Time-resolved fluorescence microscopy†
Klaus
Suhling
*a,
Paul M. W.
French
b and
David
Phillips
c
aDepartment of Physics, King's College London, Strand, London, UK WC2R 2LS. E-mail: klaus.suhling@kcl.ac.uk; Fax: +44-(0)20-7848-2420; Tel: +44-(0)20-7848 2119
bPhotonics Group, Department of Physics, Imperial College London, South Kensington Campus, London, UK SW7 2AZ
cDepartment of Chemistry, Imperial College London, South Kensington Campus, London, UK SW7 2AZ
First published on 11th November 2004
Abstract
In fluorescence microscopy, the fluorescence emission can be characterised not only by intensity and position, but also by lifetime, polarization and wavelength. Fluorescence lifetime imaging (FLIM) can report on photophysical events that are difficult or impossible to observe by fluorescence intensity imaging, and time-resolved fluorescence anisotropy imaging (TR-FAIM) can measure the rotational mobility of a fluorophore in its environment. We compare different FLIM methods: a chief advantage of wide-field time-gating and phase modulation methods is the speed of acquisition whereas for time-correlated single photon counting (TCSPC) based confocal scanning it is accuracy in the fluorescence decay. FLIM has been used to image interactions between proteins such as receptor oligomerisation and to reveal protein phosphorylation by detecting fluorescence resonance energy transfer (FRET). In addition, FLIM can also probe the local environment of fluorophores, reporting, for example, on the local pH, refractive index, ion or oxygen concentration without the need for ratiometric measurements.
 Klaus Suhling Klaus Suhling | Klaus Suhling is a lecturer in the Department of Physics at King’s College London. Trained as a physicist, he has held various post-doctoral positions in biology, chemistry and physics laboratories, most recently at Imperial College London. The common theme of most of his work has been fluorescence spectroscopy and imaging. His research at the interface of biology, chemistry and physics involves the development and use of fluorescence lifetime imaging (FLIM) and related techniques to understand the properties and interactions of macromolecules in the biological and biomedical sciences. |
 Paul M. W. French Paul M. W. French | Paul French was born in Felixstowe, UK, in 1962. He received his BSc degree in Physics and his PhD degree for work on femtosecond dye lasers from Imperial College, University of London, in 1983 and 1987, respectively. His subsequent research career at Imperial College London has concentrated on ultrafast laser technology and its applications, particularly in biomedical optics, although he worked on ultrafast all optical switching in optical fibres at AT&T Bell Laboratories from 1990 to 1991. He is currently a Professor of Physics at Imperial College London and is Head of the Photonics Group. His research portfolio includes ultrafast laser and fibre technology, high-speed 3-D imaging and multi-dimensional fluorescence imaging with a particular emphasis on fluorescence lifetime imaging. |
 David Phillips David Phillips | David Phillips is the Dean of the Faculties of Life Sciences and Physical Sciences at Imperial College London. He is a leading figure in the world of photochemistry and laser-based research and has published over 500 papers in this area. The main achievements of his group in the past decade have been in the field of photodynamic therapy (PDT): his group has synthesised new dyes, studied the binding of these dyes to proteins, and investigated the photophysics and photochemistry in detail, including measurement of singlet oxygen. To visualise the distribution of the dyes in tissue and in single cells, a variety of microscopies including confocal and picosecond time-resolved imaging has been developed in his laboratory. |
Introduction
Hiroshi Masuhara was a pioneer of the use of microscopy to study complex systems, mainly those of technological significance. In honour of his great contribution to his field, we offer here a review of fluorescence lifetime imaging (FLIM) instrumentation and highlight some cell biological studies.Optical imaging techniques, in particular fluorescence imaging techniques, are powerful tools in the biological and biomedical sciences today, because they are minimally invasive and can be applied to live cells and tissues.1,2 Microscopy, in particular, relies on the contrast that can be achieved from absorption, polarization, phase etc. in conventional microscopy, and fluorescence parameters in a fluorescence microscope. Conventionally, fluorescence intensity is measured, and three-dimensional images can be recorded using confocal [one or two-photon excitation] techniques. However, fluorescence intensity is dependent upon a variety of environmental influences, such as quenching by other molecules, aggregation, energy transfer, and refractive index effects, and can thus be difficult to quantify or interpret. FLIM provides contrast according to the fluorescence decay time, the inverse of the sum of the rate parameters for all depopulation processes.3
The fluorescence decay time, or as commonly used, lifetime τ is the average time a fluorophore remains in the excited state after excitation, and it is defined as
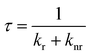 | (1) |
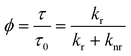 | (2) |
While the fluorescence decay time depends on the intrinsic characteristics of the fluorophore itself, it also depends in a measurable way upon the local environment. In general, the local viscosity, pH, or refractive index [see Fig. 1(a)], as well as interactions with other molecules e.g. by collision or energy transfer [Fig. 1(b)], can all affect the fluorescence lifetime.4,5 Thus, as well as being able to distinguish spectrally similar fluorophores,6 imaging of the fluorescence lifetime can be used to probe the surroundings of a fluorophore (see Table 1). In the last decade or so, since the first reports on FLIM,7,8 the technique has been improved, developed further and applied to an increasing number of studies in cell biology.9,10
![Schematic representation of the photophysical phenomena that FLIM can study, and their effect on the fluorescence decay. (a) The fluorescence decay is a function of the environment of the fluorophore. For example, the fluorescence lifetime of GFP is a function of its local refractive index.80 A low refractive index around GFP leads to a longer lifetime, whereas a high refractive index environment causes a shorter lifetime. The fluorescence lifetime of other probes can be sensitive to pH, ions, oxygen etc.
(see text). (b) FRET occurs when a suitable donor and acceptor are in close proximity, usually below 10 nm. Thus imaging of FRET can measure the proximity of fluorescent or fluorophore-tagged proteins in live cells with 10–100 times the resolution limit in far-field optical microscopes. With FLIM, FRET between a donor and acceptor can be identified by a shortened fluorescence decay of the donor. (c) Polarization-resolved FLIM to perform time-resolved fluorescence anisotropy imaging55,56 reveals the rotational mobility of a fluorophore. This is affected by the viscosity of its surroundings, or by binding and conformational changes that affect the rotational mobility. The latter is characterized by the rotational correlation time which can be calculated from the difference between the polarization-resolved fluorescence decays I∥ and I⊥. A fast rotational motion leads to a rapid depolarization. Furthermore, the initial anisotropy may be used to probe homo-FRET. In each of these applications, the fluorescence lifetime [or in (c), the rotational correlation time] is encoded in a false colour scale for each pixel of the image to generate contrast.](/image/article/2005/PP/b412924p/b412924p-f1.gif) | ||
| Fig. 1 Schematic representation of the photophysical phenomena that FLIM can study, and their effect on the fluorescence decay. (a) The fluorescence decay is a function of the environment of the fluorophore. For example, the fluorescence lifetime of GFP is a function of its local refractive index.80 A low refractive index around GFP leads to a longer lifetime, whereas a high refractive index environment causes a shorter lifetime. The fluorescence lifetime of other probes can be sensitive to pH, ions, oxygen etc. (see text). (b) FRET occurs when a suitable donor and acceptor are in close proximity, usually below 10 nm. Thus imaging of FRET can measure the proximity of fluorescent or fluorophore-tagged proteins in live cells with 10–100 times the resolution limit in far-field optical microscopes. With FLIM, FRET between a donor and acceptor can be identified by a shortened fluorescence decay of the donor. (c) Polarization-resolved FLIM to perform time-resolved fluorescence anisotropy imaging55,56 reveals the rotational mobility of a fluorophore. This is affected by the viscosity of its surroundings, or by binding and conformational changes that affect the rotational mobility. The latter is characterized by the rotational correlation time which can be calculated from the difference between the polarization-resolved fluorescence decays I∥ and I⊥. A fast rotational motion leads to a rapid depolarization. Furthermore, the initial anisotropy may be used to probe homo-FRET. In each of these applications, the fluorescence lifetime [or in (c), the rotational correlation time] is encoded in a false colour scale for each pixel of the image to generate contrast. | ||
| Advantages | Disadvantages |
|---|---|
| The fluorescence lifetime is a molecular property generally independent of variations in fluorophore concentration, illumination intensity, light pathlength, scatter, or photobleaching | Photophysical and time-resolved fluorescence spectroscopy expertise required for data interpretation (e.g. fluorescent proteins usually have complex fluorescent decays) |
| FLIM can robustly and quantitatively probe the fluorophore's local environment directly, e.g. refractive index, viscosity, pH, ions etc. without the need to compromise the cell with biochemical assays | Interpretation of specific changes in fluorescence lifetime in terms of the underlying cell biochemistry may not be straighforward |
| FLIM of FRET by imaging the fluorescence decay of the donor is more robust than fluorescence intensity-based FRET and allows to distinguish between effects due to FRET efficiency and probe concentration. Quantitative FRET studies between spectrally similar donor and acceptor, e.g. GFP and YFP are also possible88 | Complex and expensive equipment required |
| FLIM can experimentally distinguish spectrally similar probes (if their fluorescence lifetimes are different) with the same detector |
FLIM instrumentation
FLIM is a time-resolved image acquisition method, the technologies for which can be divided into two categories: (i) confocal scanning11,12 or multiphoton excitation13,14 FLIM where the image is acquired pixel-by-pixel using a non-imaging detector, e.g. a photomultiplier, and (ii) wide-field camera-based FLIM.15 The time-resolved information is obtained either in the time domain by exciting the sample with a short optical pulse and observing the decay of the fluorescence intensity (with time-correlated single photon counting (TCSPC), gating, or a streak camera), or in the frequency domain by modulating the excitation source and/or the detector to calculate the fluorescence decay time from the demodulation and the phase shift of the fluorescence.In the time domain, a fluorescence decay curve can be directly acquired after excitation of the sample with an ultrashort light pulse, usually using a sampling technique.8,16–19 In wide-field time-gated FLIM, ‘snapshots’ of the fluorescence emission are taken at various nanosecond delays after the excitation using high-speed gated image intensified cameras.15,17 This approach is fast, since all the pixels are acquired in parallel—a 100 Hz FLIM frame rate has been reported20—but it lacks single photon sensitivity and accuracy, and its temporal resolution is limited to ≈80 ps21 (see Table 2). In confocal scanning or multiphoton excitation microscopes (which provide inherent optical sectioning) FLIM is essentially a series of single channel fluorescence lifetime measurements where the fluorescence decay can be acquired by TCSPC.22,23 TCSPC is a mature and reliable technique which records the arrival time of single photons after an excitation pulse. The ease of reproducibility of measurements is due to the unique combination of advantages such as the unlimited dynamic range associated with photon counting techniques, linear recording characteristics independent of excitation intensity fluctuations and photobleaching, well-defined Poisson statistics, excellent signal to noise ratio and a high temporal (picosecond) resolution (see Table 2). As each photon is timed individually in each pixel of the image, the collection of many photons for a high statistical accuracy can be time-consuming.24 The maximum photon flux that can be timed using a single channel (one detector, time to amplitude converter (TAC) and analogue to digital converter (ADC)) is limited by photon pile-up and the dead time of the electronics to ≈106 photons s−1.
