrochloride in 10 mL of THF was added a mixture of concentrated hydrochloric acid (2.5 mL) and water (20 mL). The reaction mixture was stirred for 15 min in an ice bath, and a solution of 700 mg (10 mmol) of NaNO2 in 10 mL was added drop-wise to the solution, keeping the temperature of the reaction mixture below 5 °C. The reaction mixture was stirred for 15 min at 0 °C, and excess nitrous acid was removed by the addition of urea. To the cooled reaction mixture was added drop-wise a solution of 900 mg (13.8 mmol) of NaN3 in 10 mL of water. After the addition was complete, the reaction mixture was stirred for 1.5 h at room temperature. The organic material was extracted with diethyl ether, and the extract was washed with dilute hydrochloric acid (0.1 M) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was developed on a silica gel column with n-pentane and was then recrystallized in methanol/pentane under dry ice to give 470 mg (1.8 mmol, 33% yield) of 3,5-dichloro-2-biphenylazide as white crystals (needles). Mp 53.5–54.5 °C; IR (KBr) 2114 cm−1
(N3); HRMS calcd. for C12H7Cl2N3 263.00115, found 262.9994 m/z.
2-azido-3,5-dichlorobiphenyl-d7
The synthesis of this compound follows the same procedure as that of 3,5-dichloro-2-biphenyl azide, except that ortho-aminobiphenyl-d9 (CDN Isotopes, Inc.) was used as starting material and that the amount of starting reagent was reduced to one fifth the original quantity. 3,5-dichloro-2-biphenylazide-d7 was obtained as white crystals (needles). Mp 53.5–54.5 °C; IR (KBr) 2123 cm−1 (N3), 2286, 2358 cm−1 (C–D); HRMS calcd. for C12D7Cl2N3 270.0456, found 270.0558 m/z. The base peak in the MS was M−N2, as expected (observed 242.0368, calculated 242.0395 m/z).
Results and discussion
Laser flash photolysis of 2-azido-3,5-dichlorobiphenyl, nanosecond time scale
Laser flash photolysis (LFP, 266 nm, 5 ns) of 3,5-dichloro-2-biphenyl azide in glassy methylcyclohexane at 77 K produces the transient absorption spectra of Fig. 1. The transient spectrum, recorded immediately after the flash (spectrum 1), has λmax = 435 nm and decays with an observed rate constant of 1.4 ± 0.1 × 107 s−1 (τ = 71 ± 5 ns). The decay of this species is accompanied by the growth (kobs = 1.5 ± 0.1 × 107 s−1) of absorption, centered at 355 nm, with a weaker absorption at 413 nm (spectrum 2). The latter spectrum is identified as that of triplet nitrene 3Tb, which can be formed as a persistent species by continuous irradiation of 1b at 77 K (spectrum 3). Thus, the singlet nitrene 3Sb has been detected by LFP of 1b at 77K (λmax = 415, 435, 455 nm) and it relaxes cleanly to the lower energy triplet (3Tb) under these conditions.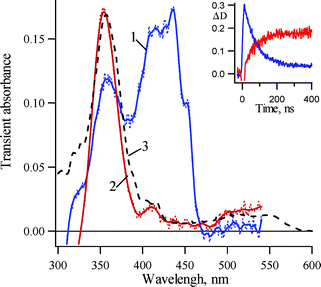 | ||
| Fig. 1 Transient absorption spectra detected over a window of 10 ns following LFP (266 nm, 5 ns) of 2-azido-3,5-dichlorobiphenyl (1b) in glassy methylcyclohexane (MCH) at 77 K (1) just after the laser pulse and (2) 200 ns after the laser pulse; (3) the persistent spectrum of triplet 3,5-dichloro-2-biphenylnitrene (3Tb) produced by brief photolysis (10 s, 254 nm) of 1b in MCH at 77 K. Insert: Kinetic traces for decay of singlet 3,5-dichloro-2-biphenyl nitrene 3Sb at 430 nm and growth of triplet nitrene 3Tb at 355 nm. | ||
The resulting transient spectra resemble those obtained upon LFP of 2-biphenyl azide (1a) at 77 K.1 The chlorine substitution has little influence on the intersystem rate constant (kISC = kobs at 77 K). Compared to the absorption maxima of unsubstituted ortho-biphenyl nitrene 3a, the chlorine substituted compound exhibits a perceptible red shift. The magnitude of the red shift is about 15 nm in singlet nitrene 3S and is about 10 nm in triplet nitrene 3T.
At ambient temperature, LFP (266 nm, 5ns) of 3,5-dichloro-2-biphenyl azide in pentane produces the transient absorption spectra of Fig. 2. A broad intense visible band ranging from 400 to 500 nm was observed 15 ns after the laser flash. This transient absorption decays exponentially at ambient temperature (Fig. 2, insert), and its disappearance proceeds with concurrent formation of the chlorinated carbazole 2b. The first order rate constants obtained by the fitting of the decay and growth observed between 290–570 nm coincide (with an accuracy of ±20%). However, there is a tendency for the rate constants in the region of 400–440 nm to be smaller than those measured in the 450–550 nm region.
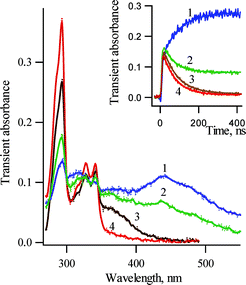 | ||
| Fig. 2 Transient absorption spectra detected over a window of 10 ns following LFP (266 nm, 5ns) of 3,5-dichloro-2-biphenyl azide 1b in pentane at ambient temperature (1) about 15 ns after the laser pulse and (2) 60 ns, (3) 1 µs and (4) 30 ms after the laser pulse. Insert: Kinetic traces (1) for the growth of carbazole absorption at 294 nm and the decay of its precursors (2) at 320 nm, (3) 440 nm and (4) 470 nm. | ||
Both the transient spectrum and the kinetics of the species absorbing in the visible region (Fig. 2) are very similar to those of the isocarbazole 6a produced by LFP of 2-biphenyl azide (1a). It is worth mentioning that the transient absorption in visible region, obtained by LFP of 1b, is much more intense than that of 1a, presumably due to a lower rate constant of azirine-formation of 3Sb, a process which competes with the formation of 6b. Thus, whereas unsubstituted carbazole formation takes place mainly on the millisecond time scale at ambient temperature with didehydroazepine (5a) serving as a reservoir of the singlet 2-biphenyl nitrene (3Sa), the chlorinated carbazole 2b is produced predominantly on the nanosecond time scale and only to a minor extent from the corresponding didehydroazepines 5b and/or 5b′.
