Determinants of cofactor binding to DNA methyltransferases: insights from a systematic series of structural variants of S-adenosylhomocysteine†
Helen M.
Cohen
ab,
Andrew D.
Griffiths
b,
Dan S.
Tawfik
c and
David
Loakes
*b
aThe MRC Centre for Protein Engineering, MRC Centre, Hills Road, Cambridge, UK CB2 2QH
bMRC Laboratory of Molecular Biology, MRC Centre, Hills Road, Cambridge, UK CB2 2QH. E-mail: david.loakes@mrc-lmb.cam.ac.uk; Fax: +44 1223 412178; Tel: +44 1223 402206
cDepartment of Biological Chemistry, The Weizmann Institute of Science, Rehovot, 76 100, Israel
First published on 26th November 2004
Abstract
S-Adenosylmethionine (AdoMet) is a commonly used cofactor, second only to ATP in the variety of reactions in which it participates. It is the methyl donor in the majority of methyl transfer reactions, including methylation of DNA, RNA, proteins and small molecules. Almost all structurally characterised methyltransferases share a conserved AdoMet-dependent methyltransferase fold, in which AdoMet is bound in the same orientation. Although potential interactions between the cofactor and methyltransferases have been inferred from crystal structures, there has not been a systematic study of the contributions of each functional group to binding. To explore the binding interaction we synthesised a series of seven analogues of the methyltransferase inhibitor S-adenosylhomocysteine (AdoHcy), each containing a single modification, and tested them for the ability to inhibit methylation by HhaI and HaeIII DNA methyltransferase. Comparison of the Ki values highlights the structural determinants for cofactor binding, and indicates which nucleoside and amino acid functional groups contribute significantly to AdoMet binding. An understanding of the binding of AdoHyc to methyltransferases will greatly assist the design of AdoMet inhibitors.
Introduction
Methyl transfer is a reaction central to cellular biochemistry, with 140 different classes of methyltransferase alkylating such diverse substrates as DNA, RNA, proteins, lipids, polysaccharides and small molecules.1S-Adenosylmethionine (AdoMet) contains a highly reactive methylthiol group and is the cofactor and methyl donor for the majority (over 90%) of these enzymes (http://www.expasy.ch/enzyme).2 With enzymes catalysing the methylation of N, O or S the substrates contain a polarisable nucleophile, which directly attacks the activated methyl group, whereas enzymes catalysing methylation of carbon atoms usually proceed via addition and subsequent elimination from a thiol group in the enzyme active site. The structures of over 50 different AdoMet-dependent methyltransferases are known and include five distinct AdoMet-binding folds.3 Despite the extensive chemical and structural diversity of methylation substrates, the great majority of known methyltransferases contain the Class I fold and share a common evolutionary origin.3,4 Currently the structures of 31 methyltransferases have been solved in complex with AdoMet or the reaction product, S-adenosylhomocysteine (AdoHcy) and the relative position of AdoMet is almost identical in each structure.Analogues of AdoMet that inhibit methyltransfer, such as the natural product sinefungin, have therapeutic properties including antifungal, antiviral, antiparasitic and antitumour activities.5,6 However the high conservation of the AdoMet-binding domain means that these analogues often inhibit a wide range of methyltransferases, leading to in vivo toxicity. Previous studies have investigated the ability of AdoMet analogues to inhibit enzymes such as the essential bacterial adenine N6-methyltransferases, involved in cell cycle regulation, and the erythromycin resistance methyltransferases, which methylate prokaryotic rRNA.7,8 Analogues of the methylation target, or mechanism-based inhibitors, are more specific than inhibitors such as sinefungin. A multisubstrate adduct synthesised by Wahnon et al., for example, inhibits adenine N6, but not cytosine C5, DNA methyltransferases.8
Despite the structural conservation of the cofactor binding site there is little sequence identity between methyltransferases in different classes. Only three motifs are conserved amongst the AdoMet-dependent methyltransferase family, located in the P loop, G loop and part of strand β4.9 Notably each of these positions is involved in cofactor binding.
There is often greater sequence conservation within methyltransferase classes and DNA and RNA methyltransferases contain particularly strongly conserved motifs. Cytosine C5 DNA methyltransferases share ten well defined and highly conserved motifs.10 Three structures of C5 DNA methyltransferases are known: two bacterial type II restriction modification system methyltransferases, M.HhaI (recognition sequence GCGC, solved in complex with AdoHcy and DNA)10 and M.HaeIII (recognition sequence GGCC, solved in complex with DNA),11 and human DNMT2, which shows homology to other prokaryotic and eukaryotic cytosine C5 DNA methyltransferases (solved in complex with AdoMet).12 Seven residues are proposed to form hydrogen bonds between M.HhaI and AdoMet, with four of these identical (six conserved) in M.HaeIII and three identical (seven conserved) in DNMT2.
In order to characterise the functional groups that allow cofactor recognition by the active site of cytosine C5 DNA methyltransferases we measured the inhibition of M.HhaI and M.HaeIII by AdoHcy analogues. AdoHcy is structurally similar to AdoMet, binding to methyltransferases in the same orientation, and with a similar Kd to AdoMet, but it lacks the reactive methyl group and acts as a competitive inhibitor with respect to AdoMet.13 A series of analogues was prepared, with single modifications to the adenine, ribose and methionine moieties (Fig. 1).
