Studies related to carba-pyranoses: synthesis of acetylated derivatives of 4-amino-2,4-dideoxy-3-O-(β-D-glucopyranosyl)-β-L-(and β-D-) altrocarba-pyranose from a D-glucose template†
Robin G.
Pritchard
a,
Richard J.
Stoodley
a and
Wai-Hung
Yuen
*b
aDepartment of Chemistry, UMIST, PO Box 88, Manchester, UK M60 1QD
bDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: yuenwh@hkusua.hku.hk; Fax: (852) 2857-1586; Tel: (852) 2859-8965
First published on 29th November 2004
Abstract
Methyl (E)-3-nitroacrylate 15, the X-ray analysis of which is reported here, is prepared from methyl acrylate by a new route involving sequential reactions with mercury(II) chloride–sodium nitrite, bromine and sodium acetate. The dienophile 15 reacts with Danishefsky's diene 17 to give, after acidic hydrolysis, a 67 : 33 mixture of the ketones rac-18 and rac-19. With (E)-1-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)-3-(trimethylsiloxy)buta-1,3-diene 1, it affords, after hydrolysis, a 42 : 18 : 28 : 12 mixture of the ketones 30, 31, 32 and 33.
The alcohol 36, obtained by sodium borohydride reduction of the ketone 30, is converted into the monocarba-disaccharide 35 by the action of aluminium amalgam, lithium aluminium hydride and acetic anhydride. Similarly, the alcohol 41, derived from the ketone 32, is transformed into the monocarba-disaccharide 40; the structure of the alcohol 41 is secured by an X-ray analysis. The isolation of a mixture of the acetoxyamino and acetylamino derivatives 38 and 39 from the reaction of the alcohol 36 with aluminium amalgam and acetic anhydride indicates that the nitro function is converted into the hydroxylamino and amino groups by the reducing agent.
The cyclohexane rings of the ketones rac-18, rac-19, 30, 31 and 33 adopt the expected chair conformations. Thus, the methoxycarbonyl, nitro and oxy substituents are equatorially orientated in the ketones rac-18 and 30; in the ketones rac-19, 31 and 33, the methoxycarbonyl and nitro groups occupy equatorial dispositions and the oxy substituent is axially orientated. The cyclohexane ring of the ketone 32 (which bears a diastereomeric relationship to that of the ketone 30) displays unexpected conformational properties, that are attributed to a significant population of the chair conformer with axial arrangements of the methoxycarbonyl, nitro and oxy groups.
Introduction
Over the past few years, we have demonstrated that anomerically linked glycopyranose units can confer a useful degree of facial reactivity on 1-oxybuta-1,3-dienes in cycloaddition reactions.1–13 For example, the diene 1 displays good Re-face‡ reactivity and undergoes highly endo-selective Diels–Alder reactions,1,2,7 with cyclic dienophiles of type 2 to give predominantly cycloadducts of type 3. As well as endeavouring to understand the basis of the stereoinduction, we have sought to employ such cycloaddition reactions in the assembly of compounds of biological importance. Within the latter context, asymmetric syntheses of anthracyclinones,1,7,13,14 bostrycins,8 dehydropiperazic acids,9 dihydropyranone,12 5-arylpentopyranoses15 and epi-shikimic acid16 have been effected.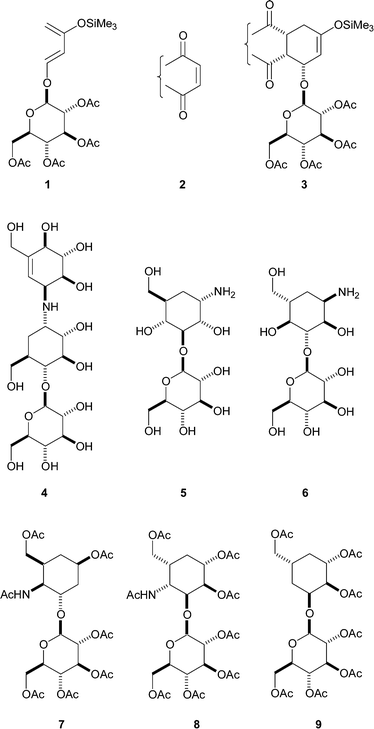
In the aforementioned syntheses, the glycopyranose moiety served as a ‘chiral auxiliary’ role, being removed from the pre-target structures by mild acidic hydrolysis. Mindful of the emerging importance of saccharides in medicinal chemistry,17–19 we have also sought to prepare oligosaccharide-like compounds that retain the glycopyranose unit. Within this framework, monocarba-disaccharides that feature a pyranose entity glycosidically linked to a carba-pyranose§ moiety have attracted our attention. Such assemblies, which are found in some aminoglycoside antibiotics, e.g. validamycin A 4,21 have been the subject of relatively few synthetic endeavours. Typically, they are prepared by glycosidation methodology in which an appropriately protected carba-pyranose (often as racemate) serves as the glycosyl acceptor.20
We planned to use Diels–Alder reactions to construct such monocarba-disaccharides and initially decided to employ the readily available diene 1.1–3 In consequence, any targets would feature the β-D-glucopyranosyl unit. Noting that few acetal-linked (1→3)-monocarba-disaccharides had been synthesised21 (examples include compounds 5 and 622), we decided to prepare further representatives of this class and have recently reported the syntheses of (1→3) linked monocarba-disaccharides 7, 823 and 9.24
Earlier,25 we showed that the cycloadduct 10 (obtained from the reaction of the diene 1 with maleic anhydride) could be readily converted into the monocarba-disaccharide 14 by way of the intermediates 11–13 as outlined in Scheme 1. It should be noted that the sequence, in which one new stereocentre had been developed, led to the generation of a 4-acetylamino-2,4-dideoxycarbahexopyranose unit¶ with the ‘β-L-galacto’ configuration.
 | ||
| Scheme 1 Reagents: i, H+, CHCl3; ii, Na(CN)BH3, HOAc; iii, (COCl)2, DMF, CH2Cl2; iv, NaN3, THF; v, Δ, PhH; vi, Et3N, THF, H2O; vii, Δ, LiAlH4, THF; viii, Ac2O, pyridine, DMAP; ix, IR-400 (OH−), MeOH. | ||
In seeking to complement this technology and provide access to relatives of compounds 7, 8 and 14 in which the 4-acetylamino-2,4-dideoxycarba-hexopyranose unit featured an anti arrangement of the 4- and 5-substituents (sugar numbering), we have undertaken a study of the reactivity of the diene 1 with methyl (E)-3-nitroacrylate 1528 and of the derived cycloadducts. We now report our findings.
Results and discussion
Previously, we had shown2,3 that the Re-face : Si-face selectivity of the diene 1 was 86 : 14 towards N-phenylmaleimide and 67 : 33 towards tetracyanoethylene in benzene at ambient temperature. Furthermore, Danishefsky had reported29 that the nitroacrylate 15 underwent reaction with the diene 17 under comparable conditions to give, after acidic work-up, the ketone rac-18 as the major product and the ketone rac-19 as the minor product.
On the basis of the foregoing information, we envisaged that the Diels–Alder reaction of the diene 1 with the nitroacrylate 15 would display reasonable-to-moderate facial selectivity, high regioselectivity and reasonable exo-nitro group selectivity. Thus, the Re-face exo-nitro cycloadduct 20 and the Re-face endo-nitro cycloadduct 21 were expected to predominate over their Si-face counterparts 22 and 23; furthermore, the total exo-nitro cycloadduct production was expected to exceed the total endo-nitro cycloadduct production (i.e.20 + 22 > 21 + 23).
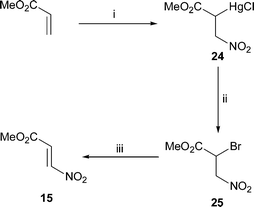
The usual method for the preparation of β-nitroacrylates is that developed by Stevens and Emmons,28 involving the formal addition of nitryl iodine to an acrylate ester using iodine and dinitrogen tetraoxide followed by dehydroiodination; the procedure is reported to give the nitroacrylate 15 in 70% overall yield30 and the nitroacrylate 16 in 81% overall yield.31 Shin prepared the latter nitroacylate in 45% overall yield using nitrosyl chloride in the first step.32 The procedure we used, which is an adaptation of that developed by Corey33 for the conversion of alkenes into nitroalkenes (but not hitherto used for the synthesis of β-nitroacrylates), is outlined in Scheme 2. Thus, methyl acrylate was converted into the nitromercuration product 24 and thence the bromide 25; dehydrobromination of compound 25 provided the nitroacrylate 15 in 67% overall yield.