As with the case of unsubstituted compound 1a, LFP of 1b in acetonitrile produces transient absorption spectra (Fig. 3), which are different from those, observed in pentane (Fig. 2). Intense absorption with a maximum at 440 nm and a shoulder at 470 nm, which is observed in pentane, is absent from the transient spectrum detected 15 ns after the laser pulse in acetonitrile. Nevertheless, a transient absorption with shoulders at 415 and 440 nm and a less intense band at 500–550 nm was detected in this solvent. Its decay, with rate constant 2.1 ± 0.1 × 107 s−1 (τ = 48 ± 3 ns), was accompanied by the growth of the substituted carbazole 2b with the same rate constant (Fig. 3, insert A). This process was not observed in the case of 1a.1 The failure to observe this process in the parent system may be due to very small transient absorption of the non-chlorinated intermediate. It is also possible, that the corresponding intermediate and process does not exist in the unsubstituted system. At this time the carrier (7b) of this spectrum with λmax = 415 and 440 nm (τ = 48 ± 3 ns) is not assigned.
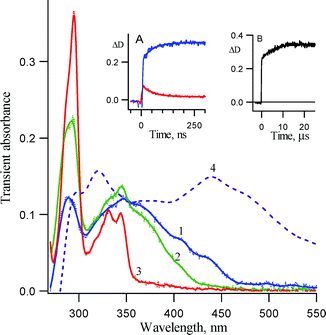 | ||
| Fig. 3 Transient absorption spectra detected over a window of 10 ns following LFP (266 nm, 5 ns) of 3,5-dichloro-2-azido biphenyl 1b in acetonitrile at ambient temperature (1) about 15 ns, (2) 250 ns and (3) 300 µs after the laser pulse. (4) For comparison, the transient absorption spectrum detected in pentane 15 ns after laser pulse is also presented. Inserts: (A) Kinetic traces for growth of carbazole 2b at 294 nm and decay of its precursor at 440 nm. (B) Growth of the carbazole absorption at 294 nm on a microsecond time scale. | ||
Additional growth of the carbazole absorption was detected on the microsecond time scale (Fig. 3, insert B). As with parent compound 1a,1 the corresponding didehydroazepines 5b and/or 5b′ eventually isomerizes to carbazole 2b on this time scale (τ = 6 ± 1 µs).
As in our previous study,1 we propose that trace quantities of water in acetonitrile catalyze the transformation of 6b into 2b. A reasonable mechanism for this catalysis is shown in Scheme 3. Fig. 4A displays the acceleration of the decay, monitored at 470 nm, in dichloromethane in the presence of ‘wet’ CH3CN. Curiously, the kinetic traces in the presence of CH3CN are clearly biexponential, which indicates that two species absorb at this wavelength, but that only one of these intermediate reacts efficiently with water. The pseudo-first order rate constant of isocarbazole 6b disappearance in ‘wet’ acetonitrile was estimated to be 9 ± 2 × 108 s−1.
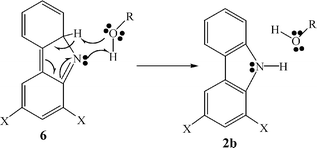 | ||
| Scheme 3 | ||
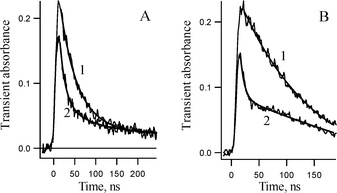 | ||
| Fig. 4 Kinetics of transient absorption decay at 470 nm at ambient temperature. (A) Kinetics detected in CH2Cl2 in the absence (1) and in the presence (2) of 1.74 M of CH3CN and (B) kinetics detected in pentane in the absence (1) and in the presence (2) of 6.7 × 10−3 mol l−1 of 1-propanol. | ||
Fig. 4B demonstrates that 1-propanol also accelerates the disappearance of 6b very efficiently. The rate constant of the reaction of isocarbazole 6b with 1-propanol is estimated to be diffusion controlled (k = 1.9 ± 0.3 × 1010 M−1 s−1). At the same time the influence of 1-propanol on the lifetime of the second intermediate (7b) absorbing at 440 nm is not pronounced and its lifetime was found to be 53 ± 3 ns in neat 1-propanol. The transient spectrum detected in neat 1-propanol 15 ns after the laser pulse is very similar to the transient spectrum in CH3CN, but is about twice as intense (Fig. 3).
Additional information on the kinetics and spectroscopy of the above-mentioned intermediates can be obtained via low temperature experiments. LFP (266 nm, 5 ns) of 1b in pentane at 161 K produces the transient absorption spectra of Fig. 5. A very intense transient absorption was observed in the visible region. The visible absorption band detected 55 ns after the laser pulse is similar to the absorption band detected at ambient temperature (Fig. 2, spectrum 1), but shows a structured shape. However, after a few hundreds of nanoseconds, it is replaced by a broad unstructured band (Fig. 5, spectrum 3), which disappears within several microseconds. Thus, after three hundred nanoseconds, the spectrum of the unassigned intermediate 7b (λmax = 425, 440 nm) disappears, and only the broad unstructured band of 6b (λmax = 470 nm, spectrum 3) is observed. Note, that dichlorinated isocarbazole 6b also has an intense absorption band with a maximum at about 320 nm, similar to the case of unsubstituted compound 6a.1
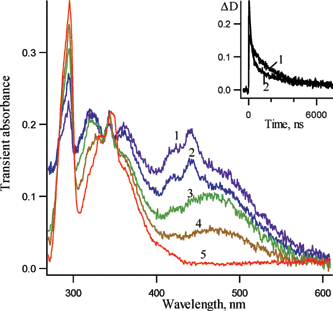 | ||
| Fig. 5 Transient absorption spectra detected over a window of 10 ns following LFP (266 nm, 5 ns) of 3,5-dichloro-2-azido biphenyl 1b in pentane at 161 K (1) 55 ns, (2) 100 ns, (3) 300 ns, (4) 1.3 µs and (5) 6 µs after the laser pulse. Insert: kinetic traces for the absorption decay at (1) 475 nm and (2) 440 nm. The best fit for the trace (1) is exponential with kobs1=5.1 ± 0.1 × 105 s−1. The best bi-exponential fit for trace (2) gives kobs1=4.8 ± 0.3 × 105 s−1 and kobs2=5.3 ± 0.2 × 106 s−1. | ||
The lifetimes of the two intermediates (6b and 7b) differ significantly (by about one order of magnitude) at 161 K, which is opposite to the case at room temperature, where they are very similar. A kinetic curve recorded at 470 nm is exponential, while a kinetic curve obtained at 440 nm is clearly bi-exponential (Fig. 6, Insert). Therefore, this data provide additional strong support for the presence of two intermediates absorbing in the visible region.