Results
Synthesis of AdoHyc analogues
A series of seven AdoHcy analogues were synthesised in order to probe the structural determinants of cofactor binding (Fig. 1). Initially we chose the N6-acetyl derivative. This analogue has the advantage of weakening the hydrogen bonding ability of the amino group, but with minimal steric effects. However, it was found that the acetyl group was both acid and base labile and it was never possible to prepare the material free from AdoHcy. We therefore chose to make the N6-benzoyl derivative (1) which has greater stability, although the benzoyl group adds steric bulk, but still weakens the hydrogen bonding capability of the amino group. In addition, the inosine derivative (SIH, 2) was prepared, which has an altered hydrogen bonding pattern. The effect of altering the nucleobase was examined by the use of SIH and also the cytosine derivative (3). Modification of the amino acid carboxyl group to alter the hydrogen bonding behaviour and probe for steric effects was carried out by conversion to the methylamide derivative 4. The remaining modifications arise from the removal of functional groups, namely the 2′-hydroxyl group of the ribose to give 5 (S2′dAH), and removal of either of the amino acid functionalities to give 5′-(3-aminopropylthio)-5′-deoxyadenosine, 6, and 5′-(3-carboxypropylthio)-5′-deoxyadenosine, 7, as described.14 | ||
| Fig. 1 AdoHcy analogues synthesized. Arrows show the functional groups that have been modified in each case. | ||
Synthesis of AdoHcy and derivatives has been previously described via coupling of 5′-O-tosyl15 or 5′-deoxy-5′-halo derivatives of adenosine16 with the di-sodium salt of L-homocysteine. Coupling of 5′-O-tosyl derivatives is generally carried out in liquid ammonia after reduction of homocysteine, whereas 5′-halo derivatives are coupled with the di-sodium salt of L-homocysteine in water.17
The first modification was the addition of a benzoyl group to the adenine N6-amino group (analogue 1, Fig. 1). The N6-benzoyl-isopropylidene adenosine derivative was converted to its 5′-O-tosylate 8, and then reaction with sodium iodide in acetone yielded the 5′-deoxy-5′-iodo derivative 9 in moderate yield (Scheme 1). This was reacted with the di-sodium salt of L-homocysteine in aqueous DMF solution to give 10. This compound partially lost the benzoyl group, but in a slower reaction than was observed with the acetyl group. Finally, removal of the isopropylidene protection was carried out in 5% aqueous TFA to give 1. N6-benzoyl-AdoHcy was stable under these conditions, and no AdoHcy was observed, as was found with the N6-acetyl route.
 | ||
| Scheme 1 Synthesis of N6-benzoyl-AdoHyc. (a) NaI–acetone, 69%. (b) Homocysteine, Na–NH3, 57%. (c) 5% aqueous TFA, 70%. | ||
S-Inosinyl-L-homocysteine (SIH, 2) has been previously described,18 prepared by coupling 5′-O-tosyl-isopropylidine inosine and the di-sodium salt of L-homocysteine in liquid ammonia. However, no characterisation of the product was provided. As we had found that coupling of 5′-iodo derivatives was more efficient, 5′-O-tosyl-isopropylidine inosine was converted to 5′-deoxy-5′-iodo-isopropylidine inosine, and then reacted with the di-sodium salt of L-homocysteine (Scheme 2). However, the product, which was not identified, did not contain homocysteine, and was possibly the cyclonucleoside. Repeating the previously described synthesis,18 it became apparent from the NMR spectra that the product was not the SIH derivative, and again may have been the cyclonucleoside; evidently the inosine-6-oxo group required protection. Using a modification of a procedure described by Grøtli et al.,19 isopropylidene inosine was converted to its 5′-O-tosylate and then reacted with tert-butyldiphenylchlorosilane. The authors stated that the silyl protecting group is unstable, in particular to column chromatography19 so the silylated inosine derivative was used without purification.
 | ||
| Scheme 2 Synthesis of S-inosyl-AdoHcy. (a) DMAP, TEA, tert-butyldiphenylchlorosilane. (b) Homocysteine, Na–NH3, 53%. (c) 5% aqueous TFA, 69%. | ||
The silylated derivative was reacted with the di-sodium salt of L-homocysteine in liquid ammonia. At the end of the reaction the product was dissolved in aqueous ethanol and acidified with acetic acid to remove the silyl protection. The product, after reverse phase purification, was shown to be the desired isopropylidene protected 11. Treatment with 5% aqueous TFA removed the isopropylidene protection to give SIH, 2.
S-Cytidinyl-L-homocysteine (SCH, 3) has also been previously described,18 but the product was not characterised. In this work SCH was prepared by an alternative route. Conversion of cytidine to 5′-deoxy-5′-chlorocytidine was carried out by reaction with thionyl chloride in hexamethylphosphoramide.16,20 This was then reacted with the di-sodium salt of L-homocysteine in water to give, after purification by reverse phase chromatography, SCH.
S-(2′-Deoxy-β-D-ribofuranosyladenosin-5′-yl)-L-homocysteine (SdAH, 5) was prepared by an alternative route to that previously described (Scheme 3).21N6-benzoyl-5′-O-tosyl-2′deoxyadenosine was converted to its 5′-iodo derivative 15, which was then treated with the di-sodium salt of L-homocysteine in aqueous DMF to give 16. Removal of the benzoyl group with ammonia solution yielded 5.
 | ||
| Scheme 3 Synthesis of 2′-deoxy-AdoHcy. (a) Homocysteine, Na–NH3, 57%. (b) 5% aqueous TFA, 89%. | ||
The final modification we required was to the carboxylic acid function, which was modified to the methylamide 4 (Scheme 4). This required the protection of most of the functional groups of AdoHcy in order to activate the carboxylic acid group. A number of different protecting group strategies were employed, primarily to avoid using acid labile protecting groups. However, it was found that acid labile protecting groups could not be avoided. The synthesis was carried out using the intermediate 10. Attempts to protect the homocysteine amino function with fluorenylmethoxycarbonyloxy-succinimide in pyridine failed, but using aqueous dioxane with sodium carbonate the desired Fmoc derivative, 12, was prepared. This was then reacted with N-hydroxysuccinimide with DCC to yield the active ester 13, which was reacted with aqueous methylamine in DMF. This yielded the desired methylamide derivative, 14, while removing the Fmoc and benzoyl protecting groups in one step. Final removal of the isopropylidene protecting group was carried out using 5% aqueous TFA as before to give 4.