Initially, we decided to re-examine the reaction of Danishefsky's diene 17 with the nitroacrylate 15 in order to quantify the exo-nitro cycloadduct : endo-nitro cycloadduct stereoselectivity.34 In dichloromethane at ambient temperature, the reaction led to mainly a 67 : 33 mixture of cycloadducts; following acidic hydrolysis, a 60 : 40 ratio of ketones resulted. As noted by Danishefsky,29 it was possible to isolate the major ketone from the mixture simply by crystallization; we obtained the material in 34% yield (compared to 48% yield reported by the Pittsburgh group). Subjection of the mother liquor to preparative HPLC gave a pure sample of the minor ketone.
A comparison of the NMR spectroscopic properties of the aforecited ketones left little doubt that they were stereoisomers. On the basis of conformational considerations, the major ketone was considered to possess the stereostructure rac-18 and the minor ketone the stereostructure rac-19, in accord with Danishefsky's assignments. Thus from the coupling constants presented in Table 1, it was clear that the major ketone adopted the chair conformation rac-26 in which the 1-, 2- and 3-substituents were equatorially orientated. Similarly, the minor ketone existed in the chair geometry rac-27, with the 1- and 2-substituents equatorial and the 3-substituent axial.

| Compound | J 1,2 | J 2,3 | J 3,4ax | J 3,4eq | J 1,6ax | J 1,6eq |
|---|---|---|---|---|---|---|
| a Ketone A. b Ketone C. c Ketone B. d Ketone D. e License is implied in the use of the axial and equatorial descriptors in this instance. f Because of the deceptively simple nature of the spectrum, only the sum of these coupling constants could be deduced (see ref. 35). | ||||||
| rac-18 | 9.5 | 7.5 | 9 | 4.5 | 11.5 | 6 |
| rac-19 | 11 | 3 | 3 | 3.5 | 13.5 | 5.5 |
| 30 a | 11.5 | 9.5 | 11.5 | 5.5 | 13.5 | 5 |
| 31 b | 11 | 3 | 3 | 3 | 13 | 5.5 |
| 32 c | 8 | 5.5 | 7 | 4.5e | ←16f→ | |
| 33 d | 11.5 | 2.5 | 2.5 | 3 | 13 | 5.5 |
In summary, the Diels–Alder reaction of the nitroacrylate 15 and the diene 17 in dichloromethane provides a 67 : 33 mixture of the exo-nitro cycloadduct rac-28 and the endo-nitro cycloadduct rac-29.
The reaction of the diene 1 with the nitroacrylate 15, carried out in dichloromethane, afforded four cycloadducts in the ratio of 43 : 30 : 18 : 9 by NMR spectroscopic analysis. Acidic hydrolysis of the cycloadducts gave a 42 : 28 : 18 : 12 mixture of four ketones, designated A–D (in order of their decreasing abundance). Following fractionation of the mixture by column chromatography and crystallisation, ketone A was isolated in 24% yield and ketone B in 18% yield. The use of preparative HPLC led to the isolation of ketone C and a 75 : 25 mixture of ketones D and C.
A comparison of the NMR spectroscopic properties of ketones A–D left little doubt that they were stereoisomers and represented by the structures 30–33. The coupling constants of the cyclohexane-ring protons of ketone A, summarised in Table 1, showed a good match to those of ketone rac-18, indicating that ketone A possessed the stereostructure 30 or 32. Similarly, the cyclohexane-ring proton coupling constants of ketones C and D (Table 1) were very similar to those of ketone rac-19, revealing that ketones C and D possessed the stereostructures 31 and 33. In view of the previously established Re-face selectivity of the diene 1, ketone A was assigned the stereostructure 30, ketone C the stereostructure 31 and ketone D the stereostructure 33. By difference, ketone B was considered to possess the stereostructure 32. As can be seen from Table 1, its cyclohexane-ring coupling constants differed significantly from those of its relative 30; this issue will be considered later.

In summary, the reaction of the diene 1 with the nitroacrylate 15 leads to a 43 : 18 : 30 : 9 mixture of the cycloadducts 20, 21, 22 and 23, corresponding to a Re-face : Si-face selectivity of 61 : 39 and an exo-nitro : endo-nitro selectivity of 73 : 27. Clearly, the former selectivity is quite similar to that (67 : 33) seen in the cycloaddition of the diene 1 with tetracyanoethylene and the latter selectivity is comparable to that (67 : 33) observed in the cycloaddition of the diene 17 with the nitroacrylate 15.
Having earlier defined the solid-state structure of the diene 1,3 we felt that a knowledge of the corresponding geometry of the nitroacrylate 15 would be a valuable input into transition-state modelling studies. Surprisingly, in spite of their wide application in synthesis, β-nitroacrylates are not represented in the Cambridge crystallographic data base. An X-ray analysis of the nitroacrylate 15, depicted in Fig. 1 with its crystallographic labelling, revealed planarity of both the nitro and ester groups. Moreover, the ester carbonyl and olefinic unit bore a syn relationship. As an example, therefore, an arrangement such as 34 is possibly relevant to the development of the transition-state geometry leading to the Re-face exo-nitro cycloadduct 20.
 | ||
| Fig. 1 Molecular structure of the nitroacrylate 15. | ||
Having shown that the ketone 30 was the major product from the hydrolysate of the reaction of the diene 1 with the nitroacrylate 15, attention was turned to effecting its conversion into the monocarba-disaccharide 35. The first task was to stereoselectively reduce the ketone function.
Treatment of the ketone 30 with sodium borohydride in methanol at −78 °C gave the alcohol 36 (74% yield after crystallisation), the stereostructure of which was established by NMR spectroscopy. The cyclohexane ring of compound 36 would be expected to adopt the chair geometry 37, in which the 5-hydroxy group is equatorial. This was borne out by the coupling constants shown in Table 2; in particular, the axial 6-H, which resonated at δ 1.52 as a double triplet, displayed three large coupling constants (J 11, 13 and 13 Hz).
| Compound | J 1,2 | J 2,3 | J 3,4ax | J 3,4eq | J 4ax,5 | J 4eq,5 | J 5,6ax | J 5,6eq | J 1,6ax | J 1,6eq |
|---|---|---|---|---|---|---|---|---|---|---|
| 35 | 10 | 11 | 11 | 4.5 | 12 | 4 | 12 | 4 | 12 | — |
| 36 | 11.5 | 10 | 11.5 | 4.5 | 12 | — | 11 | — | 13 | 4.5 |
| 38 | 11 | 11 | 12 | — | 12 | 4 | 12.5 | 4 | 13 | 4 |
| 39 | 11 | 10.5 | 12 | — | 12 | 4 | 12 | 4 | 13 | 3.5 |
| 40 | 10 | 10.5 | 11 | 4 | 11 | 4 | 11.5 | 4 | 12 | — |
| 41 | 11 | 10 | 11.5 | — | 11.5 | — | 11.5 | — | 13 | 4 |
After screening a variety of reducing agents, aluminium amalgam36 in aq. methanol was found to effect the nitro-group reduction37 of compound 36. Following acetylation of the product and column chromatographic fractionation, the hydroxylamine||38 and amine derivatives 38 and 39 were isolated in respective yields of 37 and 26%. That these reductions had occurred with retention of configuration at position 2 was demonstrated by the cyclohexane-ring proton coupling constants (Table 2), which were comparable to those of the nitro precursor 36. When the product from the aluminium amalgam reduction was subjected to the action of lithium aluminium hydride in THF and acetic anhydride in pyridine, the target carba-disaccharide 35 was obtained (37% yield after chromatography). Again, the coupling constants of the cyclohexane-ring protons (Table 2) left little doubt that the ester-reduction step had occurred with retention of configuration.