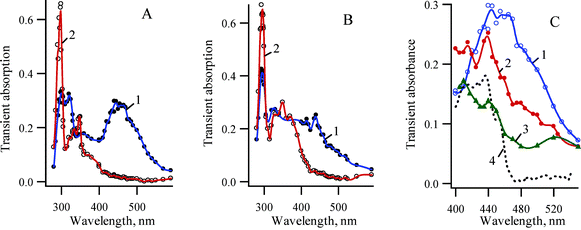 | ||
| Fig. 6 Transient absorption spectra detected following LFP of 2-azido-3,5-dichlorobiphenyl (1b) at ambient temperature, (A) in pentane using YAG laser (266 nm, 5 ns) (1) 20 ns and (2) 400 ns after the laser pulse; (B) in pentane using XeCl excimer laser (308 nm, 17 ns) (1) 35 and (2) 380 ns after the laser pulse; (C) The transient absorption spectra recorded tens of nanosecond after the laser pulse using different excitation sources and in different solvents: (1) in pentane (266 nm excitation), (2) in pentane (308 nm excitation) and (3) in acetonitrile (266 nm excitation). (4) Black line spectrum of singlet nitrene 3Sb detected at 77 K. | ||
Interesting results were obtained using a XeCl excimer laser (308 nm, 15 ns) as the excitation source. Fig. 6 presents a collection of transient absorption spectra recorded using different excitation sources (A and B) in pentane as well as in different solvents (C). The simplest way to assign these transient spectra is to propose as before, that two intermediates absorbing in the range 400–600 mn are formed upon photolysis of 1b. One of these species is isocarbazole 6b, which is the main product, upon 266 nm excitation (Fig. 6A). In acetonitrile 6b isomerizes to 2b faster than the time resolution (≈5 ns) of the experiment, and only the second intermediate 7b is detected in this solvent (Fig. 5C). This unassigned intermediate 7b is formed in greater amounts, upon 308 nm excitation, presumably at the expense of isocarbazole 6b (Fig. 6B). Upon comparing the transient spectra recorded about 400 ns after excitation with different laser sources, it is clear that similar amounts of carbazole 2b are formed in every case, but that about twice the yield of the intermediate with λmax around 360 nm is formed with longer wavelength excitation. Previously1 it was shown for parent compound 1a, that two intermediates, azepine (5a′) and an isocarbazole 8a (formed by the ‘wrong’ 1,5-hydrogen shift in 6a, Scheme 4), have absorption bands, with maxima around 360 nm. The carriers of 360 nm transient absorption produced upon LFP of 1b are likely analogous dichlorinated azepines and isocarbazoles.
 | ||
| Scheme 4 | ||
The temperature dependence of the rate constants for the disappearance of both intermediates was studied and the data are presented in Fig. 7. We measured the rate constant for the disappearance of isocarbazole 6b (kobs1) in pentane at 470 nm using the YAG laser (266 nm, 5 ns) as an excitation source. The second rate constant (kobs2) was estimated using mainly the excimer laser excitation (308 nm, 15 ns). The kinetics were measured at either 400 or 440 nm for the unassigned species 7b.
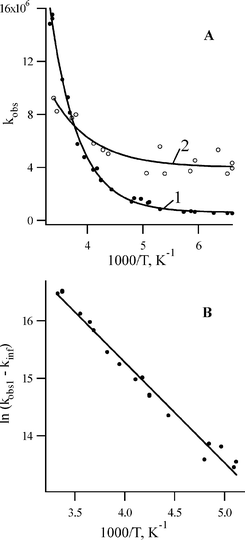 | ||
| Fig. 7 (A) The temperature dependence of the observed rate constants in pentane. (1) kobs1 of the isocarbazole 6b decay measured at 470 nm following excitation by the YAG laser (266 nm, 5 ns). (2) kobs2 of the intermediate 7b decay detected following excitation by the excimer laser (308 nm, 15 ns). (B) Arrhenius treatment of ln(kobs1−kinf) for the decay of 6b. | ||
The magnitude of kobs1 decreases with temperature until approximately 170 K whereupon a limiting value (kinf1) is reached (Fig. 7A, curve 1) in a manner reminiscent of 8aH-carbazole (6a). We previously assigned this type of temperature dependence for 6a to a change in mechanism.1 For example, a 1,5-hydrogen shift at higher temperature might be replaced by a catalyzed process at low temperature, which has a lower enthalpy of activation and a more negative entropy of activation. Alternatively, a contribution of quantum mechanical tunneling to the 1,5-hydrogen shift reaction that forms 2b from 6b could be responsible for the data obtained at low temperature. The Arrhenius treatment of ln kobs1versus 1/T is curved while a straight line is obtained by plotting ln (kobs1 − kinf1) (Fig. 7B). The activation energy Ea1 = 3.9 ± 0.2 kcal mol−1 is obtained from a plot of Fig. 7B and is similar to the value obtained with parent isocarbazole 6a.1 The pre-exponential factor was found to be A = 109.7 ± 0.1 s−1.
It is seen from Fig. 7A that the decay rate constant for the second intermediate 7b changes slightly with temperature, by a factor of about 2.5 over the temperature range 150–290 K. Unfortunately, the accuracy of these data is very poor because the transient absorption of this intermediate and 6b overlap significantly.
It must be noted that the temperature dependencies in Fig. 7A are very similar to those for the disappearance of singlet phenylnitrene and many of its derivatives.5 In addition, the structured band of the intermediate 7b with maxima at 415 and 435 nm is very similar to the absorption band of singlet nitrene 3Sb (Fig. 6C). However, assignment of the transient absorbance at 415 and 435 nm to singlet nitrene 3Sb can be excluded based on the rate of disappearance of this species observed at low temperatures. For singlet arylnitrenes the temperature dependence of the rate constant of disappearance can be described by eqn. (1) with the temperature-independent rate constant being that for intersystem crossing (kISC).5
| kobs= kR + kISC | (1) |
If the temperature dependence of the rate constant for disappearance of the species 7b is analyzed using eqn. (1), a value of kISC = (4 ± 1) × 106 s−1 is obtained. However, the rate constant for intersystem crossing in 3Sb measured at 77 K, was found to be much faster, (1.4 ± 0.1) × 107 s−1. It was shown recently for phenylnitrene (1c),18 that kISC estimated from the low temperature data in liquid pentane is the same as measured at 77 K in MCH. This argues against assigning 7b to singlet nitrene 3Sb.
This conclusion will be confirmed by picosecond spectroscopy in a later section. These studies demonstrate that 3Sb has τ = 260 ± 70 ps in cyclohexane at ambient temperature. This lifetime is about 400 times shorter than the lifetime of the unassigned transient 7b under comparable conditions.