| Implementation | Advantages | Disadvantages |
|---|---|---|
| Time-gated wide-field time domain | Fast | Low sensitivity, need strong signal |
| All pixels acquired in parallel | Consecutive acquisition of time gates vulnerable to photobleaching and sample movement | |
| Poor spatial resolution due to phosphor screen on intensified CCD camera | ||
| Need pulsed laser | ||
| Wide-field frequency domain | Fast | Cannot easily resolve two very different lifetimes |
| Easy to modulate cw laser | Vulnerable to photobleaching and sample movement | |
| Can resolve two similar lifetimes | Complex data and error analysis | |
| No deconvolution (temporal) of instrumental response and fluorescence decay necessary | Poor spatial resolution due to phosphor screen on intensified CCD camera | |
| All pixels acquired in parallel | Usually lower temporal resolution and lower signal-to-noise ratio than time domain methods | |
| Confocal/multiphoton scanning with time-correlated single photon counting | Single photon sensitivity | Slow, each photon has to be timed individually |
| Unlimited dynamic range associated with photon counting techniques | Need pulsed laser | |
| Linear recording characteristics independent of excitation intensity fluctuations and photobleaching | ||
| Easy visualization of fluorescence decays and well-defined Poisson statistics | ||
| Inherent optical sectioning | ||
| Best signal to noise ratio | ||
| High temporal resolution | ||
| Confocal/multiphoton scanning with time-binning photon detection | Fastest scanning technique | Need pulsed laser |
| Inherent optical sectioning | Slower than wide-field imaging | |
| Single photon sensitivity | Less accurate than time-correlated single photon counting | |
| Streak camera FLIM | Very high temporal resolution | Expensive |
| Fast | ||
| Easy visualization of fluorescence decays |
A similar but rather faster approach is to bin all incoming photons within preset time windows after excitation.16,25 This time-binning method is significantly faster than TCSPC because it is not necessary to reduce the fluorescence signal to the level of single photon timing. However, it is less accurate than TCSPC (see Table 2). The use of streak-camera based FLIM has also recently been reported.26–28 The technique works in line-scanning mode, is fast, has the highest temporal resolution of any FLIM technique, and it has been commercialised.
In the frequency domain periodically modulated excitation beams and detectors may be used to measure the phase shift and demodulation of fluorescence signals with respect to their excitation signals, both in wide-field microscopy using modulated intensified cameras29–32 and in confocal/multi-photon laser scanning microscopy using single channel detectors.33–37 Frequency domain techniques have been used since the 1920s to measure nanosecond fluorescence decays. With this approach a fluorescence lifetime may be calculated from both the phase shift and demodulation (at several modulation frequencies if necessary).38 For a simple mono-exponential fluorescence decay profile, both calculations should yield the same value. For more complex decays, e.g. in the case of some fluorescent proteins such a CFP, the phase shift lifetime is shorter than the demodulation lifetime.29
There is a lively debate as to the relative merits of time or frequency domain approaches to FLIM. In principle they are, of course, related by a Fourier transformation and have experimentally been demonstrated to be equivalent.39 To non-specialists, the easy visualization of fluorescence decays in the time domain may be an advantage over the frequency domain, where the analysis of complex fluorescence decay profiles, such as stretched exponentials, is less tractable than in the time domain.40 However, for some applications the frequency domain instrumentation is considered easier to implement since ultrashort pulsed laser sources are not required, especially for longer lifetimes, although practitioners are increasingly using mode-locked lasers for frequency domain measurements—particularly in multiphoton microscopes.41,42 Frequency domain techniques are more photon efficient than time-gating techniques and require no deconvolution of the instrumental response and the fluorescence decay (see Table 2). However, a recent study shows that the signal-to-noise ratio is higher for time domain measurements than for frequency domain measurements.43
One potential pitfall of the time domain approach is that there needs to be sufficient time (≈5τ) between excitation pulses for the sample fluorescence to completely decay in order to obtain accurate fluorescence lifetime values. In practice this implies using mode-locked lasers with pulse-pickers, cavity-dumpers, lower repetition rate pulsed diode lasers44 or appropriate fitting procedures to take residual fluorescence into account.45 This is not an issue for the frequency domain approach.
The FLIM techniques continue to be improved, particularly by the reduction of acquisition times,25 the extension to include spectrally-resolved imaging46,47 and rapid optical sectioning capabilities.48,49 The relative merits of the various FLIM implementations are summarized in Table 2, and it depends on the operator's preference (and finances!) for fast data acquisition or accuracy, high temporal and spatial resolution which system to choose.
Time resolved fluorescence anisotropy imaging (TR-FAIM)
Polarization-resolved FLIM captures a further parameter of the multidimensional fluorescence emission contour and allows complementary information about the fluorophore's environment to be obtained. Upon excitation with linearly polarized light, rotational diffusion of the fluorophore in its excited state results in a depolarization of the fluorescence emission.4,5 Steady-state anisotropy imaging has, for example, been used to obtain contrast between fluorescein and GFP in a cell due to their different anisotropies, i.e. their molecular sizes.50 In additon, energy migration or homo-FRET [resonance energy transfer between the same type of fluorophore] can be detected with this method, since it also leads to a depolarization of the emitted fluorescence.51,52 The technique has been used to study the proximity of isoforms of the GPI-anchored folate receptor bound to a fluorescent analogue of folic acid to detect lipid rafts.53 Fluorescence decays measured at polarizations parallel and perpendicular to that of the excitation, i.e. TR-FAIM, can measure the rotational mobility of the fluorophore in its environment [Fig. 1(c)]. Thus, the viscosity of the fluorophore's environment, binding events or hindered rotation can be examined with this method.51,54–56 The time-resolved fluorescence anisotropy r(t) can be defined as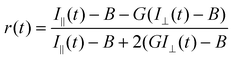 | (3) |
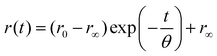 | (4) |
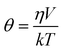 | (5) |
Clayton et al. used time-resolved fluorescence anisotropy imaging not only to map the viscosity of fluorescein solutions, but also to study the average distance between GFPs expressed in bacteria.56 They detected a reduced initial anisotropy which was thought to be caused by GFP-GFP FRET due to the close proximity of the proteins in bacteria. Moreover, as the rotational diffusion can be slowed down by binding, TR-FAIM has potential to visualise the binding of ligands and receptors in the cell. The system used in the authors’ laboratory for TR-FAIM is shown in Fig. 2.