To conclude the study, the conversion of the ketone 32 into the monocarba-disaccharide 40 was undertaken. Sodium borohydride reduction of the ketone 32 gave the alcohol 41 (73% yield after crystallisation). According to NMR spectroscopy, the cyclohexane ring of compound 41 adopted the chair conformation 42 (see Table 2). Subjection of compound 41 to the reductive acetylation sequence (Al·Hg/MeOH/H2O; LiAlH4/THF; Ac2O/pyridine) gave the monocarba-disaccharide 40 (35% yield after chromatography). On the basis of NMR spectroscopy, its cyclohexane ring adopted a chair conformation akin to that of its precursor (Table 2).

An X-ray crystallographic analysis of compound 41, shown in Fig. 2 with its crystallographic labelling, established that the cyclohexane ring possessed the absolute stereochemistry that had been assigned to it. Clearly, the chair conformation 42 observed in deuteriochloroform solution was also present in the crystalline state.
 | ||
| Fig. 2 Molecular structure of compound 41. | ||
The present work reveals that 3-O-β-D-glucopyranosyl derivatives of 4-acetylamino-2,4-dideoxycarbapyranoses with the ‘β-L-altro’ and ‘β-D-altro’ configurations can be assembled from the β-D-glucopyranosyl diene template 1. The results complement previous findings23,24 in which related monocarba-disaccharides with the ‘β-L-galacto’ and ‘β-D-galacto’ configurations were constructed from the same template.
As mentioned earlier, the differing conformational properties of the ketones 30 and 32, which feature cyclohexanone rings that bear the diastereomeric relationship, is noteworthy. Clearly, the sugar residue is responsible for the difference. In the case of the ketone 30, the cyclohexanone ring adopted the expected chair conformation 43 (in which the 1-, 2- and 3-substituents were equatorially orientated) in deuteriochloroform on the basis of the observed coupling constants (Table 1), which were close to those calculated for an equivalent conformer (see 43, Table 3). In the case of the ketone 32, the cyclohexanone ring probably existed mainly as a ca. 50 : 50 mixture of the chair conformers 43 (in which the 1-, 2- and 3-substituents occupied equatorial positions) and 44 (in which the 1-, 2- and 3-substituents adopted axial locations) (Scheme 3); thus, the observed coupling constants (Table 1) were in moderate agreement with the calculated ones (Table 3). The NOE enhancements of the cyclohexanone-ring protons of the ketones 30 and 32 (Table 4) were consistent with the conformational situations proposed. In particular, on average, the 1- and 2-protons and the 3- and 4-protons were closer together in compound 32 than in compound 30.
| Conformer | J 1,2 | J 2,3 | J 3,4ax | J 3,4eq | J 1,6ax | J 1,6eq |
|---|---|---|---|---|---|---|
| a Using Macromodel Version 5.5 (see Experimental section). | ||||||
| 43 | 12.9 | 10.3 | 11.2 | 5.1 | 12.3 | 3.7 |
| 44 | 1.8 | 3 | 3.8 | 2.2 | 2.2 | 4.5 |
| 43 : 44 (1 : 1) | 7.4 | 6.7 | 7.5 | 3.7 | 7.3 | 4.1 |


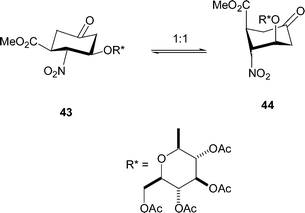 | ||
| Scheme 3 | ||
Seemingly, therefore, the sugar modifies the conformational behaviour of compound 32 by increasing the equilibrium concentration of the conformer 44 in which the 1-, 2- and 3-substituents are axially orientated. The unexpected conformational behaviour induced by the sugar residue was not observed after the reduction. Presumably, the steric and dipolar interactions between the methoxycarbonyl, nitro and oxy groups play an important role in the conformational properties of the alcohol 41. As has already been noted, the cyclohexane ring of the reduction product 41 displayed normal conformational behaviour in deuteriochloroform; thus, it adopted the chair geometry 42 (comparable to the geometry 37 adopted by the reduction product 36). Evidently, the cyclohexanone carbonyl group of compound 32 is also required for the anomalous conformational properties. A study of solvent effects on the coupling constants of the cyclohexanone-ring protons of compound 32, shown in Table 5, revealed that the atypical conformational behaviour was most pronounced in deuteriochloroform and perdeuteriobenzene. In perdeuteriodimethyl sulfoxide, the cyclohexanone ring of compound 32 existed mainly as the chair conformer 43; in perdeuteriochloromethane and perdeuteriotetrahydrofuran, an intermediary situation was in evidence.
A knowledge of the global conformations of compounds 30 and 32 is relevant to the origins of the differing conformational properties of their cyclohexane rings. That the 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloxy moieties adopted the expected 4C1 conformations was secured from vicinal coupling constants values and NOE measurements (see Experimental section). Based on exo-anomeric effect39 and torsional considerations,40 the partial geometry 45 would be expected to make a significant contribution to the global conformations of both compounds 30 and 32; this was borne out by the sizable NOE enhancements between the anomeric protons and the cyclohexanone 3-protons (8–9% for 30 and 6–7% for 32). This partial geometry can be accommodated in the conformers 46 and 47 (Scheme 4) in the case of compound 30 and in the conformers 48 and 49 (Scheme 5) in the case of compound 32.
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
For compound 30, the conformer 46 is preferred because of the equatorial arrangement of the substituents of its cyclohexanone ring. In the case of compound 32, conformers akin to 48 and 49 are considered to contribute significantly to the overall conformational situation. As represented, the conformer 48 would experience a severe destabilising interaction between the nitro group and the oxygen atom of the pyranose ring; relief of the interaction is achievable by enlargement of the O(5′)–C(1′)–O(3)–C(3) torsion angle, rotation about the O(3)–C(3) bond and expansion of the C(1′)–O(3)–C(3) bond angle.** In the conformer 49, the aforecited intra-annular interaction is absent, although there is a penalty to be paid because of the axial arrangement of the cyclohexanone-ring substituents.
It may be noted that the coupling constants of the cyclohexanone-ring protons of compound 32 in perdeuteriodichloromethane and perdeuteriotetrahydrofuran were very similar to those of compound rac-18 in deuteriochloroform, implying that the invertomer of the conformer rac-26 makes a small contribution to the conformational situation in the case of compound rac-18. Accordingly, it was of interest to determine whether the conformational properties of compound rac-18 could be influenced by solvent. From the results shown in Table 5, it is clear that they can. Thus, in perdeuteriodimethyl sulfoxide, it is evident that the conformer rac-26 is the dominant species. Possibly, intramolecular dipolar interactions between the carbonyl carbon atom of the 1-methoxycarbonyl group and a lone pair of electrons on the oxygen atom of the 3-oxy substituent contribute to the stabilisation of the axial conformer in the non-polar solvents.
Experimental
Dry solvent, referred to in the ensuing experiments were prepared as follows: diethyl ether was distilled off sodium–benzophenone; dichloromethane was distilled off phosphorus(V) oxide; pyridine was distilled off sodium hydroxide pellets. Sodium acetate was dried in an oven at 110 °C for 5 h.TLC was performed on Merck plastic or aluminium plates coated with silica gel (60 F254); chromatograms were initially examined under UV light (Mineralight UVG-58 lamp) and visualised with either iodine vapour or a p-anisaldehyde stain [plates were sprayed with EtOH : conc. H2SO4 : p-MeOC6H4CHO (95 : 4 : 1) and heated]. Column chromatography was effected, under positive pressure from a compressed-air line, employing Crossfield Sorbsil C60 flash silica. HPLC was carried out on Spherisorb S10 silica columns (25 × 0.46 cm for analytical and 25 × 0.8 cm for preparative work), using a Kontron 420 pump and Kontron 742 UV/ERC-7515A RI detectors.