The perdeuterosubstituted analogue of 1b (1b–d7) was synthesized and studied by LFP techniques. As for the undeuterated compound, the spectra and kinetics obtained from 1b–d7 depend on the excitation source and solvent. It was shown above, that isocarbazole 6b (with maxima at 320 and 470 nm) is formed mainly upon excitation of 1b with a YAG laser (266 nm) in pentane. On the contrary, only a small amount of 6b is formed upon excitation by 308 nm. The isocarbazole 6b was not detected at all in acetonitrile and 1-propanol.
Therefore, to study the dependence of the lifetime of isocarbazole 6b on deuteration, the kinetics at 320 and 470 nm was followed using the YAG laser source. The rate constant of transformation of 6b to 2b was measured at 295 K and found to be 1.53 ± 0.07 × 107 s−1 (τ = 65 ± 4 ns) and 0.38 ± 0.02 × 107 s−1 (τ = 263 ± 15 ns) for its perdeuterated analogue. The kinetic isotope effect (KIE) is deduced as kH/kD ≈ 4.0 ± 0.4, similar to that of unsubstituted 6a.1 If we deduce the effective KIE using kinetic data at 440 nm, the value drops to 3.0 ± 0.3 due to the overlap of the spectra of two intermediates. The effective KIE estimated from kinetics monitored at 294 nm was found to be 2.6 ± 0.3 and it confirms the existence of two channels of carbazole formation and that the KIE for the second reaction is smaller than that of 6b.
If the excimer laser (308 nm) is used as the excitation source, a greater yield of the unassigned intermediate 7b is observed and the apparent KIE determined at 440 nm drops to 1.4 ± 0.1, demonstrating that the KIE for the second reaction of carbazole formation is close to unity. Indeed, the KIE measured in acetonitrile and 1-propanol, where only intermediate 7b absorbs in the visible region, is equal to 0.95 ± 0.10.
LFP of 2-azido biphenyl 1a in fluid solution does not produce singlet nitrene 3Sa as a detectable species on the nanosecond time scale. Only products of its transformation, azirine 4a and isocarbazole 6a, were detected as transient intermediates in solution.1 Similarly we were unable to detect the dichlorinated singlet nitrene 3Sb in fluid solution using nanosecond LFP. Only at very low temperature (below 170 K), were we able to detect very fast decay at 440 nm (the maximum of the 3Sb absorption band, Fig. 1) and growth kinetics at 470 nm (the maximum of 6b absorption band, Fig. 5) with the same rate constant (Fig. 8). The rate constant of the decay and growth at 147 K is measured to be 5.5 ± 0.7 × 107 s−1 (τ = 18 ± 3 ns). Therefore, to accurately determine the kinetics of singlet nitrene 3Sb at room temperature, we used picosecond pump–probe techniques.
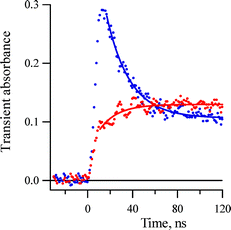 | ||
| Fig. 8 Kinetic traces of the growth of isocarbazole 6b absorption at 470 nm and decay of its precursors 3Sb at 440 nm detected after excitation (266 nm, 5 ns) of 1b at 147 K in pentane. | ||
Picosecond pump–probe experiments
The transient absorption spectra shown in Fig. 9 were recorded in pump–probe experiments. The sample solution of 1b in cyclohexane was pumped by a femtosecond laser pulse (220 fs, 265 nm) and the transient absorption spectra were recorded at different time delays between probe and pump pulses.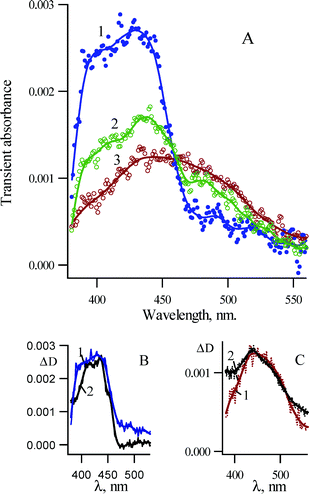 | ||
| Fig. 9 (A) Transient absorption spectra recorded in pump–probe experiments (265 nm, 220 fs) upon photolysis of 3,5-dichloro-2-azido-biphenyl 1b in cyclohexane at ambient temperature (1) 40 ps, (2) 300 ps and (3) 630 ps after excitation. (B) Spectrum 1 is the same as in A1; (2) is the spectrum of singlet 3,5-dichloro-2-biphenylnitrene (3Sb) in MCH at 77 K detected by nanosecond LFP. (C) Spectrum 1 is the same as A3; spectrum 2 was detected over a window of 10 ns following LFP (266 nm, 5 ns) of 3,5-dichloro-2-azido biphenyl 1b in pentane at ambient temperature about 15 ns after the laser pulse. | ||
Fig. 9A shows, that the initially observed transient absorption with a maximum around 430 nm and a shoulder around 400 nm decays in hundreds of picoseconds to produce a new species with a broad absorption spectrum with a maximum around 440 nm. It is seen from Fig. 9B, that the spectrum recorded 40 ps after the excitation is very similar to the spectrum of the singlet nitrene detected at 77 K by nanosecond LFP. However, the spectrum detected at 77 K has more pronounced structural features, which is typical for spectra recorded at cryogenic matrices. Therefore, we assign this spectrum with maximum at 430 nm to singlet 3,5-dichloro-2-biphenylnitrene 3Sb. The final spectrum 3 is very similar to the spectrum detected around 15 ns after the laser pulse (Fig. 9C).
Additional details can be obtained from the kinetics of the transient absorption, which are shown in Fig. 10. Fig. 10 reveals, that the decay of singlet nitrene 3Sb at 420 nm is accompanied by a growth in transient absorption with the same time constant at 470 nm, which was assigned to the isocarbazole 6b. Exponential fit of these traces yields the rate constant of singlet nitrene rearrangement kR = 3.8 ± 0.8 × 109 s−1 (τ = 260 ± 70 ps).