 | ||
| Fig. 2 (a) Experimental set-up of the wide-field TR-FAIM instrument (polarization-resolved time-gated FLIM). The Polarization-Resolved Imager (PRI) contains a polarizing beamsplitter and adjustable mirrors. Using a C-mount adapter, the PRI is mounted onto the gated optical image intensifier (GOI), the output phosphor screen of which is imaged with a CCD camera. The PRI splits a single image in image plane 1 into two spatially identical images differing only by their polarization (image plane 2), which are thus recorded simultaneously. (b) A series of such polarization-resolved fluorescence intensity image pairs are acquired at various delays after the excitation pulse to sample their fluorescence decay profiles. (c) A rotational correlation time image of a B cell stained with the fluorescein derivative CFSE staining the cytoplasm. The rotational correlation time reports on the viscosity of the cytoplasm. Its average value is 4.50 ± 0.87 ns at 20 °C, which corresponds to an average cytoplasmic viscosity of 14 cp. The image was obtained on a wide-field time-gated FLIM microscope with a ×63 water immersion objective.54 | ||
Applications of FLIM to cell biology
Förster resonance energy transfer (FRET)
The most widespread application of FLIM in cell biology is the identification of FRET upon the interaction between suitably (and stochiometrically) labelled specific proteins, lipids, enzymes, DNA and RNA, as well as cleavage of a protein, or conformational changes within a protein.57–59 FRET is a bimolecular fluorescence quenching process where the excited state energy of a donor fluorophore is non-radiatively transferred to a ground state acceptor molecule by a dipole–dipole coupling process.60 The FRET efficiency, E, varies with the inverse 6th power of the distance between donor and acceptor, and is usually negligible beyond 10 nm. FRET can therefore be used as a ‘spectroscopic ruler’ to probe intermolecular distances on the scale of the dimensions of the proteins themselves.61–63 This is a significant advantage over co-localization studies with two fluorophores which is limited by the optical resolution (approximately 200 nm laterally, 500 nm axially11,12).For FRET to occur, the emission spectrum of the donor and the absorption spectrum of the acceptor must overlap,64 and the transition dipole moments of the donor and acceptor must not be perpendicular—otherwise the transfer efficiency is zero, irrespective of the donor–acceptor distance or the spectral overlap.65,66 The critical transfer distance R0, where FRET and fluorescence emission are equally likely, can be calculated from the spectral overlap. We note here that free PhotochemCAD software to calculate the R0 for any donor/acceptor pair can be downloaded from http://chemdept.chem.ncsu.edu/%7Ejslftp/.67 Imaging FRET68 can thus be used to map interactions between proteins, lipids, enzymes, DNA and RNA, as well as follow cleavage of a protein, or conformational changes within a protein.1,2,69,70 FRET, as a fluorescence quenching process, reduces the quantum yield and the fluorescence lifetime of the donor. If the acceptor is fluorescent (which incidentally is not a necessary requirement for FRET to occur), FRET leads to sensitized acceptor emission. To identify and quantify FRET, the fluorescence decay of the donor can be measured in the absence and presence of the acceptor. The advantage of time-resolved over intensity-based measurements is the ability to directly distinguish between effects due to FRET or probe concentration. For example, a low donor fluorescence intensity can be caused by either a low donor concentration or efficient quenching—but only in the latter case is the fluorescence decay shortened [see Fig. 1(b)].
Before the availability of fluorescent proteins, intracellular fusion of endosomes was studied with FLIM of FRET.71 The endosomes were sequentially loaded using calcein as the donor, and a sulforhodamine acceptor, and FRET occurred upon the fusion of the endosomes containing the donor and those containing the acceptor. Another example before the common use of fluorescent proteins in cell biology is the study of the dimerisation of epidermal growth factor (EGF) receptors by covalently binding fluorescein and rhodamine to EGF, and observing FRET upon their dimerization.72 A similar concept was used to study the role of the protein kinase C (PKC) family of proteins in cellular signal transduction. This was done using purified PKC βI, tagging it with the fluorophore Cy3 and microinjecting it into cells.73 When the investigators co-injected an antibody to PKC βI tagged with Cy5, they found that FRET was abrogated for intranuclear PKC βI, concluding that this protein is fragmented there. A limitation of this approach is that the microinjection into cells is not a routine procedure for many cell types and is somewhat invasive.
Now, fluorescent proteins can be directly tagged to a specific protein using genetic means, which is usually minimally invasive.74,75 The excitation and emission spectra of the green fluorescent protein (GFP) are in the visible range, but the photophysics of the fluorescence proteins is complex.76 The widely used mutant enhanced GFP (F64L, S65T), for example, has at least two emitting states.77–80 Nonetheless, FLIM of GFP, and their spectral variants cyan or cerulean fluorescent protein (CFP) and yellow fluorescence protein (YFP), with average fluorescence lifetimes in the 2–3 ns region,81 has proved valuable.
FLIM of FRET between the donor PKCα-GFP and the cytoskeletal linker ezrin stained with a Cy3-labelled antibody as the acceptor showed that, on activation, PKCα colocalizes and interacts with ezrin at the plasma membrane.82 Association of the protein CD44 with ezrin upon PKC activation was observed by a similar methodology, using FLIM of FRET between GFP-tagged CD44 and a Cy3-conjugated antibody.83 These studies serve to demonstrate an approach that could be exploited for studying the constitutive or transient association of numerous other receptors with ERM proteins, other cytoskeletal linkers, or adaptor proteins.
FLIM of the GFP fluorescence decay has been used to report on the phosphorylation of GFP-tagged PKCα in live cells by identifying a shortened GFP fluorescence lifetime due to FRET between the donor PKCα-GFP and a Cy3.5-labelled phosphorylation specific antibody as the acceptor.84 A disadvantage of this application is that it requires use of an antibody against a specific phosphorylated form of the protein, which is not always available. However, this requirement was circumvented in imaging the phosphorylation of GFP-tagged ErB1 receptors, by identifying a shortened GFP fluorescence lifetime due to FRET between the donor ErB1-GFP and a Cy3-labelled antibody to phosphotyrosine.85,86 Dephosphorylation has also been studied with FRET.87
FLIM of FRET between the spectrally similar donor GFP and acceptor YFP has been used to monitor caspase activity in individual cells during apoptosis.88 Here, a tandem GFP YFP construct incorporating the DEVD caspase recognition sequence is used as a monitor of apoptosis such that FRET occurs between them. The GFP and YFP fluorescence decays are collected simultaneously, and using appropriate data analysis, FRET is identified by a rise time due to sensitised acceptor emission. As the protein undergoes proteolytic cleavage, GFP and YFP move away from each other and FRET is disrupted. In this case, detecting FRET by FLIM is particularly advantageous over detecting FRET by imaging fluorescence intensities, since the donor and acceptor emission spectra overlap so that their fluorescence intensities cannot be easily separated.
FLIM of FRET has also been used to probe NADH89,90 and the the supramolecular organization of DNA.91–94
FLIM of Ca2+, oxygen, pH concentration
Instead of using intensity-based imaging of ratiometric probes, the fluorescence lifetime of the Ca2+ sensor Quin-2,95,96 calcium crimson97 or CalciumGreen and Fluo-398 has been used to image the Ca2+ concentration in cells. The use of FLIM in these cases is more robust and reliable than fluorescence intensity-based imaging methods, since FLIM is unaffected by variations of illumination intensity, variations of fluorophore concentration or photobleaching–provided the probes do not aggregate, and the photoproducts do not fluoresce (see Table 1). Moreover, FLIM of a long-lived ruthenium-based oxygen sensor with an unquenched decay time of 760 ns has been used to map oxygen concentrations in macrophages.99 Note that intensity-based fluorescence imaging of oxygen in cells would require a calibration of the intensity of the probe unquenched by oxygen as well as knowing its concentration in the cell. This is not practically possible. FLIM has also been used to map the pH in single cells34,100,101 and skin.41,42 Here the pH sensor 2′,7′-bis-(2-carboxyethyl)-5-(and-6) carboxyfluorescein (BCECF) was used to image pH in the skin stratum corneum. The authors used two-photon excitation FLIM to non-destructively obtain pH maps at various depths, which is difficult to achieve by non-optical methods. Moreover, as the authors point out, intensity-based fluorescence imaging of the pH probe could not have been used for their study as the observation of a variation in fluorescence intensity could be ascribed to either a change in pH or a variation of the local probe concentration.FLIM of tissue
FLIM of autofluorescence has been used to provide intrinsic contrast in unstained tissue21,102–105 and teeth.106,107 The combination of multiphoton excitation for deep, sectioned, tissue imaging with FLIM yields contrast not available with fluorescence intensity-based imaging. FLIM has also been employed to study aggregation of sensitisers in photodynamic therapy.108–113Perspectives
FLIM to probe local viscosity and membrane fluidity and polarity?