Evaporations were conducted under reduced pressure (using a water-pump or an oil-pump) at ≤40 °C with a Büchi rotary evaporator. Mps were determined with a Büchi 512 melting point apparatus. Optical rotations, given in 10−1 deg cm2 g−1, were measured at ca. 20 °C using a Thorn Automation Type 243 polarimeter. Elemental analyses were performed with a Carlo-Erba Model 1108 analyser. A Perkin-Elmer Lambda 15 spectrometer was used to measure UV spectra; extinction coefficients (ε) are presented in cm2 mmol−1. IR Spectra were recorded with a Perkin-Elmer 783 spectrometer. NMR Spectra were determined using a Bruker AC 300 or a Varian VXR600S spectrometer (with DEPT editing for 13C NMR spectra); J values and separations are given in Hz. Proton assignments were supported by COSY 45° experiments. FAB Mass spectra (m-O2NC6H4CH2OH as matrix) were recorded using a Kratos MS50TC spectrometer.
Methyl (E)-3-nitroacrylate 15
Methyl acrylate (20.4 g, 0.24 mol) was added in portions to a vigorously stirred solution of mercury(II) chloride (61.2 g, 0.23 mol) and sodium nitrite (31.2 g, 0.45 mol) in water (500 cm3). After 16 h, the precipitated material was collected by filtration, washed with water followed by hexanes, and dried in vacuo to afford the nitromercurial chloride 24 (61.8 g, 74%) as a white solid.Bromine (52.7 g, 0.33 mol) was added in portions to a vigorously stirred, ice-cooled mixture of the nitromercurial chloride 24 (61.8 g, 0.17 mol) in water (200 cm3) and diethyl ether (500 cm3). After the addition was complete, the mixture was allowed to warm to room temperature and stirred overnight. Sodium hydrogen carbonate was then added in portions until effervescence ceased. The organic phase was separated and the aqueous phase was extracted with diethyl ether. Evaporation of the combined, dried (MgSO4) organic phases gave the nitro bromide 25 (33.7 g, 95%) as a yellow oil.
Dried sodium acetate (77.9 g, 0.95 mol) was added in portions to a stirred solution of nitro bromide 25
(33.7 g, 0.16 mmol) in dry diethyl ether (100 cm3). After 3 days, the mixture was diluted with diethyl ether (50 cm3) and filtered. The filtrate was washed with saturated aq. sodium hydrogen carbonate (3 ×) and water, dried (MgSO4) and concentrated. Crystallisation of the oil from diethyl ether–hexanes at low temperature gave the title compound15
(19.9 g, 95%); mp 34–35 °C (lit., 37–38 °C,28 33–35 °C)30
(Found: C, 36.8; H, 4.0; N, 10.6. Calc. For C4H5NO4: C, 36.6; H, 3.8; N, 10.7%); λmax
(EtOH)/nm 221 (ε 10 700); νmax
(KBr)/cm−1 1740 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1655 (CC) and 1550 (NO2); δH
(300 MHz; CDCl3) 3.88 (3 H, s, MeO2C) and 7.10 and 7.69 (each 1 H, d, J 13.5, 2- and 3-H); δC
(75 MHz; CDCl3) 52.92 (CH3O), 127.1 (2-CH), 149.0 (3-CH) and 163.0 (1-CO).
O), 1655 (CC) and 1550 (NO2); δH
(300 MHz; CDCl3) 3.88 (3 H, s, MeO2C) and 7.10 and 7.69 (each 1 H, d, J 13.5, 2- and 3-H); δC
(75 MHz; CDCl3) 52.92 (CH3O), 127.1 (2-CH), 149.0 (3-CH) and 163.0 (1-CO).
Cycloaddition–hydrolysis studies
Reaction of the diene 17
(a) The diene 17 (90% purity, 0.050 g, 0.26 mmol) gave rise to a yellow oil which comprised mainly a 67 : 33 mixture of the cycloadducts rac-28 and rac-29 [the ratio was estimated from the integrals of the double quartet (J 8 and 2.5) at δ 4.51 and of the double doublet (J 4 and 6) at δ 4.40, attributed to the 5-Hs of rac-28 and rac-29]. After acidic hydrolysis, a 60 : 40 mixture of the ketones rac-18 and rac-19 (the ratio was estimated from the intensities of the singlets at δ 3.38 and 3.32, attributed to the methoxy groups of the ketones rac-18 and rac-19) was isolated.(b) The diene 17
(90% purity, 4.51 g, 24 mmol) gave rise, after hydrolysis, to a brown foam. Crystallisation of the material from dichloromethane–diethyl ether–hexanes afforded methyl
(1R*,2S*,3S*)-3-methoxy-2-nitro-5-oxocyclohexane-1-carboxylate rac-18
(1.88 g, 34%); mp 110–112 °C (lit.,29 110–112 °C)
(Found: C, 47.1; H, 6.0; N, 6.2. Calc. For C9H13NO6: C, 46.8; H, 5.7; N, 6.1%); λmax
(EtOH)/nm 207 (ε 3600); νmax
(KBr) 1740br (ester CO), 1730 (ketone CO) and 1560 (NO2); δ
(300 MHz; CDCl3) 2.54 and 2.88 [each 1 H, ddd (J 1, 9 and 15) and ddd (J 1.5, 4.5 and 15), 4-Hax and 4-Heq], 2.67 and 2.76 [each 1 H, ddd, (J 1, 11.5 and 16) and ddd (J 1.5, 6 and 16), 6-Hax and 6-Heq], 3.38 (3 H, s, MeO), 3.49 (1 H, ddd, J 6, 9.5 and 11.5, 1-H), 3.75 (3 H, s, MeO2C), 4.07 (1 H, ddd, J 4.5, 7.5 and 9, 3-H) and 5.08 (1 H, dd, J 7.5 and 9.5, 2-H); m/z
(FAB) 254 (MNa+, 24%), 232 (MH+, 75) and 200 (C8H10NO5+, 100).
Evaporation of the filtrate obtained from the foregoing crystallisation gave a residue that contained a 50 : 50 mixture of the ketones rac-18 and rac-19. A portion of the mixture (0.100 g) was fractionated by HPLC [hexanes–EtOAc (2 : 1) as eluent].
The first-eluted material (0.030 g), isolated as a crystalline solid, was identified as the ketone rac-18 by NMR spectroscopy.
The second-eluted material (0.040 g) was methyl
(1S*,2R*,3S*)-3-methoxy-2-nitro-5-oxocyclohexane-1-carboxylate rac-19; mp 88–90 °C (Found: C, 47.0; H, 5.4; N, 5.9. Calc. C9H13NO6 requires C, 46.8; H, 5.7; N, 6.1%); λmax
(EtOH)/nm 206 (ε 4000); νmax
(KBr)/cm−1 1740 (ester CO), 1720 (ketone CO) and 1550 (NO2); δ
(300 MHz; CDCl3) 2.42 and 2.79 [each 1 H, dd, (J 13.5 and 15) and ddd (J 2.5, 5.5 and 15), 6-Hax and 6-Heq], 2.58 and 2.91 [each 1 H, dd (J 3 and 15.5) and ddd (J 2.5, 3.5 and 15.5), 4-Hax and 4-Heq], 3.32 (3 H, s, MeO), 3.77 (3 H, s, MeO2C), 3.85 (1 H, ddd, J 5.5, 11 and 13.5, 1-H), 4.56 (1 H, br q, separation 3, 3-H) and 5.12 (1 H, dd, J 3 and 11, 2-H); m/z
(FAB) 232 (MH+, 92%) and 200 (C8H10NO5+, 100).