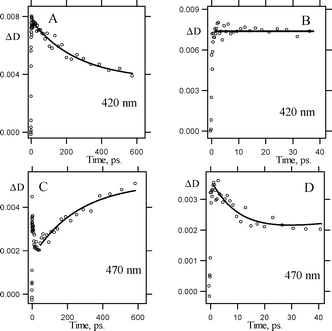 | ||
| Fig. 10 Transient absorption kinetics detected in pump–probe experiments (265 nm, 220 fs) upon excitation of 3,5-dichloro-o-biphenyl azide (1b) in cyclohexane at ambient temperature. Kinetics were recorded at 420 nm (A, B) and 470 nm (C, D) on different time scales: 0–633 ps (A, C) and 0–40 ps (B, D). | ||
Therefore, we have monitored singlet nitrene 3Sb decay in three different types of experiments. The rate constant of 3Sb decay at room temperature (kR = 3.8 ± 0.8 × 109 s−1) was measured using the picosecond pump–probe technique. The rate constant over the temperature range 147–167 K was obtained using nanosecond LFP, and eventually, the rate constant of ISC was measured at 77 K. As before, we propose, that the temperature dependence of the rate constant of the singlet nitrene decay can be described by eqn. (1) with a temperature-independent intersystem crossing rate constant (kISC).5 Thus, we were able to calculate the Arrhenius parameters for the rate constant of singlet nitrene 3Sb rearrangement (Fig. 11). The activation energy was found to be 2.7 ± 0.2 kcal mol−1 with a pre-exponential factor A = 1011.6 ± 0.2 s−1. The value of the activation energy is consistent with our recent calculations1 for 3Sa cyclization to isocarbazole 6a and computational data presented in the next section.
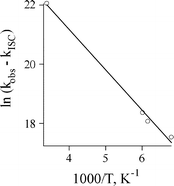 | ||
| Fig. 11 Arrhenius treatment of the decay rate constant of the singlet nitrene 3Sb in pentane. | ||
The kinetic curve recorded at room temperature at 420 nm on the time scale of few picoseconds demonstrates that singlet nitrene 3Sb is formed faster than the time resolution of our apparatus (about 220 fs). Therefore we can conclude, that dissociation of the N–N bond in the excited state of the azide 1b occurs with a rate constant kdis ≥ 5 × 1012 s−1.
However, we were able to detect a fast decay of transient absorption at longer wavelengths. A kinetic curve recorded at 470 nm is displayed in Fig. 10 as an example. The time constant for this decay was estimated to be 0.9 ± 0.2 × 1011 s−1 (τ = 11 ± 2 ps). This time constant is typical of a vibrational relaxation process, which manifests itself in a narrowing of the absorption band.19
The spectrum detected 40 ps after the pump laser pulse also has a shoulder at about 480 nm (Fig. 9, spectrum 1), which apparently is not due to the singlet nitrene. Presumably, the carrier of this transient absorption is one of the species detected on the nanosecond time scale (Fig. 9, spectrum 3). We speculate, that the carrier of transient absorption detected in the region 460–550 nm 40 ps after the pump pulse is the isocarbazole 6b, formed from a vibrationally hot state of the singlet nitrene 3Sb*.
A very similar transient absorption spectrum with a strong absorption band around 420 nm was observed in methanol 15 ps after excitation of 1b (Fig. 12, spectrum 1) and is also assigned to the singlet nitrene 3Sb. A considerable acceleration of the singlet nitrene rearrangement (presumably to 6b and 7b) was observed in methanol. The rate constant of its decay was found to be 1.6 ± 0.2 × 1010 s−1 (62 ± 10 ps). A similar acceleration of the singlet tolylnitrene rearrangement in alcohols has been observed.20 The transient spectrum detected 500 ps after the laser pulse is similar to the spectrum detected in acetonitrile and 1-propanol. Therefore, isocarbazole 6b, which should be formed due to rearrangement of 3Sb, will react very rapidly with methanol to form carbazole 2b. We believe this assumption is reasonable, after taking into account the diffusion-controlled nature of this reaction.
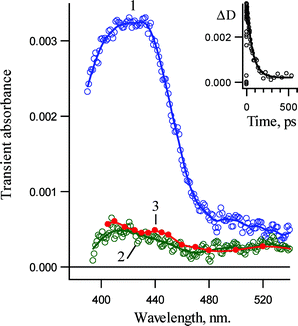 | ||
| Fig. 12 Transient absorption spectra detected in pump–probe experiments (265 nm, 220 fs) upon excitation of 3,5-dichloro-2-azido-biphenyl (1b) in methanol at ambient temperature: (1) 15 ps and (2) 500 ps after pump pulse. Curve 3 is the spectrum detected 20 ns after the laser pulse (266 nm, 5 ns) in acetonitrile recorded by convention LFP. Insert: Kinetics detected at 420 nm. | ||
Calculations of 3,5-dichloro-2-biphenylnitrene and related species
Recently,1 the stationary points on the potential energy surface of singlet 2-biphenylnitrene (3Sa) and its ring-expansion and intramolecular cyclization reactions to isocarbazole 6a were computed using the high level (14,14)CASSCF/CASPT2 procedure, in addition to more simple estimations based on the B3LYP calculations. In this work, only the latter more simple estimations were performed for the dichlorosubstituted nitrene 3Sb and its reaction.First, the absolute CASPT2(8,8)/6-31G* energies for the CASSCF(8,8)/6-31G* optimized geometries of singlet nitrene 3Sb and triplet nitrene 3Tb were determined to be −1433.750077 and −1433.778136 Eh, respectively. Thus, the singlet–triplet energy gap (ΔEST) of 3Sb/3Tb is predicted to be 17.6 kcal mol−1, which is very close to that of unsubstituted biphenylnitrene 3a and parent phenylnitrene 3c.1,10 Secondly the DFT energies of the stationary points involved in the ring-expansion reactions (Scheme 5) and intramolecular cyclization reactions (Scheme 6) of 3Sb (except for that of 3Sb itself) were performed using B3LYP/6-31G* geometry optimizations, zero-point energy calculations, and single point energy calculations using B3LYP/6-311+G(2d,p) methods. These calculated energies are given in Tables 1 and 2, respectively. Finally, the relative energies of all stationary points corresponding to 3Sb are deduced from the DFT energies and CASPT2 ΔEST by using triplet nitrene 3Tb as a common basis.