The fluorescence lifetime of so-called molecular rotors, which provide a non-radiative de-excitation pathway by internal twisting in competition with radiative de-excitation, is strongly dependent on the viscosity of their surroundings, as this affects the internal twisting.114,115 This property has been used to measure the fluidity of cell membranes116 and binding in bulk solution.117 The use of molecular rotors has been extended to imaging118 and could be combined with FLIM to image the viscosity distribution in cells.Ratiometric imaging of excited state dimer (excimer) formation of the aromatic hydrocarbon probe pyrene has also been used to study lateral diffusion in membranes.119 An excimer is only formed between an excited state and ground state molecule, and it occurs when the monomers are in close proximity. It is characterized by a red-shift in the emission spectrum, as well as a bi-exponential fluorescence decay, with one component originating from the monomer, and the other one from the excimer. As excimer formation is a diffusion-controlled process it can be used to study the membrane fluidity, and FLIM would facilitate mapping of excimer distributions. In addition, membrane probes such as Laurdan may also be used to study membrane fluidity and cholesterol content.120
FLIM to probe local refractive index?
Recently, a comprehensive comparison of the fluorescence emission of the enhanced green fluorescent protein GFP (i.e. GFP F64L, S65T) in solution and in an intracellular environment was reported.121 Careful analysis of the GFP fluorescence decays, obtained by TCSPC, revealed that the average fluorescence lifetime of GFP was about 8% shorter in the cytoplasm of rat basophilic leukemia mast cells than in aqueous solution—an experimental observation in agreement with those of other workers.81,122,123 It may be interpreted, at least in part, in terms of the refractive index of the GFP environment.In general, kr in eqn. (1) is a function of the refractive index in the vicinity of the fluorophore, recently reviewed by Toptygin.124 A simple model for this phenomenon is given by the Strickler–Berg equation125
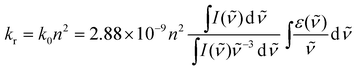 | (6) |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) the wavenumber. kr explicitly depends on the refractive index due to the polarizability of the host medium surrounding the fluorophore where the absorption and emission processes occur. Many experimental observations demonstrate the influence of the refractive index on fluorescence lifetimes, as discussed in ref. 80 and 124.
the wavenumber. kr explicitly depends on the refractive index due to the polarizability of the host medium surrounding the fluorophore where the absorption and emission processes occur. Many experimental observations demonstrate the influence of the refractive index on fluorescence lifetimes, as discussed in ref. 80 and 124.
We recently studied the fluorescence decay of GFP in aqueous solution with added glycerol, polyethylene glycol, NaCl or glucose and fructose.80 Quantitative analysis showed that τav−1 varied approximately linearly with the square of the refractive index in accordance with the Strickler–Berg equation, irrespective of whether the refractive index is increased by adding glycerol, polyethylene glycol, NaCl or glucose and fructose,80 as shown in Fig 3.
![(a) The inverse average fluorescence lifetime τav−1 of GFP versus the square of the refractive index of the solution. τav−1 varies linearly with the square of the refractive index inaccordance with the Strickler–Berg formula [eqn. (6)], irrespective of whether the refractive index is increased by adding glycerol, polyethylene glycol, NaCl or glucose and fructose. (b) Fluorescence lifetime image of GFP in mixtures of aqueous buffer and glycerol in a multiwell plate.80,144](/image/article/2005/PP/b412924p/b412924p-f3.gif) | ||
| Fig. 3 (a) The inverse average fluorescence lifetime τav−1 of GFP versus the square of the refractive index of the solution. τav−1 varies linearly with the square of the refractive index inaccordance with the Strickler–Berg formula [eqn. (6)], irrespective of whether the refractive index is increased by adding glycerol, polyethylene glycol, NaCl or glucose and fructose. (b) Fluorescence lifetime image of GFP in mixtures of aqueous buffer and glycerol in a multiwell plate.80,144 | ||
The observation that, in reverse micelles, the average GFP fluorescence lifetime is lower in the water pool than in bulk water, and that it increases with increasing water pool size79 is consistent with the decreasing refractive index of the reverse micelle as the water pool is increased.126 In biological cells, the cellular refractive index is higher to that of the surrounding medium—a fact exploited in light microscopy to generate contrast. It is therefore consistent that the fluorescent lifetime of GFP is shorter in cells than in buffer.81,121–123
Despite this consistent interpretation, a cell is a much more complex system than a carefully controlled homogeneous solution. The lifetime of GFP in cells may also be affected by other factors, but our work shows that the refractive index effect on the GFP fluorescence decay cannot be neglected.
Using the fluorescence lifetime of GFP as a refractive index sensor may allow FLIM to directly image the refractive index around specific GFP-tagged proteins in live cells. A biologically relevant and specific interpretation of a local refractive index may not be straightforward, but local refractive index variations could indicate heterogeneity. This may be of use, for example, in understanding complex supramolecular processes such as the assembly of an immunological synapse.127–129 On average, the cell membrane has a higher refractive index than the cytoplasm, nmembrane=1.46–1.60, and ncytoplasm=1.35.130,131 Different domains within a cell membrane may differ in refractive index, and contrast in the fluorescence lifetime image of GFP-tagged membrane proteins may reflect heterogeneity of the composition of the membrane. Indeed, using GFP-tagged MHC proteins in the target cell of with NK cell immune synapse, we have observed a small GFP fluorescence lifetime contrast at the intercellular contact.132
Outlook
FLIM of cell biology will not only be improved by progress in instrumentation, but also by advances in the techniques, methods and fluorescent labels used. New fluorescent proteins that can be activated by light,133 or probes that allow FRET to be switched on and off by light may allow a greater versatility in studying protein–protein interactions.134 It is likely that for imaging of cell biology, the use of quantum dots with a high fluorescence quantum yield, low photobleaching susceptibility and narrow, size-dependent emission spectra which can be excited with a single wavelength will become more widespread.135 However the extent to which they are useful for FLIM is still unclear,136 as their non-radiative rate constant appears to fluctuate.137,138 On the other hand, genetically encoded fluorophores have already been designed for a wide range of wavelengths.74 In time the complex photophysics of these probes may be fully understood. However, preferably for FLIM a fluorescent protein will be discovered or engineered to have a single excited state and a single fluorescence lifetime. The CFP variant has recently been improved along those lines with a higher quantum yield and a nearly monoexponential decay.139 Ideally, in the future this single fluorescent lifetime would be made sensitive to a biophysical parameter of choice!The application of new physical techniques to important problems in cell biology is often the path to unexpected discoveries. For example, micrometer-scale supramolecular organization of T cell receptors and integrins at immunological synapses were discovered by the use of 3-D fluorescence microscopy.127–129 There is clearly a tremendous way to go before we are close to saturating the capabilities of fluorescence imaging for cell biology. Imaging fluorescence parameters such as lifetime, spectrum and polarization, as well as imaging more rapidly in three dimensions at higher resolution, are certain to reveal exciting new aspects of inter- and intra-cellular communication.
Acknowledgements
We gratefully acknowledge David Bacon's assistance with Fig. 1, Peter Lanigan, Richard Benninger, Björn Önfeld, Bebhinn Treanor and Dan Davis in the Departments of Physics and Biological Sciences at Imperial College London for critical comments on the manuscript. The work in our laboratories is funded by the UK's Biotechnology and Biological Sciences Research Council, Medical Research Council, Engineering and Physical Sciences Research Council, a DTI Beacon Grant and the Royal Society, and an EU INTAS Grant.References
- F. S. Wouters, P. J. Verveer and P. I. Bastiaens, Imaging biochemistry inside cells, Trends Cell Biol., 2001, 11, 203–211 CrossRef CAS.
- P. I. H. Bastiaens and R. Pepperkok, Observing proteins in their natural habitat: the living cell, Trends Biochem. Sci., 2000, 25, 631–637 CrossRef CAS.
- D. Phillips, Luminescence lifetimes in biological systems, Analyst, 1994, 119, 543–550 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic–Plenum Publishers, New York, 2nd edn., 1999 Search PubMed.
- B. Valeur, Molecular Fluorescence, Wiley-VCH, New York, 2002 Search PubMed.
- H. Brismar and B. Ulfhake, Fluorescence lifetime measurements in confocal microscopy of neurons labeled with multiple fluorophores, Nat. Biotechnol., 1997, 15, 373–377 CrossRef CAS.