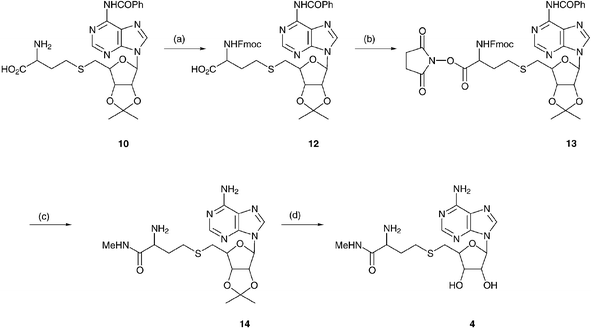 | ||
| Scheme 4 Synthesis of AdoHcy-methylamide. (a) Dioxane–H2O–Na2CO3–N-(9-Fmoc)succinimide, 94%. (b) N-Hydroxysuccinimide–DCCD, 72%. (c) Aqueous MeNH2, 62%. (d) 5% aqueous TFA, 61%. | ||
Kinetics and inhibition of methylation
The catalytic activities of M.HhaI and M.HaeIII were measured by incorporation of radiolabelled methyl groups into synthetic double stranded DNA substrates, Hhasub and Haesub, each containing a single unmethylated recognition site, GCGC and GGCC respectively. Steady state parameters, derived by fitting the data to the Michaelis–Menten model (eqn. 1) are given in Fig. 2 and Table 1.| ν0 = Vmax[S]0/([S]0 + Km) | (1) |
 | ||
| Fig. 2 Steady state kinetics of M.HaeIII. A. Plots of methyl group incorporation by M.HaeIII versus time. Methyltransferase activity of M.HaeIII was measured at 13 concentrations of AdoMet (three of which are shown), in the range 25–2000 nM. Error bars are standard error of the mean. B. Determination of the KmAdoMet of M.HaeIII. The KmAdoMet was determined by fitting the data to the Michaelis–Menten model (eqn. 1). | ||
Our measurement of KmAdoMet for M.HhaI, 110 nM, is comparable to the published value of 161 nM, measured using a similar unmethylated 30 base pair DNA substrate.22KmAdoMet and KiAdoHcy of M.HhaI have also been determined using the substrate poly(dG–dC), with values of 15 nM and 2.1 nM respectively.13 We have previously measured kinetic constants for M.HaeIII with an N-terminal FLAG tag:23 these values of KmAdoMet, 620 nM, and kcat, 0.0033 s−1, are comparable with those measured in this study, but the higher KmDNA of 130 nM may have been caused by the presence of the tag.
To determine the relative effect of each modification to AdoHcy the analogues were tested for inhibition of M.HhaI (Fig. 3) and M.HaeIII by fitting data to eqn. 2. All analogues inhibit methylation by both enzymes, with Ki values ranging from 8–3800 µM (Table 2), but none are as efficient inhibitors as AdoHcy, which was determined to have a Ki of 15 ± 2 nM for M.HhaI and 68 ± 11 nM for M.HaeIII.
| ν0 = Vmax[S]0/(Km(1 + [I]/Ki) + [S]0) | (2) |
| aInhibitor | Modification | M.HhaI | M.HaeIII | ||||
|---|---|---|---|---|---|---|---|
| K i/µM | K i/KiAdoHcy | ΔΔG/kJ mol−1 | K i/µM | K i/KiAdoHcy | ΔΔG/kJ mol−1 | ||
| a See Fig. 1. b These values are estimates only, as methylation proceeded at 35–41% of the uninhibited rate at the highest inhibitor concentrations tested. | |||||||
| AdoHcy | — | 0.015 ± 0.002 | 1.0 | — | 0.069 ± 0.011 | 1.00 | — |
| 1 | N 6-benzoyl | 8.1 ± 2.1 | 540 | 16 | 32 ± 4 | 460 | 16 |
| 2 | Inosine | b3900 ± 800 | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0000 0000 |
32 | b450 ± 80 | 6600 | 23 |
| 3 | Cytidine | 27 ± 3 | 1800 | 19 | b280 ± 60 | 4200 | 21 |
| 4 | Methylamide | 59 ± 9 | 3900 | 21 | 290 ± 46 | 4300 | 22 |
| 5 | 2′-Deoxy | 44 ± 6 | 2900 | 21 | 69 ± 10 | 1000 | 18 |
| 6 | Decarboxy | 16 ± 4 | 1100 | 18 | 37 ± 5 | 540 | 16 |
| 7 | Deamino | 13 ± 2 | 850 | 17 | 230 ± 50 | 3400 | 21 |
 | ||
| Fig. 3 Plots of M.HhaI methyltransferase activity versus inhibitor concentration for AdoHcy and two analogues with modifications to the methionine moiety, 5′-(3-aminopropylthio)-5′-deoxyadenosine, 6, and 5′-(3-carboxypropylthio)-5′-deoxyadenosine, 7. Inhibition of M.HhaI and M.HaeIII by all AdoHcy analogues were measured in a similar manner. Ki values listed in Table 2 were derived by fitting the data to eqn. 2. | ||
Effect of base modifications (analogues 1, 2 and 3)
Analogue 1 is modified at the N6 position of the adenine ring by the addition of a benzoyl group. Hydrogen bonding between N6 on the adenine ring and an acidic side chain in the loop following strand β3 is a conserved feature of Class I AdoMet-dependent methyltransferases (Asp 60 in M.HhaI, Asp 50 in M.HaeIII, Fig. 4), although in some cases serine or asparagine replace the acidic side chain. Only three of the structurally characterised type I AdoMet-dependent methyltransferases lack any side-chain mediated interaction with N6 (Table S1, ESI†). The addition of a benzoyl group at N6 disrupts hydrogen bonds and acts as a steric probe. N6-benzoyl AdoHcy, 1, inhibits M.HhaI and M.HaeIII with Ki of 8.1 µM and 32 µM respectively (Table 1). These inhibition constants are 500 fold higher than those of AdoHcy, representing changes in ΔG of 16 kJ mol−1. This is equivalent to the strength of a single hydrogen bond (typical hydrogen bond energies lie in the range of 12–38 kJ mol−1).24Despite the addition of a large hydrophobic group, analogue 1 was the best inhibitor of both methyltransferases, indicating that it is still able to compete with AdoMet for its binding site. In the structure of M.HhaI, and other methyltransferases, the edge of the AdoMet adenine ring is situated at the entrance to the AdoMet binding site where it is solvent accessible, explaining how the additional benzoyl group might be accommodated.