Reaction of the diene 1
(a) The diene 1 (0.200 g, 0.41 mmol) gave rise to a yellow oil which comprised mainly a 43 : 30 : 18 : 9 mixture of the cycloadducts 20, 22, 21, and 23 [the ratio was estimated from the intensities of the doublets (J 8 Hz) at δ 4.50, 4.59, 4.46 and 4.65, attributed to the 1′-Hs of the cycloadducts]. After hydrolysis, a foam (0.200 g) was isolated which comprised mainly a 42 : 28 : 18 : 12 mixture of the ketones 30, 32, 31 and 33 [the ratio was determined from the intensities of the doublets (J 8 Hz) at δ 4.48, 4.58, 4.45 and 4.47, attributed to the 1′-Hs of the ketones]. Subjection of the mixture to column chromatography [hexanes–EtOAc (1 : 1) as eluent] gave rise to two fractions.The first-eluted material (0.070 g) was mainly compound 30. After crystallisation from dichloromethane–diethyl ether–hexanes, methyl
(1R,2S,3S)-2-nitro-5-oxo-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy) -cyclohexane-1-carboxylate30
(0.054 g, 24%) was obtained as needles; mp 182–184 °C; [α]D
−13 (c 0.5, CH2Cl2)
(Found: C, 48.3; H, 5.0; N, 2.8. C22H29NO15 requires C, 48.3; H, 5.3; N, 2.6%); λmax
(EtOH)/nm 204 (ε 4200); νmax
(KBr)/cm−1 1760 and 1740 (ester CO), 1720 (ketone CO) and 1565 (NO2); δ
(300 MHz; CDCl3) 1.99, 2.02, 2.07 and 2.11 (each 3 H, s, 4 × MeCO2), 2.60 and 2.75 [each 1 H, dd, (J 13.5 and 15) and ddd (J 2, 5 and 15), 6-Hax and 6-Heq], 2.67 and 3.07 (each 1 H, dd, (J 11 and 15) and ddd (J 2, 5 and 15), 4-Hax and 4-Heq], 3.32 (1 H, ddd, J 5, 11 and 13.5, 1-H), 3.69 (1 H, ddd, J 2.5, 5.5 and 10, 5′-H), 3.74 (3 H, s, MeO2C), 4.11 and 4.23 [each 1 H, dd, (J 2.5 and 12.5) and dd (J 5.5 and 12.5), 6′-H2], 4.30 (1 H, ddd, J 5, 10 and 11, 3-H), 4.48 (1 H, d, J 8, 1′-H), 4.95 (1 H, dd, J 8 and 9.5, 2′-H), 5.01 (2 H, br t, J 10, 4′- and 2-H) and 5.15 (1 H, t, J 9.5, 3′-H); δ
(600 MHz; CDCl3) 1.99, 2.02, 2.08 and 2.11 (each 3 H, s, 4 × MeCO2), 2.61 and 2.75 [each 1 H, ddd, (J 0.5, 13.5 and 15.5) and ddd (J 2, 5 and 15.5), 6-Hax and 6-Heq], 2.68 and 3.06 (each 1 H, ddd (J 0.5, 11.5 and 15) and ddd (J 2, 5.5 and 15), 4-Hax and 4-Heq], 3.32 (1 H, ddd, 5, 11.5 and 13.5, 1-H), 3.69 (1 H, ddd, J 2.5, 5.5 and 10, 5′-H), 3.75 (3 H, s, MeO2C), 4.12 and 4.23 [each 1 H, dd (J 2.5 and 12.5) and dd (J 5.5 and 12.5), 6′-H2], 4.31 (1 H, ddd, J 5.5, 9.5 and 11.5, 3-H), 4.49 (1 H, d, J 8, 1′-H), 4.95 (1 H, dd, J 8 and 9.5, 2′-H), 5.01 [2 H, t (J 9.5) and dd (J 9.5 and 11.5), 4′- and 2-H] and 5.15 (1 H, t, J 9.5, 3′-H)
[NOE difference: δ 2.61→2.75 (13%), 3.32 (2%) and 5.01 (2%); δ 2.68→3.06 (13%) and 5.01 (2%); δ 2.75→2.61 (9%), 3.06 (2%) and 3.32 (4%); δ 3.06→2.68 (11%) and 4.31 (3%); δ 3.32→2.75 (3%), 4.31 (4%) and 5.01 (1%); δ 3.69→4.12 (2%), 4.23 (2%), 4.49 (7%), 5.01 (1%) and 5.15 (5%); δ 4.12→3.69 (3%), 4.23 (11%) and 5.01 (1%); δ 4.23→4.12 (9%) and 5.01 (2%); δ 4.31→3.06 (3%), 3.32 (6%), 4.49 (8%) and 5.01 (1%); δ 4.49→3.69 (7%), 4.31 (9%), 4.95 (3%) and 5.15 (5%); δ 4.95→5.15 (2%); δ 5.15→3.69 (3%), 4.49 (3%) and 5.01 (2%)]; m/z
(FAB) 570 (MNa+, 15%), 548 (MH+, 18) and 331 (C14H19O9+, 100).
The second-eluted material (0.051 g) was mainly a mixture of compounds 31 and 32, containing the latter material as the major component. After crystallisation from diethyl ether–hexanes, methyl
(1S,2R,3R)-2-nitro-5-oxo-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane-1-carboxylate32
(0.040 g, 18%) was obtained; mp 164–166 °C; [α]D
−26 (c 0.5, CH2Cl2)
(Found: C, 48.5; H, 5.3; N, 2.4); λmax
(EtOH)/nm 204 (ε 2600); νmax
(KBr)/cm−1 1750br (ester CO), 1720sh (ketone CO) and 1565 (NO2); δ
(300 MHz; CDCl3) 1.99, 2.02, 2.03 and 2.11 (each 3 H, s, 4 × MeCO2), 2.60 and 2.73 [each 1 H, dd (J 7 and 16) and dd (J 4 and 16), 4-Hax and 4-Heq], 2.70 (2 H, d, separation 8, 6-H2), 3.58 (1 H, q, separation 8, 1-H), 3.69 (1 H, ddd, J 2.5, 5 and 10, 5′-H), 3.76 (3 H, s, MeO2C), 4.13 and 4.23 [each 1 H, dd (J 2.5 and 12.5) and dd (J 5 and 12.5), 6′-H2], 4.58 (1 H, d, J 8, 1′-H), 4.69 (1H, ddd, J 4.5, 5.5 and 7, 3-H), 4.89 (1 H, dd, J 8 and 9.5, 2′-H), 5.05 (1 H, t, J 9.5 Hz, 4′-H), 5.16 (1 H, t, J 9.5, 3′-H) and 5.30 (1 H, dd, J 5.5 and 8, 2-H); δ
(600 MHz; CDCl3) 2.00, 2.025, 2.031 and 2.11 (each 3 H, s, 4 × MeCO2), 2.61 and 2.73 [each 1 H, dd, (J 7 and 16) and dd (J 4.5 and 16), 4-Hax and 4-Heq], 2.71 (2 H, d, separation 8, 6-H2), 3.58 (1 H, q, separation 8, 1-H), 3.69 (1 H, ddd, J 2.5, 5 and 10, 5′-H), 3.76 (3 H, S, MeO2C), 4.14 and 4.23 [each 1 H, dd (J 2.5 and 12.5) and dd (J 5 and 12.5), 6′-H2], 4.59 (1 H, d, J 8, 1′-H), 4.69 (1 H, ddd, J 4.5, 5.5 and 7, 3-H), 4.89 (1 H, dd, J 8 and 9.5, 2′-H), 5.06 (1 H, t, J 9.5, 4′-H), 5.17 (1 H, t, J 9.5, 3′-H) and 5.30 (1 H, dd, J 5.5 and 8, 2-H)
[NOE difference: δ 2.61→2.71 (1%), 2.73 (11%), 4.59 (1%), 4.69 (1%) and 5.30 (2%); δ 2.71→2.61 (5%), 3.58 (9%), 4.69 (1%) and 5.30 (3%); δ 2.73→2.61 (14%) and 4.69 (4%); δ 3.58→2.71 (3%), 4.69 (3%) and 5.30 (4%); δ 3.69→4.14 (2%), 4.23 (2%), 4.59 (7%), 5.06 (2%) and 5.17 (6%); δ 4.14→3.69 (4%) and 4.23 (8%); δ 4.23→3.69 (2%), 4.14 (8%) and 5.06 (3%); δ 4.59→3.69 (6%), 4.69 (6%) and 5.17 (5%); δ 4.69→2.73 (3%), 3.58 (4%), 4.59 (7%) and 5.30 (5%); δ 4.89→5.06 (5%) and 5.17 (4%); δ 5.06→3.69 (1%), 4.14 (1%), 4.23 (1%) and 4.89 (7%); δ 5.17→3.69 (4%), 4.59 (3%) and 4.89 (3%); δ 5.30→2.61 (1%), 2.71 (1%), 3.58 (3%) and 4.69 (3%)]; m/z
(FAB) 570 (MNa+, 32%), 548 (MH+, 13) and 331 (C14H19O9+, 100).
(b) The aforecited experiment was repeated and the hydrolysate was subjected to fractionation by HPLC [CH2Cl2–EtOAc (7 : 3) as eluent].
The first-eluted material (0.060 g, 27%), isolated as a crystalline solid, was identified as the ketone 30 by NMR spectroscopy.
The second-eluted material (0.039 g, 17%), also isolated as a crystalline solid, was considered to be the ketone 32 by NMR spectroscopy.
The third-eluted material (0.025 g, 11%) was methyl
(1S,2R,3S)-2-nitro-5-oxo-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane-1-carboxylate31; mp 160–162 °C; [α]D
+20 (c 0.5, CH2Cl2)
(Found: C, 48.1; H, 5.0; N, 2.5); λmax
(EtOH)/nm 204 (ε 3800); νmax
(KBr)/cm−1 1760 (ester CO), 1740 (ketone CO) and 1570 and 1545 (NO2); δ
(300 MHz; CDCl3) 1.98, 2.01, 2.03 and 2.11 (each 3 H, s, 4 × MeCO2), 2.39 and 2.83 [each 1 H, dd, (J 13 and 15.5) and ddd (J 2.5, 5.5 and 15.5), 6-Hax and 6-Heq], 2.62 and 3.00 [each 1 H, dd (J 3 and 16) and dt (J 16 and 3), 4-Hax and 4-Heq], 3.62–3.74 (2 H, m, 1- and 5′-H), 3.77 (3 H, s, MeO2C), 4.15 and 4.22 [each 1 H, dd (J 4.5 and 12.5) and dd (J 2.5 and 12.5), 6′-H2], 4.45 (1 H, d, J 8, 1′-H), 4.90 (1 H, dd, J 8 and 9.5, 2′-H), 4.92 (1 H, q, separation 3, 3-H), 5.02 (1 H, t, J 9.5, 4′-H), 5.11 (1 H, dd, J 3 and 11, 2-H) and 5.13 (1 H, t, J 9.5, 3′-H); m/z
(FAB) 570 (MNa+, 100%) and 331 (C14H19O9+, 81).
The fourth-eluted material, isolated as a foam, was mainly a 50 : 50 mixture of the ketones 31 and 33 [the ratio was estimated from the heights of the doublets (J 8 Hz) at δ 4.45 and 4.47, attributed to the 1′-Hs of the ketones 31 and 33]. It was resubjected to HPLC fractionation to give a 25 : 75 mixture of the ketones 31 and 33; δ (300 MHz; CDCl3) (for 33) 1.99, 2.00, 2.07 and 2.11 (each 3 H, s, 4 × MeCO2), 2.41 (1 H, dd, J 13 and 15, 6-Hax), 2.64 (1 H, dd, J 2.5 and 15, 4-Hax), 2.69–2.83 (2 H, m, 4- and 6-Heq), 3.63 (1 H, ddd, J 2.5, 5 and 10, 5′-H), 3.77 (3 H, s, MeO2C), 3.79 (1 H, ddd, J 5.5, 11.5 and 13, 1-H), 4.08 and 4.18 [each 1 H, dd (J 2.5 and 12.5) and dd (J 5 and 12.5), 6′-H2], 4.47 (1 H, d, J 8, 1′-H), 4.87 (1 H, dd, J 8 and 9.5, 2′-H), 5.00 (1 H, q, separation 3, 3-H), 5.01 (1 H, t, J 9.5, 4′-H), 5.10 (1 H, dd, J 2.5 and 11.5, 2-H) and 5.16 (1 H, t, J 9.5, 3′-H).
Ketone reduction studies
Methyl (1R,2S,3S,5S)-5-hydroxy-2-nitro-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane-1-carboxylate 36
Reduction of the ketone 30 gave the alcohol 36. Crystallisation of the material from diethyl ether–hexanes gave the title compound36 (0.037 g, 74%); mp 180–182 °C; [α]D −33 (c 0.5, CH2Cl2) (Found: C, 48.2; H, 5.4; N, 2.8. C22H31NO15 requires C, 48.1; H, 5.7; N, 2.5%), λmax (EtOH)/nm 205 (ε 4200); νmax (KBr)/cm−1 3500 (OH), 1755 and 1735 (ester CO) and 1560 (NO2); δ
(300 MHz; CDCl3) 1.52 and 2.30–2.39 [each 1 H, dt (J 11 and 13) and m, 6-Hax and 6-Heq], 1.58 and 2.52–2.61 [each 1 H, q (separation 12) and m, 4-Hax and 4-Heq], 1.98, 2.02, 2.07 and 2.09 (each 3 H, s, 4 × MeCO2), 3.04 (1 H, ddd, J 4.5, 11.5 and 13, 1-H), 3.63–3.69 (1 H, m, 5′-H), 3.70 (3 H, s, MeO2C), 3.76–3.88 (1 H, m, 5-H), 4.01 (1 H, ddd, J 4.5, 10 and 11.5, 3-H), 4.19–4.21 (2 H, m, 6′-H2), 4.44 (1 H, d, J 8, 1′-H), 4.63 (1 H, dd, J 10 and 11.5, 2-H), 4.94 (1 H, dd, J 8 and 9.5, 2′-H), 5.04 (1 H, t, J 9.5, 4′-H) and 5.14 (1 H, t, J 9.5, 3′-H); m/z
(FAB) 572 (MNa+, 63%), 550 (MH+, 33) and 331 (C14H19O9+, 100).
Methyl (1S,2R,3R,5R)-5-hydroxy-2-nitro-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane-1-carboxylate 41
Reduction of the ketone 32 (0.100 g, 0.18 mmol) gave the alcohol 41. Crystallisation of the material from diethyl ether–hexanes gave the title compound41 (0.073 g, 73%); mp 172–174 °C; [α]D −29 (c 0.25, CH2Cl2) (Found: C, 48.3; H, 5.9; N, 2.6. C22H31NO15 requires C, 48.1; H, 5.7; N, 2.5%), λmax (EtOH)/nm 204 (ε 4000); νmax (KBr)/cm−1 3460br (OH), 1760, 1740 and 1720 (ester CO) and 1560 (NO2); δ
(300 MHz; CDCl3) 1.49 (1 H, q, separation 11.5, 4-Hax), 1.56 (1 H, q, separation 11.5, 6-Hax), 1.99, 2.01, 2.04 and 2.10 (each 3 H, s, 4 × MeCO2), 2.31–2.43 (2 H, m, 4- and 6-Heq), 3.07 (3 H, 1 H, ddd, J 4, 11 and 13, 1-H), 3.63 (1 H, ddd, J 2.5, 4.5 and 10, 5′-H), 3.70 (3 H, s, MeO2C), 3.78–3.90 (1 H, m, 5-H), 4.10 (1 H, dd, J 2.5 and 12, 6′-H), 4.14–4.22 (2 H, m, 6′- and 3-H), 4.56 (1 H, d, J 8, 1′-H), 4.68 (1 H, dd, J 10 and 11, 2-H), 4.92 (1 H, dd, J 8 and 9.5, 2′-H), 5.05 (1 H, t, J 9.5, 4′-H) and 5.16 (1 H, t, J 9.5, 3′-H); m/z
(FAB) 572 (MNa+, 3%), 550 (MH+, 2), 331 (C14H19O9+, 44) and 169 (100).
Reductive acetylation of the nitro alcohol 36
Aq. methanol (10 vol% H2O, 1 cm3) was added to a stirred mixture of the nitro alcohol 36 (0.100 g, 0.18 mmol) and aluminium amalgam [prepared from Al foil (2.0 g) by Corey's procedure36] in methanol (100 cm3). After 4 h, Celite (5 g) was added and the mixture was filtered. Evaporation of the filtrate left a residue which was stirred with acetic anhydride (10 cm3) and pyridine (10 cm3) for 4 h. The mixture was diluted with dichloromethane and washed with water and dilute hydrochloric acid. Evaporation of the dried (MgSO4) organic phase left a foamy residue (0.114 g), which was subjected to column chromatography [hexanes–EtOAc (1 : 2→neat EtOAc) as eluent] to give two fractions.The first-eluted material (0.042, 37%), isolated as a crystalline solid, was methyl
(1R,2S,3S,5S)-5-acetoxy-2-acetoxyamino-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane-1-carboxylate38; mp 104–106 °C; [α]D
+13 (c 0.25, CH2Cl2)
(Found: C, 50.4; H, 5.9; N, 2.6. C26H37NO16 requires C, 50.4; H, 6.0; N, 2.3%), λmax
(EtOH)/nm 204 (ε 5100); νmax
(KBr)/cm−1 1750 (ester CO); δ
(300 MHz; CDCl3) 1.61 and 2.14–2.21 [each 1 H, q (separation 12.5) and m, 6-Hax and 6-Heq], 1.64 and 2.48–2.54 [each 1 H, q (separation 12) and m, 4-Hax and 4-Heq], 2.00, 2.02, 2.03, 2.05, 2.06 and 2.08 (each 3 H, s, 6 × MeCO2), 2.60 (1 H, ddd, J 4, 11 and 13, 1-H), 3.17 (1 H, dt, J 2 and 11, 2-H), 3.60–3.73 (2 H, m, 3- and 5′-H), 3.71 (3 H, s, MeO2C), 4.11 and 4.21 [each 1 H, dd (J 2.5 and 12.5) and dd (J 5.5 and 12.5), 6′-H2], 4.62 (1 H, d, J 8, 1′-H), 4.72 (1 H, tt, J 4 and 11.5, 5-H), 4.99 (1 H, dd, J 8 and 9.5, 2′-H), 5.03 (1 H, t, J 9.5, 4′-H), 5.19 (1 H, t, J 9.5, 3′-H) and 7.76 (1 H, d, J 2, NHOAc); m/z
(FAB) 642 (MNa+, 13%), 620 (MH+, 82) and 331 (C14H19O9+, 46 and 169 (100).
The second-eluted material (0.029 g, 26%), isolated as a crystalline solid, was methyl
(1R,2S,3S,5S)-5-acetoxy-2-acetylamino-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane-1-carboxylate39; mp 192–194 °C; [α]D
−17 (c 0.25, CH2Cl2)
(Found: C, 51.8; H, 5.9; N, 2.6. C26H37NO15 requires C, 51.7; H, 6.2; N, 2.3%), λmax
(EtOH)/nm 205 (ε 4700); νmax
(KBr)/cm−1 1760 (ester CO) and 1660br (amide CO); δ
(300 MHz; CDCl3) 1.54 and 2.18–2.27 [each 1 H, q (separation 12) and m, 6-Hax and 6-Heq], 1.58 and 2.39–2.46 [each 1 H, q (separation 12) and m, 4-Hax and 4-Heq], 1.94, 1.99, 2.02, 2.03, 2.04 and 2.09 (each 3 H, s, 5 × MeCO2 and MeCON), 3.03 (1 H, ddd, J 3.5, 11 and 13, 1-H), 3.47 (1 H, dt, J 7.5 and 10.5, 2-H), 3.67 (3 H, s, MeO2C), 3.67–3.71 (1 H, m, 5′-H), 4.10 and 4.22 [each 1 H, d, (J 2.5 and 12.5) and dd (J 5 and 12.5), 6′-H2], 4.12–4.21 (1 H, m, 3-H), 4.60 (1 H, d, J 8, 1′-H), 4.79 (1 H, tt, J 4 and 11.5, 5-H), 4.94 (1 H, dd, J 8 and 9.5, 2′-H), 5.02 (1 H, t, J 9.5, 4′-H), 5.15 (1 H, t, J 9.5, 3′-H) and 5.58 (1 H, d, J 7.5, NHAc); m/z
(FAB) 626 (MNa+, 46%), 604 (MH+, 46), 331 (C14H19O9+, 79) and 169 (100).
Nitro ester reductions and acetylation studies
(1R,2S,3S,5S)-5-Acetoxy-1-acetoxymethyl-2-acetylamino-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane 35
Compound 36 gave rise to the title compound35 (0.042 g, 37%) as a crystalline solid; mp 210–212 °C; [α]D −16 (c 0.75, CH2Cl2) (Found: C, 52.2; H, 6.3; N, 2.4. C27H39NO15 requires C, 52.5; H, 6.4; N, 2.3%), λmax (EtOH)/nm 208 (ε 3900); νmax (KBr)/cm−1 3320br (NH), 1750 (ester CO) and 1660 (amide CO); δ
(300 MHz; CDCl3) 1.35 (1 H, q, separation 12, 6-Hax), 1.58 and 2.40–2.43 [each 1 H, q (separation 12) and m, 4-Hax and 4-Heq], 1.991, 1.993, 2.02, 2.03, 2.056, 2.061 and 2.09 (each 3 H, s, 6 × MeCO2 and MeCON), 3.37 (1 H, br q, separation 10, 2-H), 3.68 (1 H, ddd, J 2.5, 5 and 10, 5′-H), 3.88 (1 H, dt, J 4.5 and 11, 3-H), 4.02 and 4.08 [each 1 H, dd, (J 3 and 11) and dd (J 5 and 11), 1-CH2OAc], 4.11 and 4.23 [each 1 H, dd, (J 2.5 and 12.5) and dd (J 5 and 12.5), 6′-H2], 4.61 (1 H, d, J 8, 1′-H), 4.77 (1 H, tt, J 4 and 11, 5-H), 4.94 (1 H, dd, J 8 and 9.5, 2′-H), 5.03 (1 H, t, J 9.5, 4′-H), 5.16 (1 H, t, J 9.5, 3′-H) and 5.42 (1 H, d, J 8.5, NH)
(1-H and 6-Heq were located at ca. δ 2.10 in a COSY 45° experiment); m/z
(FAB) 640 (MNa+, 9%), 618 (MH+, 39) and 331 (C14H19O9+, 100).
(1S,2R,3R,5R)-5-Acetoxy-1-acetoxymethyl-2-acetylamino-3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)cyclohexane 40
The nitro ester 41 gave rise to the title compound40 (0.039 g, 35%) as a crystalline solid; mp 218–220 °C; [α]D +5 (c 0.5, CH2Cl2) (Found: C, 52.8; H, 6.7; N, 2.3. C27H39NO15 requires C, 52.5; H, 6.4; N, 2.3%), λmax (EtOH)/nm 206 (ε 5000); νmax (KBr)/cm−1 3400br (NH), 1740 (ester CO) and 1670 (amide CO); δ
(300 MHz; CDCl3) 1.38 (1 H, q, separation 12, 6-Hax), 1.43 and 2.28–2.35 [each 1 H, q (separation 11) and m, 4-Hax and 4-Heq], 1.96, 1.99, 2.02, 2.03, 2.05 and 2.10 (3, 3, 3, 3, 6 and 3 H, each s, 6 × MeCO2 and MeCON), 3.37 (1 H, dt, J 8 and 10, 2-H), 3.67 (1 H, ddd, J 2, 5 and 9.5, 5′-H), 3.74 (1 H, dt, J 4 and 10.5, 3-H), 4.04 and 4.12 [each 1 H, dd (J 6 and 11) and dd (J 2.5 and 11), 1-CH2OAc], 4.08 and 4.35 [each 1 H, dd (J 2 and 12.5) and dd (J 5 and 12.5), 6′-H2], 4.51 (1 H, d, J 8, 1′-H), 4.74 (1 H, tt, J 4 and 11, 5-H), 4.88 (1 H, dd, J 8 and 9.5, 2′-H), 5.04 (1 H, t, J 9.5, 4′-H), 5.17 (1 H, t, J 9.5, 3′-H) and 5.63 (1 H, d, J 7.5, NH)
(1-H and 6-Heq were located at δ 2.06 in a COSY 45° experiment); m/z
(FAB) 656 (MK+, 79%), 618 (MH+, 24) and 331 (C14H19O9+, 100).
Computational methods
The conformational searches of 43 and 44 were obtained by Monte Carlo searches in MacroModel version 5.542 on a Silicon Graphics IRIS workstations using the MM2 force field. The searches were performed with 2000 random structures generated per rotatable bond followed by energy minimization. The coupling constants (Hz) of the cyclohexanone-ring protons was calculated based on the two lowest energy conformations using the modified Karplus equation.43Acknowledgements
We thank Dr. C. M. Raynor for carrying out the preparative HPLC, Mr. C. Evans for measuring the NMR spectra, Mr. K. Walking for recording the IR and UV spectra, Mr. R. Perkins for the mass spectral determinations and Dr. R. Perry for the elemental analyses. We are also grateful to Dr. J. A. Parkinson (Edinburgh University Ultra high Field NMR Service) for measuring the 600 MHz NMR spectra and for performing the NOE difference studies on compound 30 and 32.References
- R. C. Gupta, P. A. Harland and R. J. Stoodley, Tetrahedron, 1984, 40, 4657 CrossRef CAS.
- R. C. Gupta, C. M. Raynor, R. J. Stoodley, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1988, 1773 RSC.
- R. C. Gupta, D. S. Larsen, R. J. Stoodley, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1989, 739 RSC.
- D. S. Larsen and R. J. Stoodley, J. Chem. Soc., Perkin Trans. 1, 1989, 1841 RSC.
- D. S. Larsena and R. J. Stoodley, J. Chem. Soc., Perkin Trans. 1, 1990, 1339 RSC.
- B. Beagley, D. S. Larsen, R. G. Pritchard and R. J. Stoodley, J. Chem. Soc., Perkin Trans. 1, 1990, 3113 RSC.
- W. D. Edwrads, R. C. Gupta, C. M. Raynor and R. J. Stoodley, J. Chem. Soc., Perkin Trans. 1, 1991, 1913 RSC.
- D. S. Larsen and R. J. Stoodley, Tetrahedron, 1990, 46, 4711 CrossRef CAS.
- I. H. Aspinall, P. M. Cowley, G. Mitchell and R. J. Stoodley, J. Chem. Soc., Chem. Commun., 1993, 1179 RSC.
- I. H. Aspinall, P. M. Cowley, R. J. Stoodley and G. Mitchell, Tetrahedron Lett., 1994, 35, 3397 CrossRef CAS.
- P. M. Cowley, R. J. Stoodley and G. Mitchell, Tetrahedron Lett., 1994, 35, 7853 CrossRef CAS.
- R. F. Lowe and R. J. Stoodley, Tetrahedron Lett., 1994, 35, 6351 CrossRef CAS.
- S. Kotha and R. J. Stoodley, Bioorg. Med. Chem., 2002, 10, 621 CrossRef CAS.
- L. M. S. Bourghli and R. J. Stoodley, Bioorg. Med. Chem., 2004, 12, 2863 CrossRef CAS.
- M. Helliwell, I. M. Philips, R. G. Pritchard and R. J. Stoodley, Tetrahedron Lett., 1999, 40, 8651 CrossRef CAS.
- S. Pornpakakul, R. G. Pritchard and R. J. Stoodley, Tetrahedron Lett., 2000, 41, 2691 CrossRef CAS.
- J. H. Musser, Annu. Rep. Med. Chem., 1992, 27, 301 Search PubMed.
- Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry, ed. H. Ogura, A. Hasegawa and T. Suami, Kodansha, Tokyo and VCH, Weinheim, 1992 Search PubMed.
- Carbohydrate-based Drug Discovery, ed. C.-H. Wong, Wiley-VCH, Weinheim, 2003 Search PubMed.
- T. Suami, Pure Appl. Chem., 1987, 59, 1509 CAS; T. Suami and S. Ogawa, Adv. Carbohydr. Chem. Biochem., 1990, 48, 21 CAS; T. Suami, Top. Curr. Chem., 1990, 154, 257 CAS; T. Suami, ref. 18, p. 136.
- Dictionary of Antibiotics and Related Substances, ed. B. W. Bycroft, Chapman and Hall, London and New York, 1988, p. 722 Search PubMed.
- S. Ogawa, Y. Shibata, N. Chida and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 494 CAS.
- D. S. Larsen, R. J. Lins, R. J. Stoodley and N. S. Trotter, J. Chem. Soc., Perkin Trans. 1, 2001, 2204 RSC.
- D. S. Larsen, R. J. Lins, R. J. Stoodley and N. S. Trotter, Org. Biomol. Chem., 2004, 2, 1934 RSC.
- D. S. Larsen, N. S. Trotter and R. J. Stoodley, Tetrahedron Lett., 1993, 34, 8151 CrossRef CAS.
- J. G. Buchanan and R. H. Wightman, Topics in Antibiotic Chemistry, vol. 6, ed. P. G. Sammes, Horwood, Chichester, 1982, p. 291 Search PubMed; J. F. Kennedy and C. A. White, Bioactive Carbohydrate; in Chemistry, Biochemistry and Biology, Horwood, Chichester, 1983, ch. 12 Search PubMed.
- S. Ogawa, T. Takai and A. Isaka, Carbohydr. Res., 1989, 191, 154 CrossRef CAS.
- T. E. Stevens and W. D. Emmons, J. Am. Chem. Soc., 1958, 80, 338 CrossRef CAS.
- S. Danishefsky, M. P. Prisbylla and S. Hiner, J. Am. Chem. Soc., 1978, 100, 2918 CrossRef CAS.
- J. E. McMurry, J. H. Musser, I. Fleming, J. Fortunak and C. Nubling, Org. Synth., 1988, Coll. Vol. VI, 799.
- J. E. McMurry and J. H. Musser, Org. Synth., 1977, 56, 65 CAS.
- C.-g. Shin, M. Yamaura, E. Inui, Y. Ishida and J. Yoshimura, Bull. Chem. Soc. Jpn., 1978, 51, 2618 CAS.
- E. J. Corey and H. Estreicher, J. Am. Chem. Soc., 1978, 100, 6294 CrossRef CAS.
- Preliminary communication R. J. Stoodley and W.-H. Yuen, Chem. Commun., 1997, 1371 Search PubMed.
- R. J. Abraham and H. J. Bernstein, Can. J. Chem., 1961, 39, 216 CAS.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1964, 86, 1639 CrossRef CAS.
- E. J. Corey, N. H. Andersen, R. M. Carlson, J. Paust, E. Vedejs, I. Vlattas and R. E. K. Winter, J. Am. Chem. Soc., 1968, 90, 3245 CrossRef CAS; P. Camps and D. Muñoz-Torrero, Tetrahedron Lett., 1994, 35, 3187 CrossRef CAS.
- K. Healey and I. C. Calder, Aust. J. Chem., 1979, 32, 1307 CAS.
- E. Juaristi and G. Cuevas, Tetrahedron, 1992, 48, 5019 CrossRef CAS.
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1990, 29, 823 CrossRef; H. Serderowitz and W. C. Still, J. Org. Chem., 1997, 62, 1427 CrossRef.
- SHELX97 (includes SHELXS97, SHELXL97 and CIFTAB), Programs for Crystal Structure Analysis (Release 97-2), G. M. Sheldrick, Institüt für Anorganische Chemie der Universität, Tammamstrasse 4, D-3400 Göttingen, Germany, 1998.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caulfield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440 CrossRef CAS.
- L. M. Kackmann and S. Sternhill, Applications of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry, Pergamon, Oxford, 2nd edn., 1969, p. 334 Search PubMed.
Footnotes |
| † The work presented in this article was carried out at UMIST. |
| ‡ The stereodescriptor refers to the carbon atom of the diene bearing the 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloxy unit. |
| § Carba-pyranoses—pyranoses in which the ring oxygen atom is replaced by a methylene group—are examples of carbasugars (also called pseudo-sugars); for review, see ref. 20. |
| ¶ Although hexopyranoses with an amino or substituted amino group at position 4 feature in numerous compounds of biological relevance (ref. 26), only two such carba-hexopyranose representatives appear to have been synthesised (ref. 27). |
| || Hydroxylamines are the usual products of aluminium amalgam reductions of nitro compounds when the reactions are conducted in moist diethyl ether (ref. 38). |
| ** It is worth nothing that for compound 41 in the crystal state, the O(5′)–C(1′)–O(3)–C(3) torsion angle was 92.3°, the H(1′)⋯H(3) interatomic distance was 2.257 Å and the C(1′)–O(3)–C(3) bond angle was 113.3°. |
| †† CCDC reference numbers 245218 (15) and 245217 (41). See http://www.rsc.org/suppdata/ob/b4/b410556g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2005 |