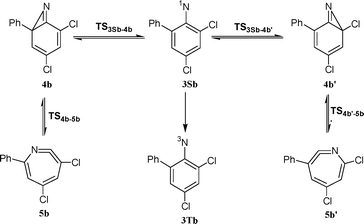 | ||
| Scheme 5 | ||
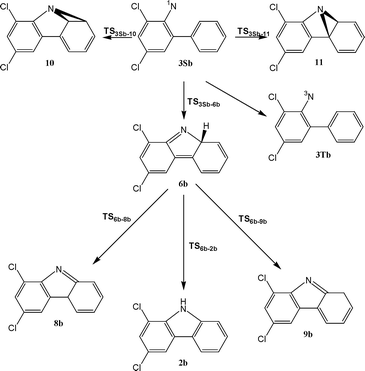 | ||
| Scheme 6 | ||
| Species | B3LYP/6-31G* | ZPEa | B3LYP/6-311+G(2d,p) | Relative energyb |
|---|---|---|---|---|
| a B3LYP/6-31G* zero point vibrational energies. b (U)B3LYP/6-311+G(2d,p)//(U)B3LYP/6-31G* + ZPE relative energies to 3Sb combined with CASPT2 ΔEST. | ||||
| 3Tb | −1436.551411 | 96.21 | −1436.751619 | −17.6 |
| TS3Sb–4b′ | −1436.505894 | 95.51 | −1436.708620 | 8.7 |
| 4b′ | −1436.530208 | 96.65 | −1436.730184 | −3.7 |
| TS4b′–5b′ | −1436.519476 | 95.60 | −1436.720341 | 1.4 |
| 5b′ | −1436.536804 | 96.62 | −1436.740096 | −10.0 |
| TS3Sb–4b | −1436.506421 | 95.61 | −1436.708907 | 8.6 |
| 4b | −1436.520764 | 96.46 | −1436.721656 | 1.4 |
| TS4b–5b | −1436.514554 | 95.67 | −1436.715655 | 4.4 |
| 5b | −1436.533471 | 96.81 | −1436.736250 | −7.4 |
| Species | B3LYP/6-31G* | ZPEa | B3LYP/6-311+G(2d,p) | Relative energyb |
|---|---|---|---|---|
| a B3LYP/6-31G* zero point vibrational energies. b DFT/CASPT2 relative energies, derived from the B3LYP/6-311+G(2d,p)//B3LYP/6-31G*+ZPE energies relative to 3Tb minus the CASPT2 ΔEST of 3Sb/3Tb. c Broken symmetry B3LYP calculations (UB3LYP) were applied. | ||||
| 3Tb | −1436.551411 | 96.21 | −1436.751619 | −17.6 |
| TS3Sb–6 c | −1436.520573 | 95.66 | −1436.721421 | 0.8 |
| 6b | −1436.571772 | 97.47 | −1436.772699 | −29.6 |
| TS3Sb–11b c | −1436.501351 | 96.12 | −1436.701589 | 13.7 |
| 11b | −1436.511404 | 97.21 | −1436.710426 | 9.2 |
| TS3S–10b c | −1436.490029 | 96.16 | −1436.689676 | 21.2 |
| 10b | −1436.495407 | 97.59 | −1436.693861 | 20.0 |
| TS6b–2b | −1436.556187 | 95.58 | −1436.758405 | −22.5 |
| 2b | −1436.661711 | 98.98 | −1436.863362 | −85.0 |
| TS6b–9b | −1436.552369 | 95.49 | −1436.754227 | −20.0 |
| 9b | −1436.611798 | 98.00 | −1436.810754 | −52.9 |
| TS6b–8b | −1436.553003 | 95.60 | −1436.755212 | −20.5 |
| 8b | −1436.598348 | 97.91 | −1436.799081 | −45.7 |
These relative energies are listed in Tables 1 and 2 as well, and the calculated potential energy surfaces for the ring-expansion and intramolecular cyclization reactions of 1b are depicted in Fig. 13 and 14, respectively.
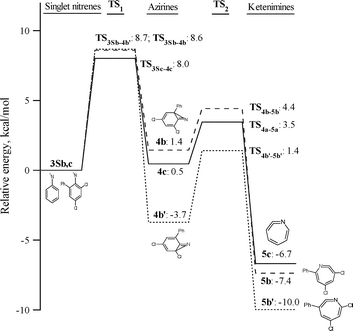 | ||
| Fig. 13 DFT/CASPT2 energies relative to singlet nitrenes for the species involved in the ring-expansion reactions of singlet 3,5-dichloro-2-nitrenebiphenyl 3Sb and phenylnitrene (3Sc). | ||
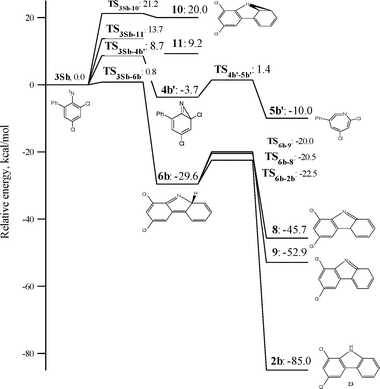 | ||
| Fig. 14 DFT/CASPT2 energies relative to the singlet 3,5-dichloro-2-biphenylnitrene 3Sb for species involved in the intramolecular cyclization and ring-expansion reactions. | ||
Inspection of the data in Table 1 and Fig. 13 reveals that chlorine substitution does raise the barrier to ring-expansion of 3Sb relative to singlet phenylnitrene. As depicted in Fig. 13, the barrier to ring-expansion by cyclization toward either the ortho-chlorine substituted carbon or the ortho-phenyl substituted carbon (Scheme 5) are similar and about 1 kcal mol−1 higher than that of unsubstituted phenylnitrene. In comparison with the barrier to ring-expansion of unsubstituted ortho-biphenylnitrene 3Sa, chlorine substitution raises the barrier to ring-expansion of 3Sb by 3.8 kcal mol−1.
Note, that theory predicts that the rate-determining step in the two-step ring-expansion reaction sequence of 3Sb is the first step, the formation of an azirine. The opposite situation was observed in unsubstituted o-biphenylnitrene 3Sa, for which the second step is the rate determining reaction.1 Therefore unsubstituted azirine 4a was detected as transient intermediate over the entire temperature range studied (150–298 K).
Examination of the data in Table 2 and Fig. 14 reveals that the calculated potential energy surfaces for several intramolecular cyclization reactions of 3Sb are very similar to those of singlet ortho-biphenyl nitrene.1 The results demonstrate that chlorine substitution does not distinctly influence any of the intramolecular cyclization reactions of singlet ortho-biphenylnitrene other than azirine formation. Thus, 3Sb should cyclize effectively to dichloroisocarbazole 6b, and with an elementary rate constant analogous to the parent 2-biphenyl nitrene system.
More sophisticated (14,14)CASSCF/CASPT2 calculations, which were done for the parent nitrene 3Sa, predicted the barrier for the cyclization to isocarbazole 6a to be 6 kcal mol−1. Taking into account the fact, that this procedure usually overestimates the barrier for the cyclization of singlet arylnitrenes by about 3 kcal mol−1,5b,c,3,9 we can estimate barrier for cyclization of 3Sa to 6a (as well as 3Sb to 6b) to be about 3 kcal mol−1. This value is in perfect agreement with experimentally estimated activation energy for the rearrangement of the singlet nitrene 3Sb.