Analogues 2 and 3, S-inosinyl-L-homocysteine and S-cytidinyl-L-homocysteine, contain alternative nucleobases. There is no conserved interaction between methyltransferases and the N1, N3, and N7 atoms of the adenine ring. Hydrogen bonds to N1 and N3 are suggested in about half the structures, with hydrogen bonds predicted more frequently to N1 than N3 (Table S1†) and hydrogen bonds to N7 only predicted in two methyltransferase structures. Where such hydrogen bonds are suggested they always involve main chain atoms, often using the same residues that form hydrophobic interactions with the adenine and ribose rings. The interaction with N1 is often provided by the main chain amino group of a hydrophobic residue immediately following the conserved Asp or Glu that contacts N6. Similarly N3 is most frequently contacted by a main chain amino group of the residue immediately following or immediately preceding the residue that contacts the sugar hydroxyl groups. In the structure of M.HhaI these residues are Ile 61 and Trp 41 respectively. Ile 61 is conserved in M.HaeIII whereas Trp 41 aligns with Tyr 30.
Analogues 2 and 3 have substantial changes to the base. The modifications to analogue 2 would disrupt the hydrogen bonding interactions normally formed by the methyltransferase to N6 and N1. In the case of the cytidine derivative, analogue 3, the positions of all the potential hydrogen bond-forming groups are altered. Despite the large structural differences from AdoHcy, the cytidine derivative inhibits both M.HhaI and M.HaeIII, albeit with higher Ki values than AdoHcy. The Ki of analogue 2 for M.HhaI, 3.9 mM, is 260000 fold higher than that of AdoHcy, indicating poor competition with AdoMet for the cofactor binding site. The Ki for analogue 2 for M.HaeIII is lower (0.45 mM), perhaps indicating some difference between M.HhaI and M.HaeIII in the hydrophobic pocket that binds the adenosine moiety.
Effect of sugar modification (analogue 5)
Compound 5, 2′-deoxy-AdoHcy, lacks one of the hydroxyl groups on the sugar ring. With only one exception (the mycolic acid synthetase family) methyltransferases with the type I fold have a conserved glutamate or aspartate residue (Glu 40 in M.HhaI, Glu 29 in M.HaeIII, Fig. 3) at the end of strand β2 that forms a pair of hydrogen bonds to the 2′- and 3′-ribose hydroxyl groups (Table S1†). This conserved pair of hydrogen bonds cannot be formed with 5. The Ki of 5 for M.HhaI is 2900 fold higher than that of AdoHcy, allowing an estimate of the strength of the interaction between M.HhaI and the 2′-OH of AdoHcy of 21 kJ mol−1. Similarly, the interaction between M.HaeIII and the 2′-OH of AdoHcy is estimated as 18 kJ mol−1.Effects of amino acid modifications (analogues 4, 6 and 7)
Interactions with the methionine moiety are not conserved between methyltransferases in different sub-classes within E.C. 2.1.1.- and there is some variation even within the DNA cytosine C5 methyltransferases. M.HhaI is thought to make a number of side chain and main chain contacts with the cofactor carboxyl group but only water-mediated contacts to the cofactor amino group. Main chain contacts may be conserved in M.HaeIII, but Ser 305, which makes a hydrogen bond to the AdoHcy carboxyl group in the crystal structure of M.HhaI, is not conserved (Table S1†).Three of the analogues were modified at the amino acid moiety; analogue 6 lacks the carboxyl group, analogue 7 lacks the amino group, and analogue 4 is modified at the carboxyl group by conversion to the methylamide. Each of these alterations caused a substantial increase in Ki compared to that of AdoHcy, (values of Ki were 540 to 4300 fold higher), evidence of the important role of these functional groups in cofactor binding and recognition by both enzymes.
Discussion
This study analyses the inhibition of two cytosine C5 DNA methyltransferases, M.HhaI and M.HaeIII, by a series of AdoHcy analogues (Fig. 1) to characterise quantitatively the contribution of individual functional groups to affinity. All modifications to AdoHcy increase Ki, suggesting that multiple protein–ligand interactions are involved in binding and recognition. We found evidence of significant interactions made by both methyltransferases to the ribose 2′-hydroxyl group, the adenine N6 amino group and the methionine amino and carboxyl groups of AdoHcy. Additionally we found that a bulky substituent on the adenine N6 amino group, or even replacement of the adenine with hypoxanthine or cytosine, do not preclude binding of AdoHcy analogue 1, 2 and 3 to these enzymes.The dissociation constant (Kd) of four analogues of AdoHcy for M.HhaI has previously been determined14 with a view to understanding the role of the methionine moiety in protein–cofactor interaction in DNA methyltransferases and other methyltransferases. Two of these analogues were also tested in this study (compounds 6 and 7).
In the previous study of M.HhaI compounds 6 and 7 were found to have Kd values 78 and 17 fold higher than that of AdoHcy respectively,14 whereas we observed that Ki values 1100 and 850 fold higher than that of AdoHcy. The conclusion of the previous study is that the adenosyl moiety acts as a “molecular anchor” for cofactor binding, whereas our results emphasise the involvement of multiple groups on both the adenosyl and methionine moieties in cofactor binding.
K d values were determined in the absence of DNA, by measuring the quenching of tryptophan fluorescence upon AdoHcy binding.14 It has been previously reported that AdoHcy binds with high affinity to the M.HhaI–DNA complex but not to the free enzyme or the binary product complex (enzyme–methylated DNA),13 in fact AdoMet is bound in a different orientation in the binary M.HhaI–AdoMet complex compared to the ternary complex of M.HhaI, AdoMet and DNA.25,26 Our values of Ki are measurements of the competition of each analogue with AdoMet for occupation of the cofactor binding site in the catalytically competent enzyme–DNA complex. It is likely that our choice to measure Ki instead of Kd accounts for the differences in our estimated ΔΔG values.
Crystal structures of HhaI and HaeIII in complex with AdoHcy indicate hydrogen bonds with each of the nucleobase, sugar and amino acid moieties, as shown in Fig. 4. However, we compared the data from 31 crystal structures of methyltransferases in complex with AdoHcy or analogues and found that the only conserved hydrogen bonds are between acidic side chains and the adenine N6 amino group and the ribose hydroxyls (Table S1†). Hydrogen bonds to the nitrogens N1 and N3 in the adenine ring were made by the main chain amino groups of hydrophobic residues, and were only predicted in about half the methyltransferases studied. Hydrogen bonds to N7 were predicted in two structures. In contrast to the interactions with the adenosine moiety, the residues making hydrogen bonds to the methionine moiety are not conserved. Interactions with both the amino and carboxyl functional groups are predicted in 27 of the structures, yet these hydrogen bonds are a mixture of main chain, side chain and water mediated contacts made by a wide variety of residues from two regions of the consensus fold or from outside the consensus fold. Examination of the phylogenetic data alone might suggest that the interactions with the methionine moiety are less significant than those with other parts of the molecule. Our results indicate that, despite the lack of conservation of hydrogen bond-forming residues, the methionine functional groups are an important determinant of cofactor binding by both M.HhaI and M.HaeIII. We observe that modification of either the amino or the carboxyl group of AdoHcy leads to an increase in Ki comparable to the change caused by modification of the 2′-hydroxyl or N6 amino groups.
 | ||
| Fig. 4 Schematic diagram of the proposed hydrogen bonds between M.HhaI and AdoHcy. M.HhaI amino acids are written in grey, with the equivalent residues in M.HaeIII below. Water-mediated hydrogen bonds are omitted for clarity. | ||
Our findings suggest that recognition of AdoMet by M.HhaI and M.HaeIII relies on multiple interactions with the adenine, ribose and methionine moieties. This pattern of methyltransferase-cofactor recognition is likely to be conserved amongst other cytosine C5 DNA methyltransferases and may also be conserved in other proteins with the type I AdoMet-dependent methyltransferase fold. However the residues involved in cofactor binding are highly variable, particularly residues predicted to contact the methionine moiety, therefore the strength of interactions with this moiety may differ significantly. These AdoHcy analogues should prove useful tools for further study of methyltransferase function.
Experimental
General methods
1H-NMR spectra were obtained on a Bruker DRX-300 spectrometer in d6-DMSO unless otherwise stated. Mass spectra were recorded on a Bruker FTICR Bioapex II instrument. Ultraviolet spectra were recorded on a Perkin Elmer Lambda 40 spectrophotometer in 10% aqueous methanol unless otherwise stated. TLC was carried out on pre-coated F254 or Merck RP-18 F254s silica plates, and column chromatography with Merck Kieselgel 60, or Merck RP-18 silica. Reactions were worked up as follows, unless otherwise stated. Reaction mixtures were evaporated to dryness and the products dissolved in chloroform and washed with saturated aqueous sodium bicarbonate solution. The organic fractions were combined and dried over anhydrous sodium sulfate, filtered and then evaporated to dryness. HPLC was carried out using a Phenomenex Luna 10 µm C-18 reverse phase column. Unless otherwise stated, HPLC conditions are buffer A, 0.1% aqueous TFA; buffer B, 0.1% aqueous TFA, 25% MeCN. 0% to 100% buffer B over 20 minutes, 100% buffer B 10 minutes.400), λmin/nm 245; pH 1, λmax/nm 290 (21800), λmin/nm 240; pH 12, λmax/nm 302 (12300), λmin/nm 258. m/z 544 (70%, M + Na)+, 522 (15%, M + H)+, 394 (100%, M − I)+. Accurate mass measurement on C20H20N5O4INa gives 544.04600, deviation 0.36ppm.