- C. G. Morgan, A. C. Mitchell and J. G. Murray, Nanosecond Time-Resolved Fluorescence Microscopy: Principles and Practice, Proc. R. Microsc. Soc., 1990, 1, 463–466 Search PubMed.
- I. Bugiel, K. König and H. Wabnitz, Investigation of cells by fluorescence laser scanning microscopy with subnanosecond time resolution, Lasers Life Sci., 1989, 3, 47–53 Search PubMed.
- P. Herman, H.-J. Lin and J. R. Lakowicz, Lifetime-based imaging, in Biomedical Photonics Handbook, CRC Press, Boca Raton, FL, 2003, pp. 9-1–9-30 Search PubMed.
- R. M. Clegg, O. Holub and C. Gohlke, Fluorescence lifetime-resolved imaging: measuring lifetimes in an image, in Methods in Enzymology, Academic Press, New York, 2003, pp. 509–542 Search PubMed.
- W. B. Amos, Instruments for fluorescence imaging, in Protein localization by fluorescence microscopy. A practical approach, Oxford University Press, Oxford, 2000, pp. 67–108 Search PubMed.
- C. J. R. Sheppard, Scanning confocal microscopy, in Encyclopedia of Optical Engineering, Marcel Dekker, New York, 2003, pp. 22–41 Search PubMed.
- P. T. C. So, C. Y. Dong, B. R. Masters and K. M. Berland, Two-Photon Excitation Fluorescence Microscopy, Annu. Rev. Biomed. Eng., 2000, 2, 399–430 Search PubMed.
- K. König, Multiphoton microscopy in life sciences, J. Microsc., 2000, 200, 83–104 CrossRef CAS.
- S. E. D. Webb, Y. Gu, S. Lévěque-Fort, J. Siegel, M. J. Cole, K. Dowling, R. Jones, P. M. W. French, M. A. A. Neil, R. Juškaitis, L. O. D. Sucharov, T. Wilson and M. J. Lever, A wide-field time-domain fluorescence lifetime imaging microscope with optical sectioning, Rev. Sci. Instrum., 2002, 73, 1898–1907 CrossRef CAS.
- E. P. Buurman, R. Sanders, A. Draaijer, H. C. Gerritsen, J. J. F. van Ween, P. M. Houpt and Y. K. Levine, Fluorescence lifetime imaging using a confocal laser scanning microscope, Scanning, 1992, 14, 155–159 Search PubMed.
- K. Dowling, S. C. W. Hyde, J. C. Dainty, P. M. W. French and J. D. Hares, 2-D fluorescence lifetime imaging using a time-gated image intensifier, Opt. Commun., 1997, 135, 27–31 CrossRef CAS.
- X. F. Wang, A. Periasamy, B. Herman and D. M. Coleman, Fluorescence lifetime imaging microscopy (FLIM): Instrumentation and applications, Crit. Rev. Anal. Chem., 1992, 23, 369–395 CAS.
- A. Periasamy, P. Wodnicki, X. F. Wang, S. Kwon, G. W. Gordon and B. Herman, Time-resolved fluorescence lifetime imaging microscopy using a picosecond pulsed tunable dye laser system, Rev. Sci. Instrum., 1996, 67, 3722–3731 CrossRef CAS.
- A. V. Agronskaia, L. Tertoolen and H. C. Gerritsen, High frame rate fluorescence lifetime imaging, J. Phys. D: Appl. Phys., 2003, 36, 1655–1662 CrossRef.
- K. Dowling, M. J. Dayel, M. J. Lever, P. M. W. French, J. D. Hares and A. K. L. Dymoke-Bradshaw, Fluorescence lifetime imaging with picosecond resolution for biomedical applications, Opt. Lett., 1998, 23, 810–812 Search PubMed.
- D. J. S. Birch and R. E. Imhof, Time-domain Fluorescence Spectroscopy Using Time-Correlated Single Photon Counting, in Topics in Fluorescence Spectroscopy: Techniques, Plenum Press, New York, 1991 Search PubMed.
- D. V. O'Connor and D. Phillips, Time-correlated single-photon counting, Academic Press, New York, 1984 Search PubMed.
- W. Becker, A. Bergmann, M. A. Hink, K. König, K. Benndorf and C. Biskup, Fluorescence Lifetime Imaging by Time-Correlated Single-Photon Counting, Microsc. Res. Tech., 2004, 63, 58–66 CrossRef CAS.
- H. C. Gerritsen, N. A. H. Asselbergs, A. V. Agronskaia and W. G. J. H. M. Van Sark, Fluorescence lifetime imaging in scanning microscopes: acquisition speed, photon economy and lifetime resolution, J. Microsc., 2002, 206, 218–224 CrossRef CAS.
- R. V. Krishnan, E. Biener, J. H. Zhang, R. Heckel and B. Herman, Probing subtle fluorescence dynamics in cellular proteins by streak camera based fluorescence lifetime imaging microscopy, Appl. Phys. Lett., 2003, 83, 4658–4660 CrossRef CAS.
- R. V. Krishnan, A. Masuda, V. E. Centonze and B. Herman, Quantitative imaging of protein–protein interactions by multiphoton fluorescence lifetime imaging microscopy using a streak camera, J. Biomed. Opt., 2003, 8, 362–367 CrossRef CAS.
- R. V. Krishnan, H. Saitoh, H. Terada, V. E. Centonze and B. Herman, Development of a multiphoton fluorescence lifetime imaging microscopy system using a streak camera, Rev. Sci. Instrum., 2003, 74, 2714–2721 CrossRef CAS.
- T. W. J. Gadella, T. M. Jovin and R. M. Clegg, Fluorescence lifetime imaging microscopy (FLIM) - spatial resolution of structures on the nanosecond timescale, Biophys. Chem., 1993, 48, 221–239 CrossRef CAS.
- J. R. Lakowicz and K. Berndt, Lifetime-selective fluorescence imaging using an rf phase-sensitive camera., Rev. Sci. Instrum., 1991, 62, 1727–1734 CrossRef CAS.
- J. R. Lakowicz, H. Szmacinski, K. Nowaczyk, K. W. Berndt and M. Johnson, Fluorescence lifetime imaging, Anal. Biochem, 1992, 202, 316–330 CAS.
- C. G. Morgan, A. C. Mitchell and J. G. Murray, In situ fluorescence analysis using nanosecond decay time imaging, Trends Anal. Chem., 1992, 11, 32–41 CrossRef CAS.
- S. Andersson-Engels, I. Rokahr and J. Carlsson, Time-Resolved and Wavelength-Resolved Spectroscopy in 2-Photon-Excited Fluorescence Microscopy, J. Microsc., 1994, 176, 195–203 CAS.
- K. Carlsson, A. Liljeborg, R. M. Andersson and H. Brismar, Confocal pH imaging of microscopic specimens using fluorescence lifetimes and phase fluorometry: influence of parameter choice on system performance, J. Microsc., 2000, 199, 106–114 CrossRef CAS.
- T. French, P. T. C. So, D. J. Weaver, T. Coelho-Sampaio, E. Gratton, E. W. Voss and J. Carrero, Two-photon fluorescence lifetime imaging microscopy of macrophage-mediated antigen processing, J. Microsc., 1997, 185, 339–353 CrossRef CAS.
- K. Carlsson and A. Liljeborg, Simultaneous confocal lifetime imaging of multiple fluorophores using the intensity-modulated multiple-wavelength scanning (IMS) technique, J. Microsc., 1998, 191, 119–127 CrossRef CAS.
- M. J. Booth and T. Wilson, Low-cost, frequency-domain, fluorescence lifetime confocal microscopy, J. Microsc., 2004, 214, 36–42 CrossRef CAS.
- A. Squire, P. J. Verveer and P. I. H. Bastiaens, Multiple frequency fluorescence lifetime imaging microscopy, J. Microsc., 2000, 197, 136–149 CrossRef.
- J. Hedstrom, S. Sedarus and F. G. Prendergast, Measurements of fluorescence lifetimes by use of a hybrid time-correlated and multifrequency phase fluorometer, Biochemistry, 1988, 27, 6203–6208 CrossRef CAS.
- K. C. B. Lee, J. Siegel, S. E. D. Webb, S. Lévěque-Fort, M. J. Cole, R. Jones, K. Dowling, M. J. Lever and P. M. W. French, Application of the stretched exponential function to fluorescence lifetime imaging, Biophys. J., 2001, 81, 1265–1274 CAS.
- M. J. Behne, J. W. Meyer, K. M. Hanson, N. P. Barry, S. Murata, D. Crumrine, R. W. Clegg, E. Gratton, W. M. Holleran and P. M. Elias, NHE1 Regulates the Stratum Corneum Permeability Barrier Homeostasis. Microenvironment acidification assessed with fluorescence lifetime imaging, J. Biol. Chem., 2002, 277, 47399–47406 CrossRef CAS.
- K. M. Hanson, M. J. Behne, N. P. Barry, T. M. Mauro, E. Gratton and R. M. Clegg, Two-photon fluorescence lifetime imaging of the skin stratum corneum pH gradient, Biophys. J., 2002, 83, 1682–1690 CrossRef CAS.
- J. Philip and K. Carlsson, Theoretical investigation of the signal-to-noise ratio in fluorescence lifetime imaging, J. Opt. Soc. Am. A, 2003, 20, 368–379 Search PubMed.
- D. S. Elson, J. Siegel, S. E. D. Webb, S. Lévěque-Fort, M. J. Lever, P. M. W. French, K. Lauritsen, M. Wahl and R. Erdmann, Fluorescence lifetime system for microscopy and multi-well plate imaging with a blue picosecond diode laser, Opt. Lett., 2002, 27, 1409–1411 CAS.
- Y. Sakai and S. Hirayama, A Fast Deconvolution Method to Analyze Fluorescence Decays When the Excitation Pulse Repetition Period Is Less Than the Decay Times, J. Lumin., 1988, 39, 145–151 CrossRef CAS.
- Q. S. Hanley, D. J. Arndt-Jovin and T. M. Jovin, Spectrally resolved fluorescence lifetime imaging microscopy, Appl. Spectrosc., 2002, 56, 155–166 CrossRef CAS.
- P. Tinnefeld, D. P. Herten and M. Sauer, Photophysical dynamics of single molecules studied by spectrally-resolved fluorescence lifetime imaging microscopy (SFLIM), J. Phys. Chem. A, 2001, 105, 7989–8003 CrossRef CAS.
- M. Straub and S. W. Hell, Fluorescence lifetime three-dimensional microscopy with picosecond precision using a multifocal multiphoton microscope, Appl. Phys. Lett., 1998, 73, 1769–1771 CrossRef CAS.
- J. Siegel, D. S. Elson, S. E. D. Webb, D. Parsons-Karavassilis, S. Lévěque-Fort, M. J. Cole, M. J. Lever, P. M. W. French, M. A. A. Neil, R. Juškaitis, L. O. Sucharov and T. Wilson, Whole-field five-dimensional fluorescence microscopy combining lifetime and spectral resolution with optical sectioning, Opt. Lett., 2001, 26, 1338–1340 Search PubMed.
- C. E. Bigelow, D. L. Conover and T. H. Foster, Confocal fluorescence spectroscopy and anisotropy imaging system, Opt. Lett., 2003, 28, 695–697.
- D. S. Lidke, P. Nagy, B. G. Barisas, R. Heintzmann, J. N. Post, K. A. Lidke, A. H. A. Clayton, D. J. Arndt-Jovin and T. M. Jovin, Imaging molecular interactions in cells by dynamic and static fluorescence anisotropy (rFLIM and emFRET), Biochem. Soc. Trans., 2003, 31, 1020–1027 CrossRef CAS.
- A. Squire, P. J. Verveer, O. Rocks and P. I. Bastiaens, Red-edge anisotropy microscopy enables dynamic imaging of homo-FRET between green fluorescent proteins in cells, J. Struct. Biol., 2004, 147, 62–69 CrossRef CAS.
- R. Varma and S. Mayor, GPI-anchored proteins are organized in submicron domains at the cell surface, Nature, 1998, 394, 798–801 CrossRef CAS.
- K. Suhling, J. Siegel, P. M. P. Lanigan, S. Lévěque-Fort, S. E. D. Webb, D. Phillips, D. M. Davis and P. M. W. French, Time-resolved fluorescence anisotropy imaging applied to live cells, Opt. Lett., 2004, 29, 584–586 CrossRef.
- J. Siegel, K. Suhling, S. Lévěque-Fort, S. E. D. Webb, D. M. Davis, D. Phillips, Y. Sabharwal and P. M. W. French, Wide-field time-resolved fluorescence anisotropy imaging (TR-FAIM): Imaging the rotational mobility of a fluorophore, Rev. Sci. Instrum., 2003, 74, 182–192 CrossRef CAS.
- A. H. A. Clayton, Q. S. Hanley, D. J. Arndt-Jovin, V. Subramaniam and T. M. Jovin, Dynamic Fluorescence Anisotropy Imaging Microscopy in the Frequency Domain (rFLIM), Biophys. J., 2002, 83, 1631–1649 CrossRef CAS.
- M. Parsons, B. Vojnovic and S. Ameer-Beg, Imaging protein–protein interactions in cell motility using fluorescence resonance energy transfer (FRET), Biochem. Soc. Trans., 2004, 32, 431–433 CrossRef CAS.
- M. Peter and S. M. Ameer-Beg, Imaging molecular interactions by multiphoton FLIM, Biol. Cell, 2004, 96, 231–236 CrossRef CAS.
- P. R. Selvin, The renaissance of fluorescence resonance energy transfer, Nat. Struct. Biol., 2000, 7, 730–734 CrossRef CAS.
- T. Förster, Zwischenmolekulare Energiewanderung und Fluoreszenz, Ann. Phys., 1948, 2, 55–75 Search PubMed.
- L. Stryer and R. P. Haugland, Energy Transfer: A spectroscopic ruler, Proc. Natl. Acad. Sci. USA, 1967, 58, 719–726 CAS.
- L. Stryer, Fluorescence energy transfer as a spectroscopic ruler, Annu. Rev. Biochem., 1978, 47, 819–846 CrossRef CAS.
- C. G. Dos Remedios and P. D. J. Moens, Fluorescence Resonance Energy Transfer Spectroscopy is a Reliable ‘Ruler’ for Measuring Structural Changes in Proteins. Dispelling the problem of the unknown orientation factor, J. Struct. Biol., 1995, 115, 175–185 CrossRef CAS.
- R. P. Haugland, J. Yguerabide and L. Stryer, Dependence of kinetics of singlet-singlet energy transfer on spectral overlap, Proc. Natl. Acad. Sci. USA, 1969, 63, 23–30 CAS.
- R. E. Dale, J. Eisinger and W. E. Blumberg, The orientational freedom of molecular probes, Biophys. J., 1979, 26, 161–194 CrossRef CAS.
- J. Eisinger and R. E. Dale, Interpretation of intramolecular energy transfer experiments, J. Mol. Biol., 1974, 84, 643–647 CAS.
- H. Du, R. C. A. Fuh, J. Li, L. A. Corkan and J. S. Lindsey, PhotochemCAD: A Computer-Aided Design and Research Tool in Photochemistry, Photochem. Photobiol., 1998, 68, 141–142 CAS.
- E. A. Jares-Erijman and T. M. Jovin, FRET imaging, Nat. Biotechnol., 2003, 21, 1387–1396 CrossRef CAS.
- P. I. H. Bastiaens and A. Squire, Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell, Trends Cell Biol., 1999, 9, 48–52 CrossRef CAS.
- P. van Roessel and A. H. Brand, Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins, Nat. Cell Biol., 2002, 4, E15–20 CrossRef CAS.
- T. Oida, Y. Sako and A. Kusumi, Fluorescence lifetime imaging microscopy (flimscopy). Methodology development and application to studies of endosome fusion in single cells, Biophys. J., 1993, 64, 676–685 CAS.
- T. W. J. Gadella and T. M. Jovin, Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation, J. Cell Biol., 1995, 129, 1543–1558 CrossRef CAS.
- P. I. H. Bastiaens and T. M. Jovin, Microspectroscopic imaging tracks the intracellular processing of a signal transduction protein: fluorescent-labeled protein kinase C βI, Proc. Natl. Acad. Sci. USA, 1996, 93, 8407–8412 CrossRef CAS.
- J. Lippincott-Schwartz and G. H. Patterson, Development and use of fluorescent protein markers in living cells, Science, 2003, 300, 87–91 CrossRef.
- R. Y. Tsien, The green fluorescent protein, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS.
- M. Zimmer, Green fluorescent protein (GFP): Applications, structure and related photophysical behavior, Chem. Rev., 2002, 102, 759–781 CrossRef CAS.
- M. Cotlet, J. Hofkens, M. Maus, T. Gensch, M. van der Auweraer, J. Michiels, G. Dirix, M. van Guyse, J. Vanderleyden, A. J. W. G. Visser and F. C. de Schryver, Excited state dynamics in the enhanced green fluorescent protein mutant probed by picosecond time-resolved single photon counting spectroscopy, J. Phys. Chem. B, 2001, 105, 4999–5006 CrossRef CAS.
- A. A. Heikal, S. T. Hess and W. W. Webb, Multiphoton molecular spectroscopy and excited-state dynamics of enhanced green fluorescent protein (EGFP): acid–base specificity, Chem. Phys., 2001, 274, 37–55 CrossRef CAS.
- M. A. Uskova, J. Borst, M. A. Hink, A. van Hoek, A. Schots, A. L. Klyachko and A. J. W. G. Visser, Fluorescence dynamics of green fluorescent protein in AOT reversed micelles, Biophys. Chem., 2000, 87, 73–84 CrossRef CAS.
- K. Suhling, J. Siegel, D. Phillips, P. M. W. French, S. Lévěque-Fort, S. E. D. Webb and D. M. Davis, Imaging the environment of green fluorescent protein, Biophys. J., 2002, 83, 3589–3595 CAS.
- R. Pepperkok, A. Squire, S. Geley and P. I. H. Bastiaens, Simultaneous detection of multiple green fluorescent proteins in live cells by fluorescence lifetime imaging microscopy, Curr. Biol., 1999, 9, 269–272 CrossRef CAS.
- T. Ng, M. Parsons, W. E. Hughes, J. Monypenny, D. Zicha, A. Gautreau, M. Arpin, S. Gschmeissner, P. J. Verveer, P. I. H. Bastiaens and P. J. Parker, Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility, EMBO J., 2001, 20, 2723–2741 CrossRef CAS.
- J. W. Legg, C. A. Lewis, M. Parsons, T. Ng and C. M. Isacke, A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility, Nat. Cell Biol., 2002, 4, 399–407 CrossRef CAS.
- T. Ng, A. Squire, G. Hansra, F. Bornancin, C. Prevostel, A. Hanby, W. Harris, D. Barnes, S. Schmidt, H. Mellor, P. I. Bastiaens and P. J. Parker, Imaging protein kinase C alpha activation in cells, Science, 1999, 283, 2085–2089 CrossRef CAS.
- P. J. Verveer, F. S. Wouters, A. R. Reynolds and P. I. H. Bastiaens, Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane, Science, 2000, 290, 1567–1570 CrossRef CAS.
- F. S. Wouters and P. I. Bastiaens, Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells, Curr. Biol., 1999, 9, 1127–1130 CrossRef CAS.
- F. G. Haj, P. J. Verveer, A. Squire, B. G. Neel and P. I. H. Bastiaens, Imaging Sites of Receptor Dephosphorylation by PTP1B on the Surface of the Endoplasmic Reticulum, Science, 2002, 1708–1710 CrossRef CAS.
- A. G. Harpur, F. S. Wouters and P. I. Bastiaens, Imaging FRET between spectrally similar GFP molecules in single cells, Nat. Biotechnol., 2001, 19, 167–169 CrossRef CAS.
- J. R. Lakowicz, H. Szmacinski, K. Nowaczyk and M. L. Johnson, Fluorescence lifetime imaging of free and protein-bound NADH, Proc. Natl. Acad. Sci. USA, 1992, 89, 1271–1275 CAS.
- Q. Zhang, D. W. Piston and R. H. Goodman, Regulation of Corepressor Function by Nuclear NADH, Science, 2002, 1895–1897 CAS.
- S. Murata, P. Herman and J. R. Lakowicz, Texture analysis of fluorescence lifetime images of AT- and GC-rich regions in nuclei, J. Histochem. Cytochem., 2001, 49, 1443–1451 Search PubMed.
- S. Murata, P. Herman and J. R. Lakowicz, Texture analysis of fluorescence lifetime images of nuclear DNA with effect of fluorescence resonance energy transfer, Cytometry, 2001, 43, 94–100 CrossRef CAS.
- S. Murata, P. Herman, H. J. Lin and J. R. Lakowicz, Fluorescence Lifetime Imaging of Nuclear DNA: Effect of Fluorescence Resonance Energy Transfer, Cytometry, 2000, 41, 178–185 CrossRef CAS.
- M. van Zandvoort, C. J. de Grauw, H. C. Gerritsen, J. L. V. Broers, M. Egbrink, F. C. S. Ramaekers and D. W. Slaaf, Discrimination of DNA and RNA in cells by a vital fluorescent probe: Lifetime Imaging of SYTO13 in healthy and apoptotic cells, Cytometry, 2002, 47, 226–235 CrossRef CAS.
- J. R. Lakowicz, H. Szmacinski, K. Nowaczyk and M. L. Johnson, Fluorescence lifetime imaging of calcium using Quin-2, Cell Calcium, 1992, 13, 131–147 CrossRef CAS.
- J. R. Lakowicz, H. Szmacinski, K. Nowaczyk and W. J. Lederer, Fluorescence lifetime imaging of intracellular calcium in COS cells using Quin-2, Cell Calcium, 1994, 15, 7–27 CrossRef CAS.
- B. Herman, P. Wodnicki, S. Kwon, A. Periasamy, G. W. Gordon, N. Mahajan and W. Xue Feng, Recent Developments in Monitoring Calcium and Protein Interactions in Cells Using Fluorescence Lifetime Microscopy, J. Fluoresc., 1997, 7, 85–92 CAS.
- R. Sanders, H. C. Gerritsen, A. Draaijer, P. M. Houpt and Y. K. Levine, Fluorescence Lifetime Imaging of Free Calcium in Single Cells, Bioimaging, 1994, 2, 131–138 CrossRef CAS.
- H. C. Gerritsen, R. Sanders, A. Draaijer, C. Ince and Y. K. Levine, Fluorescence Lifetime Imaging of Oxygen in Living Cells, J. Fluoresc., 1997, 7, 11–16 CAS.
- H. J. Lin, P. Herman and J. R. Lakowicz, Fluorescence lifetime-resolved pH imaging of living cells, Cytometry, 2003, 52A, 77–89 CrossRef.
- R. Sanders, A. Draaijer, H. C. Gerritsen, P. M. Houpt and Y. K. Levine, Quantitative pH imaging in cells using confocal fluorescence lifetime imaging microscopy, Anal. Biochem., 1995, 227, 302–308 CrossRef CAS.
- R. Cubeddu, A. Pifferi, P. Taroni, A. Torricelli, G. Valentini, F. Rinaldi and E. Sorbellini, Fluorescence lifetime imaging: An application to the detection of skin tumors, IEEE J. Sel. Top. Quantum Electron., 1999, 5, 923–929 CrossRef CAS.
- P. J. Tadrous, J. Siegel, P. M. W. French, S. Shousha, E. N. Lalani and G. W. Stamp, Fluorescence lifetime imaging of unstained tissues: early results in human breast cancer, J. Pathol., 2003, 199, 309–317 CrossRef.
- D. S. Elson, J. Requejo-Isidro, I. Munro, F. Reavell, J. Siegel, K. Suhling, P. J. Tadrous, R. Benninger, P. M. P. Lanigan, J. McGinty, C. Talbot, B. Treanor, S. Webb, A. Sandison, A. Wallace, D. M. Davis, J. Lever, M. A. A. Neil, D. Phillips, G. W. Stamp and P. M. W. French, Time-domain fluorescence lifetime imaging applied to biological tissue, Photochem. Photobiol. Sci., 2004, 3, 795–801 RSC.
- J. Siegel, D. S. Elson, S. E. D. Webb, K. C. B. Lee, A. Vlandas, G. L. Gambaruto, S. Leveque-Fort, M. J. Lever, P. J. Tadrous and G. W. H. Stamp, Studying biological tissue with fluorescence lifetime imaging: microscopy, endoscopy and complex decay profiles, Appl. Opt., 2003, 42, 2995–3004.
- J. J. Birmingham, Frequency-domain lifetime imaging methods at Unilever Research, J. Fluoresc., 1997, 7, 45–54 CAS.
- K. König, H. Schneckenburger and R. Hibst, Time-gated in vivo autofluorescence imaging of dental caries, Cell. Mol. Biol., 1999, 45, 233–239 Search PubMed.
- J. P. Connelly, S. W. Botchway, L. Kunz, D. Pattison, A. W. Parker and A. J. MacRobert, Time-resolved fluorescence imaging of photosensitiser distributions in mammalian cells using a picosecond laser line-scanning microscope, J. Photochem. Photobiol. A Chem., 2001, 142, 169–175 CrossRef CAS.
- M. Kress, T. Meier, R. Steiner, F. Dolp, R. Erdmann, U. Ortmann and A. Rück, Time-resolved microspectrofluorometry and fluorescence lifetime imaging of photosensitizers using picosecond pulsed diode lasers in laser scanning microscopes, J. Biomed. Opt., 2003, 8, 26–32 CrossRef CAS.
- A. C. Rück, M. Kress, F. Dolp, N. Akgün, T. Meier and R. Steiner, New Microscopic techniques to investigate intracellular localization and reactions of photosensitizers. Laser scanning fluorescence lifetime imaging (LS-FLIM), Med. Laser Appl., 2002, 41–47 Search PubMed.
- A. D. Scully, A. J. Mac Robert, S. Botchway, P. O'Neill, A. W. Parker, R. B. Ostler and D. Phillips, Development of a laser-based fluorescence microscope with subnanosecond time resolution, J. Fluoresc., 1996, 6, 119–125 CAS.
- A. D. Scully, R. B. Ostler, A. J. MacRobert, A. W. Parker, C. d. Lara, P. O'Neill and D. Phillips, Laser line-scanning confocal fluorescence imaging of the photodynamic action of aluminium and zinc phthalocyanines in V79-4 Chinese hamster fibroblasts, Photochem. Photobiol., 1998, 68, 199–204 CrossRef CAS.
- A. D. Scully, R. B. Ostler, D. Phillips, P. O'Neill, A. W. Parker and A. J. MacRobert, Application of fluorescence lifetime imaging microscopy to the investigation of intracellular PDT mechanisms., Bioimaging, 1997, 5, 9–18 CrossRef.
- T. Förster and G. Hoffmann, Die Viskositätsabhängigkeit der Fluoreszenzquantenausbeuten einiger Farbstoffsysteme, Z. Phys. Chem. (Munich), 1971, 75, 63–76.
- B. Wilhelmi, Influence of solvent viscosity on excited state lifetime and fluorescence quantum yield of dye molecules, Chem. Phys., 1982, 66, 351–355 CrossRef CAS.
- C. E. Kung and J. K. Reed, Microviscosity measurements of phospholipid bilayers using fluorescent dyes that undergo torsional relaxation, Biochemistry, 1986, 25, 6114–6121 CrossRef CAS.
- T. Iwaki, C. Torigoe, M. Noji and M. Nakanishi, Antibodies for Fluorescent Molecular Rotors, Biochemistry, 1993, 32, 7589–7592 CrossRef CAS.
- M. A. Haidekker, T. Ling, M. Anglo, H. Y. Stevens, J. A. Frangos and E. A. Theodorakis, New fluorescent probes for the measurement of cell membrane viscosity, Chem. Biol., 2001, 8, 123–131 CrossRef CAS.
- J. A. Dix and A. S. Verkman, Pyrene excimer mapping in cultured fibroblasts by ratio imaging and time-resolved microscopy, Biochemistry, 1990, 29, 1949–1953 CrossRef CAS.
- M. Viard, J. Gallay, M. Vincent and M. Paternostre, Origin of Laurdan Sensitivity to the Vesicle-to-Micelle Transition of Phospholipid-Octylglucoside System: A Time-Resolved Fluorescence Study, Biophys. J., 2001, 80, 347–359 CrossRef CAS.
- S. T. Hess, E. D. Sheets, A. Wagenknecht-Wiesner and A. A. Heikal, Quantitative Analysis of the Fluorescence Properties of Intrinsically Fluorescent Proteins in Living Cells, Biophys. J., 2003, 85, 2566–2580 CrossRef CAS.
- S. Jakobs, V. Subramaniam, A. Schonle, T. M. Jovin and S. W. Hell, EGFP and DsRed expressing cultures of Escherichia coli imaged by confocal, two-photon and fluorescence lifetime microscopy, FEBS Lett., 2000, 479, 131–135 CrossRef CAS.
- R. Swaminathan, C. P. Hoang and A. S. Verkman, Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion, Biophys. J., 1997, 72, 1900–1907 CAS.
- D. Toptygin, Effects of the Solvent Refractive Index and Its Dispersion on the Radiative Decay Rate and Extinction Coefficient of a Fluorescent Solute, J. Fluoresc., 2003, 13, 201–219 CrossRef CAS.
- S. J. Strickler and R. A. Berg, Relationship between absorption intensity and fluorescence lifetime of molecules, J. Chem. Phys., 1962, 37, 814–820 CrossRef CAS.
- K. Suhling, D. M. Davis, Z. Petrášek, J. Siegel and D. Phillips, The influence of the refractive index on EGFP fluorescence lifetimes in mixtures of water and glycerol, Proc. Soc. Photo. Opti. Instrum. Eng., 2001, 4259, 92–101 Search PubMed.
- D. M. Davis, Assembly of the immunological synapse for T cells and NK cells, Trends Immunol., 2002, 23, 356–363 Search PubMed.
- D. M. Davis and M. L. Dustin, What is the importance of the immunological synapse?, Trends Immunol., 2004, 25, 323–327 Search PubMed.
- D. M. Davis, Molecular recognition of disease at natural killer cell immune synapses, Sci. Prog., 2000, 83, 303–316 Search PubMed.
- J. Beuthan, O. Minet, J. Helfmann, M. Herrig and G. Müller, The spatial variation of the refractive index in biological cells, Phys. Med. Biol., 1996, 41, 369–382 CrossRef CAS.
- S. Johnsen and E. A. Widder, The physical basis of transparency in biological tissue: Ultrastructure and the minimization of light scattering, J. Theor. Biol., 1999, 199, 181–198 CrossRef.
- B. Treanor, P. M. P. Lanigan, K. Suhling, T. Schreiber, I. Munro, M. A. A. Neil, D. Phillips, D. M. Davis and P. M. W. French, Imaging fluorescence lifetime heterogeneity applied to GFP-tagged MHC protein at an immunological synapse, J. Microsc., 2004, in press Search PubMed.
- G. H. Patterson and J. Lippincott-Schwartz, A Photoactivatable GFP for Selective Photolabeling of Proteins and Cells, Science, 2002, 297, 1873–1876 CrossRef CAS.
- L. Giordano, T. M. Jovin, M. Irie and E. A. Jares-Erijman, Diheteroarylethenes as thermally stable photoswitchable acceptors in photochromic fluorescence resonance energy transfer (pcFRET), J. Am. Chem. Soc., 2002, 124, 7481–7489 CrossRef CAS.
- M. Green, Semiconductor Quantum Dots as Biological Imaging Agents, Angew. Chem., 2004, 43, 4129–4131.
- T. M. Jovin, Quantum dots finally come of age, Nat. Biotechnol., 2003, 21, 32–33 CrossRef CAS.
- G. Schlegel, J. Bohnenberger, I. Potapova and A. Mews, Fluorescence Decay Time of Single Semiconductor Nanocrystals, Phys. Rev. Lett., 2002, 88, 137401–137900 CrossRef.
- B. R. Fisher, H. J. Eisler, N. E. Stott and M. G. Bawendi, Emission Intensity Dependence and Single-Exponential Behavior In Single Colloidal Quantum Dot Fluorescence Lifetimes, J. Phys. Chem. B, 2004, 108, 143–148 CrossRef CAS.
- M. A. Rizzo, G. H. Springer, B. Granada and D. W. Piston, An improved cyan fluorescent protein variant useful for FRET, Nat. Biotechnol., 2004, 22, 445–449 CrossRef CAS.
- M. J. Cole, J. Siegel, S. E. D. Webb, R. Jones, K. Dowling, M. J. Dayel, D. Parsons-Karavassilis, P. M. W. French, M. J. Lever, L. O. D. Sucharov, M. A. A. Neil, R. Juškaitis and T. Wilson, Time-domain whole-field fluorescence lifetime imaging with optical sectioning, J. Microsc., 2001, 203, 246–257 CrossRef CAS.
- M. J. Cole, J. Siegel, S. E. D. Webb, R. Jones, K. Dowling, P. M. W. French, M. J. Lever, L. O. D. Sucharov, M. A. A. Neil, R. Juškaitis and T. Wilson, Whole-field optically sectioned fluorescence lifetime imaging, Opt. Lett., 2000, 25, 1361–1363.
- K. Suhling, R. W. Airey and B. L. Morgan, Minimization of fixed pattern noise in photon event counting imaging, Rev. Sci. Instrum., 2002, 73, 2917–2922 CrossRef CAS.
- K. Suhling, R. W. Airey and B. L. Morgan, Optimisation of centroiding algorithms for photon event counting imaging, Nucl. Instrum. Methods, 1999, 437, 393–418 Search PubMed.
- F. E. McCann, K. Suhling, L. M. Carlin, K. Eleme, S. B. Taner, K. Yanagi, B. Vanherberghen, P. M. W. French and D. M. Davis, Imaging immune surveillance by T cells and NK cells, Immunol. Rev., 2002, 189, 179–192 CrossRef CAS.
Footnote |
| † Dedicated to Professor Hiroshi Masuhara on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2005 |