Previously,1 the electronic absorption spectra of the intermediates (4a′, 5a′, 6a and 8a–11a), proposed for the rearrangement of unsubstituted 2-biphenyl azide, were calculated using time-dependent (TD) DFT theory with the random phase approximation.10 The data of these calculations were used to support the assignment of the transient spectra.1 It is clear that chlorine substituents do not significantly change the spectra of these intermediates and the data obtained in previous calculations can model the intermediates of photolysis of 1b.
The unidentified intermediate 7b has an absorption with λmax = 415, 435 and 530 nm. Among all the proposed intermediates (Schemes 5 and 6), which can be formed due to the 3Sb cyclization reactions only 10b and azepine 5b, produced via cyclization toward the phenyl ring, have absorption bands in the visible region. The spectrum of 10a was found to have the following absorption bands (λmax and oscillator strength): 462 (0.007), 322 (0.031) and 275 (0.097).1 The spectrum of didehydroazepine 5b was found to have λmax 432 nm (0.023), 390 nm (0.017) and 304 nm (0.259). However, according to the calculations the formation of these intermediates is unfavorable due to the very high barriers for these rearrangements (Fig. 13, Table 1).
It was noted before, that the absorption spectrum of 7b (415, 435 nm) resembles the spectrum of 3Sb and the temperature dependence of its kinetics is typical of singlet arylnitrenes. Therefore, at this time the only tentative assignment of this intermediate (7b) is to a complex between the nitrene and the pendant phenyl ring. More calculations and experiments must be performed to support or exclude this assignment.
Conclusions
Laser flash photolysis (LFP) of 3,5-dichloro 2-biphenyl azide releases the corresponding singlet nitrene 3Sb. At 77 K, nitrene 3Sb (415, 435, 455 nm) has a lifetime of 71 ± 3 ns controlled by intersystem crossing to the lower energy triplet state (3Tb; λmax = 355 nm). CASPT2 (8,8) 6-31G* calculations predict that triplet nitrene 3Tb is 17.6 kcal mol−1 lower in energy than singlet nitrene 3Sb, an amount similar to that of parent singlet phenylnitrene. LFP of 3,5-dichloro-2-biphenyl azide at ambient temperature releases singlet nitrene 3Sb, which rearranges to isocarbazole 6b (λmax = 470 nm) and unidentified intermediate 7b with λmax = 415, 435 and 530 nm. The lifetime of 3Sb at room temperature was found to be 260 ± 70 ps in cyclohexane and 62 ± 70 ps in methanol. The activation energy of the 3Sb rearrangement (2.7 ± 0.2 kcal mol−1) is in excellent agreement with the computed barrier for cyclization to isocarbazole.The lifetime of isocarbazole 6b is 65 ± 4 ns in pentane at ambient temperature. The lifetime of 6b–d7 is 263 ± 15 ns under the same conditions corresponding to a KIE of 4.0 ± 0.4. This value is very reasonable for the dichlorinated isocarbazole isomerization to the corresponding carbazole by 1,5-hydrogen shift. The rearrangement reaction is both alcohol and water catalyzed.
The unidentified intermediate 7b with λmax = 415, 435 and 530 nm also rearranges to the dichlorinated carbazole 2b. The rate of its rearrangement in pentane at 295 K is about 1.5 times slower than that of 6b and is about seven times faster at temperatures below 180 K. The lifetime of the unidentified intermediate 7b is about 400 times longer than that of singlet nitrene 3Sb in fluid solution at ambient temperature. Moreover, the lifetime of this intermediate does not depend on deuteration and is not shortened in the presence of water and alcohol. At this time, the only tentative assignment of the intermediate 7b is to a complex between the nitrene and the pendant phenyl ring. More calculations and experiments must be performed to support or exclude this assignment.
All experimental results are in excellent agreement with CASPT2 and density functional theoretical calculations.
Acknowledgements
Support of this work in Columbus by the National Science Foundation and by the Ohio Supercomputer Center is gratefully acknowledged. One of us (N.P.G.) gratefully acknowledges the support of the NSF, Ministry of Education of Russian Federation (grant E02-5.0-27) and the Division of Chemistry and Material Sciences of Russian Academy of Sciences. MLT thanks the Ohio State University for a Presidential Fellowship. The authors wish to thank Professor Bern Kohler for the use of his femtosecond pump–probe spectrometer.References
- M.-L. Tsao, N. P. Gritsan, T. R. James, M. S. Platz, D. Hrovat and W. T. Borden, ‘Study of the Chemistry of ortho- and para-Biphenylnitrenes by Laser Flash Photolysis and Time-Resolved IR Experiments and by B3LYP and CASPT2 Calculations’, J. Am. Chem. Soc., 2003, 125, 9343 CrossRef CAS.
- (a) P. A. S. Smith and B. B. Brown, ‘The reaction of aryl azides with hydrogen halides’, J. Am. Chem. Soc., 1951, 73, 2438 CrossRef CAS; (b) P. A. S. Smith and B. B. Brown, ‘The synthesis of heterocyclic compounds from aryl azides. I. Bromo and nitro carbazoles’, J. Am. Chem. Soc., 1951, 73, 2435 CrossRef CAS; (c) P. A. S. Smith and J. H. Hall, ‘Kinetic evidence for the formation of azene (electron-deficient nitrogen) intermediates from aryl azides’, J. Am. Chem. Soc., 1962, 84, 1632 CrossRef; (d) P. A. S. Smith, in ‘Azides and Nitrenes’, ed. E. F. U. Scriven, Academic Press, 1984, p. 95 Search PubMed; (e) P. Walker and W. A. Waters, ‘Pyrolysis of organic azides: a mechanistic study’, J. Am. Chem. Soc., 1961, 84, 408.
- (a) R. J. Sundberg, M. Brenner, S. R. Suter and B. P. Das, ‘Reactions of arylnitrenes. Bond reorganizations in o-biphenylnitrene and phenylnitrene’, Tetrahedron Lett., 1970, 31, 2715 CrossRef; (b) R. J. Sundberg and R. W. Heintzelman, ‘Reactivity of aryl nitrenes. Competition between carbazole formation and internal bond reorganization in biphenylnitrenes’, J. Org. Chem., 1974, 39, 2546 CrossRef CAS; (c) R. J. Sundberg, D. W. Gillespie and B. A. DeGraff, ‘Mechanism of photolytic conversion of 2-azidobiphenyl to carbazole’, J. Am. Chem. Soc., 1975, 97, 6193 CrossRef CAS.