The di-sodium salt of L-homocysteine from above was dissolved in DMF (20 cm3) and water (10 cm3), and to this was added potassium iodide (10 mg) and (9)27
(6.50 g, 12.5 mmol), and the solution heated at 100 °C for 45 minutes. The solution was neutralized (acetic acid) and evaporated to dryness to give a yellow–brown gum, which, by reverse phase TLC, showed two products. The mixture was chromatographed (Merck RP-18 silica, water to 25% acetonitrile in water gradient). The first product eluted in 10–15% MeCN, was shown to be the de-benzoylated product, S-(O-2′,O-3′-isopropylidene-adenosin-5′-yl)-L-homocysteine. Yield 1.25 g, 24%. δH 1.30, 1.51 (6H, 2 × s, isopropylidene CH3), 1.78–1.99 (2H, m, CH2CH2S), 2.57–2.61 (2H, m, CH2S), 2.63–2.82 (2H, m, H5′, H5″), 3.30–3.34 (1H, m, COCHNH2), 4.22–4.26 (1H, m, H4′), 4.96–4.99 (1H, m, H3′), 5.46–5.49 (1H, m, H2′), 6.14–6.15 (1H, m, H1′), 7.39 (2H, br. s, NH2), 8.17 (1H, s, H2), 8.35 (1H, s, H8). UV λmax/nm 259 (13000), λmin/nm 227; pH 1 λmax/nm 257 (13700), λmin/nm 228. m/z 425 (100%, M + H)+, 381 (10%, M − CO2)+. Accurate mass measurement on C17H24N6SO5 gives 425.1603, deviation −0.98 ppm.
The second product, eluting in 20–25% MeCN, was shown to be the title product as a white foam. Yield 3.75 g, 57%. δH
(D2O) 1.34, 1.55 (6H, 2 × s, isopropylidene CH3), 1.73–1.99 (2H, m, CH2CH2S), 2.58–2.63 (2H, m, CH2S), 2.70–2.83 (2H, m, H5′, H5″), 3.25–3.29 (1H, m, COCHNH2), 4.28–4.30 (1H, m, H4′), 5.02–5.04 (1H, m, H3′), 5.56–5.58 (1H, m, H2′), 6.29 (1H, d, J 1.9, H1′), 7.44–7.65 (3H, m, Ph–CH), 7.92–8.08 (2H, m, Ph–CH) 8.69 (1H, s, H2), 8.79 (1H, s, H8). UV λmax/nm 281 (22000), λmin/nm 246; pH 1, λmax/nm 288 (23300), λmin/nm 263; pH 12, 303 (13100), λmin/nm 260. m/z 529.19 (70%, M + H)+, 439 (10%, M − COPh)+, 412 (10%, M − COPh − CO)+. Accurate mass measurement on C24H29N6SO6 gives 529.18570, deviation −2.330 ppm.
100), λmin/nm 245; pH 1, λmax/nm 290 (23600), λmin/nm 242; pH 12, λmax/nm 301 (12800), λmin/nm 258. ε260 (µM) 11.2, ε280 (µM) 19. m/z 511.14 (50%, M + Na)+, 489.17 (40%, M + H)+, 425 (5%, M − Ph)+, 399 (5%, M − CoPh)+. Accurate mass measurement on C21H24N6SO6Na gives 511.13990, deviation 4.49ppm.
Homocystine (0.4 g, 1.5 mmol) in liquid ammonia (approximately 50 cm3) was converted to its di-sodium salt as described above. Once the blue colour had dissipated the reaction was cooled to −78 °C and the above silyl protected inosine derivative (0.4 g, 0.5 mmol) added, and the solution stirred at −78 °C for 6 hours. The reaction was allowed to warm to room temperature. Under a nitrogen atmosphere ethanol (20 cm3) was added followed by water (20 cm3) and the solution acidified (HOAc) and stirred at room temperature overnight to remove the silyl protection. The solution was evaporated, dissolved in water and purified on RP-C18 silica eluting with water and then a gradient of 0–10% acetonitrile in water to elute the product as an off-white foam. Yield 128 mg, 53%. δH
(D2O) 1.24, 1.45 (6H, 2 × s, isopropylidene CH3), 1.85–2.00 (2H, m, CH2CH2S), 2.49 (2H, t, J 7.2, CH2S), 2.64–2.67 (2H, m, H5′, H5″), 3.68 (1H, t, J 5.7, NH2CHCO2H), 4.25 (1H, br. s, H4′), 4.90 (1H, br. s, H3′), 5.24–5.27 (1H, m, H2′), 5.98 (1H, s, H1′), 7.99 (1H, s, H2), 8.05 (1H, s, H8). UV λmax/nm 249 (11600), λmin/nm 222; pH 12 λmax/nm 254 (12100), λmin/nm 232. m/z 448 (70%, M + Na)+, 399 (10%, M − CO)+. Accurate mass measurement on C17H23N5SO6Na gives 448.12670, deviation −0.01 ppm.
000), λmin/nm 223; pH 12 λmax/nm 254 (12500), λmin/nm 231. m/z 408.09 (100%, M + Na)+, 386.09 (20%, M + H)+. Accurate mass measurement on C14H19N5SO6Na gives 408.0957, deviation 0.72ppm.
250), λmin/nm 240; pH 12 λmax/nm 272 (7800), λmin/nm 251. m/z 383 (100%, M + Na)+. Accurate mass measurement on C13H20N4SO6Na 383.09980, deviation −0.93 ppm.
000), λmin/nm 245; pH 1, λmax/nm 290 (25400), 253 (10800), λmin/nm 261, 240; pH 12, λmax/nm 301 (13900), λmin/nm 258. m/z 473.2 (100%, M + H)+, 408 (10%, M − Ph)+. Accurate mass measurement on C21H25N6SO5 gives 473.16010, deviation −1.30 ppm.
200), λmin/nm 227; pH 1 λmax/nm 258 (14900), λmin/nm 229; pH 12 λmax/nm 260 (14300), λmin/nm 234. m/z 369.1 (40%, M + H)+, 391.1 (100%, M + Na)+. Accurate mass measurement on C14H20N6SO4Na gives 391.11620, deviation −0.71ppm.
200), λmin/nm 234; pH 12, λmax/nm 255 (28100), λmin/nm 240. m/z 773.23 (80%, M + Na)+. Accurate mass measurement on C39H38N6SO8Na gives 773.23720, deviation 0.28ppm.
200), 209 (69000), λmin/nm 245; pH 1, λmax/nm 281 (36600), λmin/nm 245; pH 12, λmax/nm 300 (21100), 255 (31300), λmin/nm 285, 240. m/z 870 (100%, M + Na)+, 848 30%, M + H)+. Accurate mass measurement on C43H41N7SO10Na gives 870.2533, deviation −2.60 ppm.
100), λmin/nm 227; pH 1, λmax/nm 258 (10000), λmin/nm 227. m/z 460.2 (100%, M + Na)+, 438 (80%, M + H)+. Accurate mass measurement on C18H27N7SO4Na gives 460.17280, deviation −3.31 ppm.
200), λmin/nm 228. m/z 398.16 (80%, M + H)+. Accurate mass measurement on C15H23N7SO4 gives 398.1610, deviation −0.13 ppm.
For electrophoretic mobility shift assays a substrate with a single hemimethylated HaeIII site was prepared by annealing complementary oligonucleotides Gsub (5′-TTCGAGTGACTGAGGCCGGTGTACCTGGAG-3′) and Gsub-meth (5′-CTCCAGGTACACCGGMCTCAGTCACTCGAA-3′), where M represents C5 methylcytosine. The substrate was 32P end labeled.
The concentration of M.HaeIII active sites was estimated by electrophoretic mobility shift assay as described previously.31 Reactions contained 1–150 nM DNA, 2–5 nM enzyme, 200 µM AdoHcy and 10 mM EDTA in M.HaeIII reaction buffer. Reactions were incubated at 37 °C for 30 minutes before addition of glycerol to 5% of the final volume and electrophoresis using 10% TBE gels (Novex, Invitrogen). Gels were fixed and dried according to the manufacturer's instructions, exposed for 18 hours and scanned using a phosphorimager (Typhoon 8600, Molecular Dynamics). Results were analysed using Imagequant software (Molecular Dynamics).
Plots of reaction velocity versus inhibitor concentration were fit to the curve in eqn. 2 using the Levenberg–Marquardt algorithm, where [I] is the concentration of AdoHcy or analogue. Inhibition is assumed to be competitive with AdoMet, as observed for inhibition of M.HhaI by AdoHcy (13), ΔΔG was calculated from the inhibition constants for AdoHcy and analogues using eqn. 3.
| ΔΔG = −RTln(Kianalogue/ KiAdoHcy) | (3) |
Acknowledgements
We wish to acknowledge the use of the EPSRC's Chemical Database Service at Daresbury.32References
- S-Adenosylmethionine-dependent methyltransferases: structures and functions, X. Cheng and R. M. Blumenthal, eds., World Scientific, Singapore, 1999 Search PubMed.
- G. L. Cantoni, Biological methylation: selected aspects, Annu. Rev. Biochem., 1975, 44, 435–451 CrossRef CAS.
- H. L. Schubert, R. M. Blumenthal and X. Cheng, Many paths to methyltransfer: a chronicle of convergence, Trends Biochem. Sci., 2003, 28, 329–335 CrossRef CAS.
- J. L. Martin and F. M. McMillan, SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold, Curr. Opin. Struct. Biol., 2002, 12, 783–793 CrossRef CAS.
- A. K. Ghosh and W. Liu, Total synthesis of (+)-sinefungin, J. Org. Chem., 1997, 62, 2299 CrossRef.
- A. K. Ghosh and W. M. Liu, Total synthesis of (+)-sinefungin, J. Org. Chem., 1996, 61, 6175–6182 CrossRef CAS.
- S. Hanessian and P. W. M. Sgarbi, Design and synthesis of mimics of S-adenosyl-L-homocysteine as potential inhibitors of erythromycin methyltransferases, Bioorg. Med. Chem. Lett., 2000, 10, 433–437 CrossRef CAS.
- D. C. Wahnon, V. K. Shier and S. J. Benkovic, Mechanism-based inhibition of an essential bacterial adenine DNA methyltransferase: Rationally designed antibiotics, J. Am. Chem. Soc., 2001, 123, 976–977 CrossRef CAS.
- R. M. Kagan and S. Clarke, Widespread occurrence of 3 sequence motifs in diverse S-adenosylmethionine-dependent methyltransferases suggests a common structure for these enzymes, Arch. Biochem. Biophys., 1994, 310, 417–427 CrossRef CAS.
- S. Kumar, X. D. Cheng, S. Klimasauskas, S. Mi and J. Posfai, The DNA (cytosine-5) methyltransferases, Nucleic Acids Res., 1994, 22, 1–10 CAS.
- K. M. Reinisch, L. Chen, G. L. Verdine and W. N. Lipscomb, The crystal-structure of HaeIII methyltransferase covalently complexed to DNA - an extrahelical cytosine and rearranged base-pairing, Cell, 1995, 82, 143–153 CrossRef CAS.
- A. P. Dong, J. A. Yoder, X. Zhang, L. Zhou and T. H. Bestor, Structure of human DNMT2, an enigmatic DNA methyltransferase homolog that displays denaturant-resistant binding to DNA, Nucleic Acids Res., 2001, 29, 439–448 CrossRef CAS.
- J. C. Wu and D. V. Santi, Kinetic and catalytic mechanism of HhaI methyltransferase, J. Biol. Chem., 1987, 262, 4778–4786 CAS.
- M. Pignot, G. Pljevaljcic and E. Weinhold, Efficient synthesis of S-adenosyl-L-homocysteine natural product analogues and their use to elucidate the structural determinant for cofactor binding of the DNA methyltransferase M HhaI, Eur. J. Org. Chem., 2000, 3, 549–555 CrossRef.
- J. Baddiley and G. A. Jamieson, Synthesis of S-(5′-deoxyadenosine-5′)-homocysteine, a product from enzymatic methylations involving “active methionine”, J. Am. Chem. Soc., 1955, 1, 1085–1089.
- R. T. Borchardt, J. A. Huber and Y. S. Wu, Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 4. Further modifications of the amino acid and base portions of S-adenosyl-L-homocysteine, J. Med. Chem., 1976, 19, 1094–1103 CrossRef CAS.
- K. Ramalingam and R. W. Woodward, Preparation of S-(N6,N6-dimethyladenosyl)-L-methionine, Carbohydr. Res., 1985, 142, 123–126 CrossRef CAS.
- R. T. Borchardt, J. A. Huber and Y. S. Wu, Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 2. Modification of the base portion of S-adenosylhomocysteine, J. Med. Chem., 1974, 17, 868–873 CrossRef CAS.
- M. Grøtli, M. Douglas, B. Beijer, R. G. Garcia and R. Eritja, Protection of the guanine residue during synthesis of 2′-O-alkyl-guanosine derivatives, J. Chem. Soc., Perkin Trans. 1, 1997, 18, 2779–2788 RSC.
- K. Ramalingam and R. W. Woodward, An improved synthesis of S-adenosylhomocysteine and related compounds, J. Org. Chem., 1984, 49, 1291–1293 CrossRef CAS.
- R. T. Borchardt and Y. S. Wu, Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 3. Modifications of the sugar portion of S-adenosylhomocysteine, J. Med. Chem., 1975, 18, 300–304 CrossRef CAS.
- W. M. Lindstrom, J. Flynn and N. O. Reich, Reconciling structure and function in HhaI DNA cytosine-C-5 methyltransferase, J. Biol. Chem., 2000, 275, 4912–4919 CrossRef CAS.
- H. M. Cohen, D. S. Tawfik and A. D. Griffiths, Altering the sequence specificity of HaeIII methyltransferase by directed evolution using in vitro compartmentalization, Protein Eng., Des. Sel., 2004, 17, 3–11 Search PubMed.
- A. Fersht, Structure and mechanism in protein science, Freeman, San Francisco, 1999 Search PubMed.
- X. Cheng, S. Kumar, S. Klimasauskas and R. J. Roberts, Crystal-structure of the HhaI DNA methyltransferase, Cold Spring Harbor Symp. Quant. Biol., 1993, 58, 331–338 Search PubMed.
- S. Klimasauskas, S. Kumar, R. J. Roberts and X. D. Cheng, HhaI methyltransferase flips its target base out of the DNA helix, Cell, 1994, 76, 357–369 CrossRef CAS.
- A. R. Maguire, W. D. Meng, S. M. Roberts and A. J. Willetts, Synthetic approaches towards nucleocidin and selected analogs - anti-HIV activity in 4′-fluorinated nucleoside derivatives, J. Chem. Soc., Perkin Trans. 1, 1993, 15, 1795–1808 RSC.
- A. Hampton, M. Bayer, V. S. Gupta and S. Y. Chu, Synthesis of 5′-substituted derivatives of inosine, J. Med. Chem., 1968, 11, 1229–1232 CrossRef CAS.
- A. Romieu, D. Gasparutto, D. Molko and J. Cadet, Site-specific introduction of (5′ S)-5′,8-cyclo-2′-deoxyadenosine into oligodeoxyribonucleotides, J. Org. Chem., 1998, 63, 5245–5249 CrossRef CAS.
- H. M. Cohen, D. S. Tawfik and A. D. Griffiths, Promiscuous methylation of non-canonical DNA sites by HaeIII methyltransferase, Nucleic Acids Res., 2002, 30, 3880–3885 CrossRef CAS.
- G. Vilkaitis, E. Merkiene, S. Serva, E. Weinhold and S. Klimasauskas, The mechanism of DNA cytosine-5 methylation - Kinetic and mutational dissection of HhaI methyltransferase, J. Biol. Chem., 2002, 276, 20924–20934 CrossRef.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, The United Kingdom Chemical Database Service, J. Chem. Inf. Comput. Sci., 1996, 36, 746–749 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: proposed hydrogen bonds between methyltransferase and cofactor based on the crystal structure of each methyltransferase with bound AdoHcy. See http://www.rsc.org/suppdata/ob/b4/b415446k/ |
| This journal is © The Royal Society of Chemistry 2005 |