- (a) R. J. Sundberg, S. R. Suter and M. Brenner, ‘Photolysis of 0-substituted aryl azides in diethylamine. Formation and autoxidation of 2-diethylamino-1H-azepine intermediates’, J. Am. Chem. Soc., 1972, 94, 513 CrossRef CAS; (b) E. Leyva and R. Sagredo, ‘Photochemistry of fluorophenyl azides in diethylamine. Nitrene reaction versus ring expansion’, Tetrahedron, 1998, 54, 736 CrossRef CAS.
- (a) N. P. Gritsan, A. D. Gudmundsdóttir, D. Tigelaar and M. S. Platz, ‘Laser flash photolysis study of methyl derivatives of phenyl azide’, J. Phys. Chem. A, 1999, 103, 3458 CrossRef CAS; (b) N. P. Gritsan, A. D. Gudmundsdottir, D. Tigelaar, Z. Zhu, W. L. Karney, C. M. Hadad and M. S. Platz, ‘A laser flash photolysis and quantum chemical study of the fluorinated derivatives of singlet phenylnitrene’, J. Am. Chem. Soc., 2001, 123, 1951 CrossRef CAS; (c) N. P. Gritsan and M. S. Platz, ‘Kinetics and spectroscopy of substituted phenylnitrenes’, Adv. Phys. Org. Chem., 2001, 36, 255 CAS.
- C. B. Martin, X. Shi, M.-L. Tsao, D. Karweik, J. Brooke, C. M. Hadad and M. S. Platz, ‘The Photochemistry of Riboflavin Tetraacetate and Nucleosides. A Study Using Density Functional Theory, Laser Flash Photolysis, Fluorescence, UV-Vis, and Time Resolved Infrared Spectroscopy’, J. Phys. Chem. B, 2002, 106, 10263 CrossRef CAS.
- J. Peon, G. C. Hess, J.-M. L. Pecourt, T. Yuzawa and B. Kohler, ‘Ultrafast Photoionization Dynamics of Indole in Water’, J. Phys. Chem. A, 1999, 103, 2460 CrossRef CAS.
- (a) B. O. Roos, in ‘Ab Initio Methods in Quantum Chemistry’, ed. K. P. Lawley, Wiley: New York, 1987, vol. 2, p. 399 Search PubMed; (b) B. O. Roos, ‘The complete active space self-consistent field method and its applications in electronic structure calculations’, Adv. Chem. Phys., 1987, 69, 399 CAS; (c) B. O. Roos, ‘The complete active space SCF method in a Fock-Matrix-based super-CI formulation’, Int. J. Quant. Chem. Symp., 1980, 14, 175 Search PubMed.
- P. C. Hariharan and J. A. Pople, ‘Influence of polarization functions on MO hydrogenation energies’, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- W. L. Karney and W. T. Borden, ‘Ab Initio Study of the Ring Expansion of Phenylnitrene and Comparison with the Ring Expansion of Phenylcarbene’, J. Am. Chem. Soc., 1997, 119, 1378 CrossRef CAS.
- (a) K. Anderson, P.-Å. Malmqvist, B. O. Roos, A. J. Sadlej and K. Wolinski, ‘Second-order perturbation theory with a CASSCF reference function’, J. Phys. Chem., 1990, 94, 5483 CrossRef CAS; (b) K. Anderson, P.-Å. Malmqvist and B. O. Roos, ‘Second-order perturbation theory with a complete active space self-consistent field reference function’, J. Chem. Phys., 1992, 96, 1218 CrossRef CAS; (c) K. Anderson and B. O. Roos, ‘Multiconfigurational second-order perturbation theory: a test of geometries and binding energies’, Int. J. Quant. Chem., 1993, 45, 591 CrossRef CAS.
- B. O. Roos, K. Anderson, M. P. Fulscher, L. Serrano-Andres, K. Pierloot, M. Merchank and V. Molinac, ‘Application of level shift corrected perturbation theory in electronic spectroscopy’, J. Mol. Struct. (THEOCHEM), 1996, 388, 257 CrossRef CAS.
- K. Andersson, M. Barysz, A. Bernhardsson, M. R. A. Blomberg, D. L. Cooper, T. Fleig, M. P. Fülscher, C. de Graaf, B. A. Hess, G. Karlström, R. Lindh, P.-Å. Malmqvist, P. Neogrády, J. Olsen, B. O. Roos, A. J. Sadlej, M. Schütz, B. Schimmelpfennig, L. Seijo, L. Serrano-Andrés, P. E. M. Siegbahn, J. Stålring, T. Thorsteinsson, V. Veryazov andP.-O. Widmark, MOLCAS, Version 5, Lund University, Sweden, 2000 Search PubMed.
- (a) A. D. Becke, ‘Density-functional thermochemistry. III. The role of exact exchange’, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee and W. Yang, R. G. Parr, ‘Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density’, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, ‘Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr’, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- (a) M. J. Frisch, J. A. Pople and J. S. Binkley, ‘Self-consistent molecular orbital methods. 25. Supplementary functions for Gaussian basis sets’, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS; (b) T. Clark, J. Chandrasekhar and P. v. R. Schleyer, ‘Efficient diffuse function-augmented basis sets for anion calculations. III. The 3–21+G basis set for first-row elements, lithium to fluorine’, J. Comp. Chem., 1983, 4, 294 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, ‘Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: characterization and correction of the time-dependent local density approximation ionization threshold’, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.9), Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed.
- M.-L. Tsao and M. S. Platz, ‘Photochemistry of Ortho, Ortho′ Dialkyl Phenyl Azides’, J. Am. Chem. Soc., 2003, 125, 12014 CrossRef CAS.
- (a) S. A. Kovalenko, R. Schanz, H. Hennig and N. P. Ernsting, ‘Cooling dynamics of an optically excited molecular probe in solution from femtosecond broadband transient absorption spectroscopy’, J. Chem. Phys., 2001, 115, 3256 CrossRef CAS; (b) R. Knochenmuss, V. Karbach, C. Wickleder, S. Graf and S. Leutwyler, ‘Vibrational-energy redistribution and vibronic coupling in 1-naphthol×water complexes’, J. Phys. Chem. A, 1998, 102, 1935 CrossRef CAS; (c) V. Wong and M. Gruebele, ‘How Does Vibrational Energy Flow Fill the Molecular State Space?’, J. Phys. Chem. A, 1999, 103, 10664 CrossRef CAS; (d) N. E. Levinger, P. H. Davis and M. D. Fayer, ‘Vibrational relaxation of the free terminal hydroxyl stretch in methanol oligomers: Indirect pathway to hydrogen bond breaking’, J. Chem. Phys., 2001, 115, 9352 CrossRef CAS; (e) M. Gruebele, ‘Vibrational energy flow: a state space approach’, Adv. Chem. Phys., 2001, 114, 193 CAS.
- N. P. Gritsan and M. S. Platz, unpublished results.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |