Chemo-enzymatic synthesis of 4-methylumbelliferyl β-(1→4)-D-xylooligosides: new substrates for β-D-xylanase assays
Elena V.
Eneyskaya
a,
Dina R.
Ivanen
a,
Konstantin A.
Shabalin
a,
Anna A.
Kulminskaya
*a,
Leon V.
Backinowsky
b,
Harry
Brumer III
c and
Kirill N.
Neustroev†
a
aPetersburg Nuclear Physics Institute, Russian Academy of Science, Molecular and Radiation Biology Division, Gatchina, 188300, Russia. E-mail: kulm@omrb.pnpi.spb.ru; Fax: +7 81371 32 303; Tel: +7 81371 32 014
bN.D.Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky prosp. 47, Moscow, 119991, Russia
cDepartment of Biotechnology, Royal Institute of Technology (KTH), AlbaNova University Centre, S-106 91, Stockholm, Sweden
First published on 26th November 2004
Abstract
Transglycosylation catalyzed by a β-D-xylosidase from Aspergillus sp. was used to synthesize a set of 4-methylumbelliferyl (MU) β-1→4-D-xylooligosides having the common structure [β-D-Xyl-(1→4)]2–5-β-D-Xyl-MU. MU xylobioside synthesized chemically by the condensation of protected MU β-D-xylopyranoside with ethyl 2,3,4-tri-O-acetyl-1-thio-β-D-xylopyranoside was used as a substrate for transglycosylation with the β-D-xylosidase from Aspergillus sp. to produce higher MU xylooligosides. The structures of oligosaccharides obtained were established by 1H and 13C NMR spectroscopy and electrospray tandem mass spectrometry. MU β-D-xylooligosides synthesized were tested as fluorogenic substrates for the GH-10 family β-D-xylanase from Aspergillus orizae and the GH-11 family β-D-xylanase I from Trichoderma reesei. Both xylanases released the aglycone from MU xylobioside and the corresponding trioside. With substrates having d.p. 4 and 5, the enzymes manifested endolytic activities, splitting off MU, MUX, and MUX2 primarily.
Introduction
β-D-Xylanase (1,4-β-xylanase, 1,4-β-D-xylan xylanohydrolase, E.C.3.2.1.8) is a glycoside hydrolase, which hydrolyses β-1→4-xylosidic linkages in the xylan component of plant cell walls. These enzymes have been isolated mainly from microorganisms and are of a great interest because of numerous biotechnological applications.1,2 Xylanases are currently used widely in pulp and paper manufacture, alcohol, brewery, and food industries, and the need for these enzymes continuously grows.3,4 Extensive use of β-D-xylanases stimulates both the search for new sources of the enzymes, and detailed studies of their enzymatic properties. Assay methods for β-D-xylanases based on tracking the liberation of reducing ends during xylan hydrolysis have some restrictions. Xylans obtained from natural sources are polydisperse and contain arabinose and glucuronic acid side-chains.5 Furthermore, these methods have insufficient sensitivity for detailed mechanistic analysis and use less desirable stopped assay conditions.6,7 The use of polymeric substrates, such as xylans and glucans, precludes studies of the transglycosylating activities of β-D-xylanases due to difficulties in distinguishing transglycosylation and hydrolysis products.8 For these reasons, detailed kinetic and thermodynamic analyses and investigations into the action patterns of xylanases require more convenient substrates which enable high sensitivity assays. For such purposes, a range of xylooligosides bearing chromogenic or fluorescent groups at the reducing end of a chain can be employed; analogous substrates are widely used for detailed enzymatic investigations of various glucanases. For example, the modes of action of α-amylases from different origins have been studied with p-nitrophenyl (PNP) maltooligosides with degree of polymerization (d.p.) 2–7.9 A set of 4-methylumbelliferyl (MU) β-1,3(4)-glucosides were successfully applied for subsite mapping of the active site of β-1,3(4)-glucanases based on the Hiromi approach.10 Kinetic properties of an endo-β-galactosidase were studied with the aid of PNP β-acetyllactosamine-repeating oligosides.11 Fluorogenic substrates for glycosidases have certain advantages over chromogenic analogues: they are both more sensitive and can be used for samples in highly colored media.12–14 For example, MU xylobioside enables detection of β-xylanase activity on agar plates.15 Since synthetic organic methods are difficult for the production of MU xylosides with a d.p. more than 2,15 an enzymatic approach can serve as an alternative and effective way to manufacture such compounds. In our previous related work, we described the enzymatic synthesis of a set of PNP β-D-xylooligosides with d.p. up to 7 produced with the aid of a β-D-xylosidase from Aspergilus sp.16 The chromophore-bearing substrates were produced in good yields and absolute stereoselectivity; only β-1→4-D-xylosidic linkages were formed.Here, we used the same β-D-xylosidase for the enzymatic synthesis of new β-D-xylanase substrates, namely, 4-methylumbelliferyl β-D-xylooligosides (MUXn) with d.p. 3–6, and tested two β-D-xylanases with the fluorescent xylosides produced.
Results and discussion
Synthesis of MU β-D-xylooligosides and the structure of the transxylosylation products
To produce MU β-D-xylooligosides (MUXn), we took advantage of the transglycosylating ability of Aspergillus sp. β-D-xylosidase, which had previously been used for the synthesis of PNP β-D-xylooligosides of various lengths.16 MU xyloside (MUX) is a substrate for the β-D-xylosidase, which splits the glycoside with a catalytic efficiency comparable with that for the well-known PNPX (the values for kcat/Km are 220 and 480 s mM−1 for MUX and PNPX, respectively). However, it appeared impossible to use MUX in the synthesis of MU β-D-xylooligosides due to low solubility of the substrate in aqueous solutions (<5 mM), which significantly limits the use of this compound in transglycosylation reactions. Transglycosylation reactions catalyzed by glycosidases result from involvement of a glycosyl acceptor molecule other than water, and are thus favored by high concentrations of the alternative acceptor. Previously, the optimum concentration of PNPX in the synthesis of PNPXn by the Aspergillus β-D-xylosidase was found to be 110 mM.16 The addition of up to 30% (v/v) DMF or DMSO to increase the solubility of MUX led to inactivation of the enzyme. Therefore, to produce a set of extended MU xylooligosaccharides, we chose the more water-soluble 4-methylumbelliferyl β-D-xylopyranosyl-(1→4)-β-D-xylopyranoside (MUX2) as a transglycosylation acceptor (Scheme 1).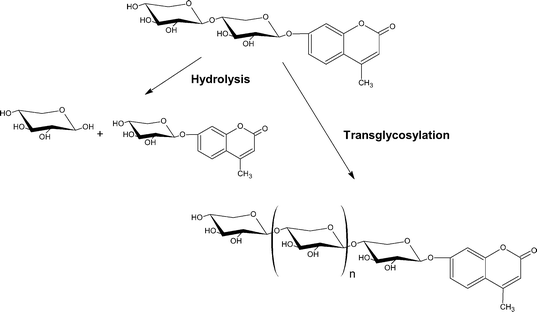 | ||
| Scheme 1 | ||
MU xylobioside (MUX2) was synthesized by the reaction of the known ethyl 2,3,4-tri-O-acetyl-1-thio-β-D-xylopyranoside 1 (glycosyl donor) as described17 with a suitably protected acceptor and subsequent deacylation (Scheme 2). The NMR data strongly support the β-configuration of the (1→4)-xylosidic bond formed. Selective, dibutyltin oxide-mediated chloroacetylation of commercially available MUX (2→3), subsequent benzoylation (→4), and dechloroacetylation with thiourea in methanol resulted in the target xyloside acceptor (5) with the free OH group at C-4. Its coupling with the donor, ethyl 1-thioxyloside 1, (methyl fluorosulfonate as a promoter18), afforded the peracylated MU xylobioside 6 in a good yield. This was deacylated into the target 4-methylumbelliferyl β-D-xylobioside (MUX2, 7). Compared with the previously reported protocol of the synthesis of MUX2 from a mixture of xylose, xylobiose, and xylotriose,15 our procedure avoids the need of isolation of a rather moisture-sensitive per-O-acetylxylobiosyl bromide by crystallization.
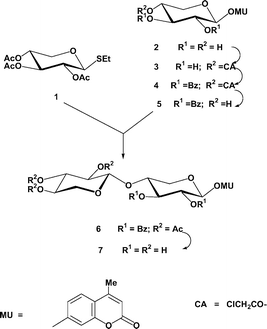 | ||
| Scheme 2 | ||
The transglycosylation reaction conditions which gave rise to the maximum yields of MUX3–6 were selected using analytical HPLC. Under optimum reaction conditions (pH 5.5–7.0; 37 °C; acceptor concentration, 140–160 mM), MU xylooligosides of d.p. 3–6 were obtained in 21, 12, 6, and 3%, respectively, at 50–60% conversion of MUX2. The structures of individual transglycosylation products isolated by HPLC were established by MS and NMR.
Assignment of the signals in the 1H and 13C NMR spectra followed the same methodology as described in ref. 16 with reference to the spectra of PNPXn. Thus, a comparative analysis of 1H and 13C NMR spectroscopic data for MUX3 and MUX4 and those for PNPX2–416,19 showed that the transglycosylation catalyzed by Aspergilus β-xylosidase occurs with absolute stereospecificity giving only β-1→4-linkages. Furthermore, the stoichiometry of xylose residues in all isolated transglycosylation products, MUX3–6, was unambiguously confirmed by CID MS/MS analysis (Fig. 120). Trace amounts of longer MU xylooligosides produced by the enzyme were also detected by TLC (not shown). The ability to produce higher oligosaccharides with high stereospecificity is rather unique; oligosaccharide chains produced by enzymatic transglycosylation often contain two or three monosaccharide residues with different linkage configurations.21,22 Only one successful synthesis of a highly stereoregular product, viz., PNP β-D-galactopyranosyl-(1→4)-β-D-galactopyranoside, by the β-galactosidase from Bacillus circulans has been reported.23 Other attempts to prepare MU oligosides using various exo- or endo-glycosidases have rarely been effective. This can be explained by poor acceptor properties of MU glycosides in transglycosylation reactions due to their low solubility and poor binding affinity. Therefore, chemical methods are traditionally used for the synthesis of MU oligosaccharides with d.p. more than 2.17,24 To our knowledge, only one enzyme, namely, the laminarinase from Oerskovia sp., has been reported, which was able to produce a range of chromo/fluorogenic oligosides (in this case, MU β-(1→3)-D-glucooligosides).25 In the present work, we expanded this repertoire by employing an exo-glycoside hydrolase, viz., β-D-xylosidase from Aspergillus sp., to synthesize MU 1→4-β-xylooligosides in good yields.
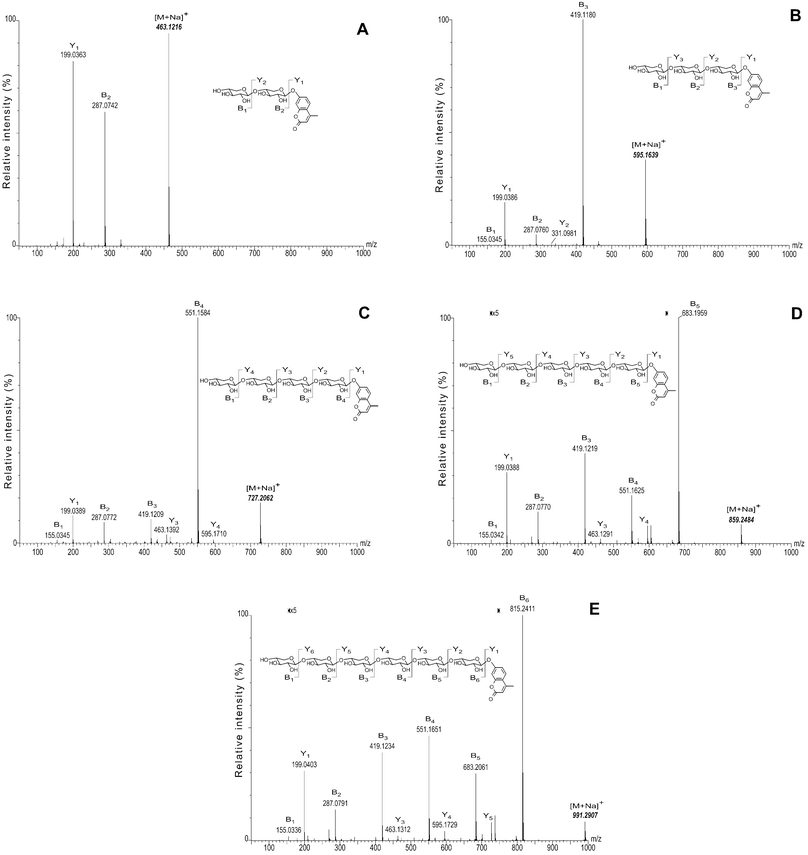 | ||
| Fig. 1 CID MS/MS spectra of monosodiated adducts of MU xylooligosaccharides. A. MUX2; B. MUX3; C. MUX4; D. MUX5; E. MUX6. Calculated m/z values of the monosodiated MU xylooligosaccharide adducts (bold italics) were used as reference values to recalibrate all spectra. | ||
| Product | Yield (%) |
|---|---|
| MUX3 | 21.0 |
| MUX4 | 12.0 |
| MUX5 | 6.0 |
| MUX6 | 3.0 |
| Substrate | K m/mM | |
|---|---|---|
| XynA (GH10) | XynT (GH11) | |
| MUX2 | 0.1 | 0.15 |
| MUX3 | 0.054 | 0.083 |
| PNPX2 | 0.25 | 0.3 |
| PNPX3 | 0.162 | 0.189 |
MUX4 and MUX5 also serve as substrates for both β-D-xylanases, which produce lower MU β-xylooligosides along with trace quantities of free MU upon hydrolysis of these compounds. Reproducible values for the yields of MU xylooligosides and free MU for all the specimens tested are given in Table 3 and Table 4. The xylanase subsite structure can be characterized with the aid of MU xylooligosides since one can easily separate the hydrolysis products by HPLC.
Thus, the synthesised MU β-D-xylooligosides (d.p. 2–6) are promising tools for studies of subsite architecture of the active centre of β-xylanases on the basis of procedures developed for other glycosidases with artificial substrates.9,10,28 Since β-D-xylanase splits off the aglycon moiety from MUX2 and MUX3, these compounds are ideal substrates for detailed kinetic studies of the enzyme due to a higher sensitivity of detection when compared with the methods of detecting reducing sugars released during the β-D-xylan hydrolysis or the use of alternative substrates.6,7
Experimental
Substrates and enzymes
β-D-Xylan from birchwood and MUX (2) were purchased from Sigma Chemical Co., St. Louis, USA; D-xylopyranosyl-(1→4)-β-D-xylopyranose was prepared by enzymatic digestion of xylan by β-D-xylanase from Trichoderma reesei followed by chromatographic purification on a Biogel P2 column (25 × 200 mm) equilibrated in water. p-Nitrophenyl β-D-xylobioside (PNPX2) and p-nitrophenyl β-D-xylotrioside (PNPX3) were synthesized enzymatically by the β-D-xylosidase from Aspergillus sp. as described earlier.16The β-D-xylosidase from Aspergillus sp. was purified according to ref. 16. β-Xylanase I from Trichoderma reesei (XynT) of the glycoside hydrolase family 11, was kindly donated by Dr A. Miasnikov, Danisco Global Innovation, Finland, and separated from contaminating β-xylosidase activity as reported previously.16 The specific activity of the purified protein was about 300 ± 5 U mg−1. The Aspergillus orizae β-xylanase (XynA) was isolated from culture liquid according to the procedures described by Kitamoto et al.28 resulting in 250 ± 5 U mg−1 activity. Both xylanase preparations contained less than 0.01% of extrinsic β-D-xylosidase activity.
Analytical methods
Protein concentrations were measured following the Lowry procedure with BSA as a standard.29All 1H and 13C NMR spectra were recorded with an AMX-500 Bruker spectrometer (1H at 500.13 MHz, 13C at 125.13 MHz) at ambient temperature for solutions in different solvents. Me4Si was used as an external standard and calibration was performed using the signal of the residual protons of the solvents. Chemical shifts (δ in ppm) are given relative to those for CDCl3 (δH 7.25), 30% solution of CD3CN in D2O (δH 2.21), 30% solution of (CD3)2CO in D2O (δH 2.19); J-values are given in Hz.
The oligosaccharides obtained upon enzymatic hydrolysis and enzymatic synthesis were analyzed qualitatively by TLC on Kieselgel 60 plates (Merck) with a butan-1-ol–acetic acid–water (3 : 1 : 1, v/v) solvent system.
Mass spectrometric analysis was performed with a Q-Tof™ 2 mass spectrometer fitted with a nanoflow ion source (Waters Corporation, Micromass MS Technologies, Manchester, U.K.). Calibration of the TOF analyser (single-reflectron mode, resolution >10000 FWHM) was obtained over the m/z range 50–1000 using a solution of NaI (1.5 g L−1) in 1 : 1 propan-2-ol–water. A scan time of 2.5 s with an interscan delay of 0.1 s was used in all MS modes. Solutions of MU xylooligosides (typical concentration 10–20 µM in 1 : 1 MeOH–water containing 0.5 mM NaCl) were infused into the ion source at 200 nL min−1 (syringe pump). The cone voltage was set at 45 V and the electrospray capillary voltage was varied (2.2 kV to 3.0 kV) to obtain less than 500 counts s−1 for the base peak ion signal. These conditions favored the production of [M + Na]+ adducts; the formation of the [M + 2Na]2+ adduct was only significant for Xyl6-MU. For accurate mass determinations in the TOF MS mode, centroid spectra were generated from continuum spectra using the (M + Na)+ adduct of raffinose (α-Galp-(1→6)-α-Glcp-(1↔2)-β-Fruf) as an internal standard (calc'd. mass 527.1588 u). The raffinose (raffinose pentahydrate, R-0250, Sigma-Aldrich, minimum purity 99%) concentration in each sample was adjusted empirically to give a peak intensity similar to that of the MU xylooligoside analyte. For MS/MS experiments, the collision energy was varied (30–55 V), depending on the resilience of the (M + Na)+ ion, to achieve an optimal balance between parent and fragment ion intensities. Data were collected until an acceptable signal-to-noise ratio was achieved after the combination of individual spectra (typical 15–30 spectra). Centroid MS/MS spectra were produced from continuum spectra using the calculated m/z of the monosodiated adduct of the appropriate MU xylooligosaccharide as a reference mass.
Enzyme assay
The β-xylosidase activity towards PNPX was determined at 37 °C in 50 mM sodium acetate buffer, pH 4.5.16 One unit of β-D-xylosidase was defined as the amount of the enzyme releasing 1 µmol of p-nitrophenol per min in the above conditions. β-Xylanase activity was evaluated by the Somogyi–Nelson method30 using xylan as a substrate. One unit of the β-xylanase activity was defined as the amount of the enzyme capable of releasing 1 µmol of reducing sugar from xylan with concentration of 5 mg mL−1 at 37 °C in 20 mM sodium acetate buffer, pH 4.0, per min.Modes of action of XynT and XynA
The action patterns of both xylanases were investigated in 20 mM sodium acetate buffer, pH 4.0, at 37 °C, using substrate (MUX2–5) concentrations in the range from 0.8 to 1.2 mM, the course of the reactions being followed by TLC. To initiate the reaction, about 0.01 U of the xylanase preparation was added to the reaction mixture. The reaction was terminated by freezing and lyophilization. Analysis of MU-containing hydrolysis products was performed on a Waters Hypersil ODS column (200 × 4.8 mm) using a linear gradient (0–90%) of MeCN in water with spectrophotometric detection at 254 nm.Determination of kinetic parameters of the hydrolysis of MU-β-D-xylooligosides by XynT and XynA
The activities of the XynT and XynA in the hydrolysis of MUX2 and MUX3 were evaluated spectrofluorometrically after incubating the reaction mixture in 50 mM sodium acetate buffer, pH 4.0, by measuring the released MU in accordance with ref. 25. The Michaelis constants were determined for each substrate from the Michaelis-Menten equation by non-linear regression analysis.31 The rates were determined for at least eight different substrate concentrations ranging from about 0.1 × the Km value determined to 4–6 × Km.4-Methylumbelliferyl β-D-xylopyranosyl-(1→4)-β-D-xylopyranoside (MUX2)
MU xylobioside was synthesized by the condensation of suitably protected MUX derivative 5, having 4-OH unsubstituted, with ethyl 2,3,4-tri-O-acetyl-1-thio-β-D-xylopyranoside (1) using methyl fluorosulfonate as a thiophilic activator (Scheme 2).Ethyl 2,3,4-tri-O-acetyl-1-thio-β-D-xylopyranoside 1
Compound 1 was prepared by boron trifluoride-catalyzed condensation of β-D-xylose peracetate with thiourea as described.174-Methylumbelliferyl 4-O-chloroacetyl-β-D-xylopyranoside 3
Compound 2 (10 mmol; 3.24 g) and dibutyltin oxide (15 mmol; 3.72 g) were suspended in PriOH (200 ml) and heated under reflux for 3 h, and the solvent was evaporated. The residue was dissolved in dry CH2Cl2 (100 ml), cooled to 0 °C, and a solution of ClCH2COCl (11 mmol; 876 µl) in CH2Cl2 (10 ml) was added dropwise. The mixture was stirred for 1 h at 0 °C and concentrated. Chromatography (toluene : acetone, 9 : 1→1 : 1) of the residue on a silica gel afforded 3 (2.55 g, 68%); δH(500 MHz, CD3CN): 7.63 (1 H, d, J5,6 8.8, H-5 MU), 6.99 (1 H, dd, J6,8 2.28, H-6 MU), 6.97 (1 H, d, H-8 MU), 6.14 (1 H, m, J3,4-Me 1.1, H-3 MU), 5.06 (1 H, d, J1,2 7.24, H-1), 4.85 (1H, ddd, J3,4 9.14, J4,5a 5.35, J4,5b 9.55, H-4), 4.23 (2H, s, ClH2CO), 4.03 (1 H, dd, J5a,5b 11.6, H-5a), 3.91 (0.7 H, s, OH), 3.77 (0.7 H, s, OH), 3.67 (1 H, dd, J2,3 8.96, H-3), 3.55 (1 H, dd, H-2), 3.52 (1 H, dd, H-5b), 2.38 (3 H, d, 4-Me MU).4-Methylumbelliferyl 2,3-di-O-benzoyl-4-O-chloroacetyl-β-D-xylopyranoside 4
Compound 3 (10 mmol; 3.82 g) was dissolved in dry CH2Cl2 (100 ml) containing pyridine (0.1 mol; 8 ml) and cooled to 0 °C. Benzoyl chloride (0.03 mol; 3.51 ml) was added dropwise and stirring was continued overnight at room temperature. The solution was washed with cold dil HCl, aq NaHCO3, H2O, dried, and concentrated. Chromatography (toluene : acetone, 20 : 1→4 : 1) of the residue on a column with silica gel gave the peracylated xyloside 4 (5.84 g, 93%); δH (CDCl3): 8.06 (2 H, dd, J 8.06, m-OBz), 8.05 (2 H, dd, J 7.9, m-OBz), 7.50 (1 H, d, J5,6 8.78, H-5 MU), 7.46 (2 H, d, J3,4 7.6, o-OBz), 7.43 (2 H, d, J3,4 7.8, o-OBz), 7.24 (1 H, dd, p-OBz), 7.16 (1 H, dd, p-OBz), 7.01 (1 H, d, J6,8 2.45, H-8 MU), 6.95 (1 H, dd, H-6 MU), 6.17 (1 H, m, J3,4-Me 1.44, H-3 MU), 5.64 (1 H, dd, J2,3 5.83, J3,4 5.83, H-3), 5.60 (1 H, d, J1,2 4.03, H-1), 5.55 (1 H, dd, H-2), 5.23 (1 H, ddd, J4,5a 3.53, J4,5b 5.54, H-4), 4.40 (1 H, dd, J5a,5b 12.8, H-5a), 4.11 (1 H, d, JCH2-a, CH2-b 14.83, ClCH2CO-a), 4.06 (1 H, d, ClCH2CO-b), 3.86 (1 H, dd, H-5b), 2.38 (3 H, 4-Me-MU).4-Methylumbelliferyl 2,3-di-O-benzoyl-β-D-xylopyranoside 5
A mixture of peracylated glycoside 4 (10 mmol; 4.66 g) and thiourea (50 mmol; 3.8 g) in MeOH (100 ml) was boiled under reflux for 6 h. The mixture was concentrated, the residue was dissolved in CH2Cl2 and subjected to a column chromatography (9 : 1 → 2 : 1 PhMe–acetone) to give dibenzoate 5 (4.47 g; 87%); δH (CDCl3): 8.04 (2 H, d, J2,3 8.06, m-OBz), 7.99 (2 H, d, J2,3 7.91, m-OBz), 7.57 (2 H, dd, J3,4 7.48, o-OBz), 7.54 (2 H, dd, J3,4 7.48, o-OBz), 7.48 (1 H, d, J5,6 8.78, H-5 MU), 7.43 (1 H, dd, p-OBz), 7.40 (1 H, dd, p-OBz), 7.00 (1 H, d, J6,8 2.45, H-8 MU), 6.94 (1 H, dd, H-6-MU), 6.16 (1 H, s, H-3 MU), 5.60 (1 H, dd, J1,2 5.25, J2,3 7.12, H-2), 5.49 (1 H, d, H-1), 5.40 (1 H, dd, J3,4 6.9, H-3), 4.33 (1 H, dd, J4,5a 4.13, J5a,5b 12.23, H-5a), 4.09 (1 H, ddd, J4,5b 6.4, H-4), 3.73 (1 H, dd, H-5b), 3.21 (1H, s, OH-4), 2.37 (3 H, s, 4-Me MU).4-Methylumbelliferyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-benzoyl-β-D-xylopyranoside 6
A solution of ethyl 1-thioxyloside 1 (2 mmol; 640 mg) and the dibenzoate 5 (1 mmol; 514 mg) in dry CH2Cl2 (25 ml) was stirred with freshly activated MS 4 Å (1.5 g) for 1 h, methyl fluorosulfonate (10 mmol; 1.14 g) was added, and stirring was continued for 20 h at room temperature. Triethylamine (5 ml) was added, the mixture was filtered through a layer of Celite into a mixture of ice–H2O and CH2Cl2. The organic layer was washed with 10% aq HCl, water, aq NaHCO3, water, dried, and concentrated. Column chromatography (20 : 1 → 9 : 1 PhMe–acetone) of the residue gave the protected bioside 6 (613 mg; 79%); δH (CDCl3): 8.06–7.97 (4 H, m, o-OBz), 7.58–7.51 (4 H, m, m-OBz), 7.47 (1 H, d, J5,6 8.78, H-5-MU), 7.45–7.38 (2 H, m p-OBz), 6.97 (1 H, d, J6,8 2.45, H-8 MU), 6.91 (1 H, dd, H-6 MU), 6.15 (1 H, m, J3,4-Me 1.44, H-3 MU), 5.66 (1 H, dd, J2,3 6.96, J3,4 6.76, H-3), 5.50 (1 H, dd, J1,2 5.15, H-2), 5.45 (1 H, d, H-1), 5.05 (1 H, dd, J2′,3′ 7.98, J3′,4′ 7.74, H-3′), 4.86 (1 H, dd, H1′,2′ 6.19, H-2′), 4.68 (1 H, ddd, J4′,5a′ 4.75, J4′,5b′ 7.87, H-4′), 4.66 (1 H, d, H-1′), 4.22 (1 H, dd, J4,5a 4.17, J5a,5b 12.38, H-5a), 4.07 (1 H, ddd, J4,5b 6.65, H-4), 3.79 (1H, dd, J5a′,5b′ 12.05, H-5a′), 3.71 (1 H, dd, H-5b), 3.20 (1 H, dd, H-5b′), 2.36 (3 H, d, 4-Me MU), 2.04 (3 H, s, OAc), 1.98 (6H, s, OAc).4-Methylumbelliferyl β-D-xylopyranosyl-(1→4)-β-D-xylopyranoside (MUX2, 7)
A solution of 6 (500 mg) in dry MeOH (10 ml) was treated with methanolic NaOMe (100 µl) according to ref. 32 and purified by reverse-phase chromatography on an INERTSIL PREP-ODS column (20 × 250 mm) using a linear gradient (0–90%) of MeCN in water to afford the bioside 7. 1H NMR (D2O) δH 5.12 (1 H, d, J1,2 7.52, H-1), 4.51 (1 H, d, J1,2 7.84, H-1′), 4.19 (1 H, dd, J5a,5b 11.72, J4,5a 5.27, H-5a), 4.01 (1 H, dd, J5a,5b 11.60, J4,5a 5.40, H-5a′), 3.88 (1H, ddd, J4,6b 10.1, J3,4 8.8, H-4), 3.74 (1 H, t, J2,3 9.4, H-3), 3.66 (1H, ddd, J4,6b 10.3, J3,4 9.2, H-4′), 3.64 (1H, dd, H-2), 3.62 (1 H, dd, H-5b), 3.48 (1 H, t, J2,3 9.2, H-3′), 3.35 (1 H,dd, H-5b′), 3.32 (1 H, dd, H-2′); 13C NMR (D2O) δC 103.98 (C-1′), 102.15 (C-1), 78.25 (C-4′), 77.68 (C-3′), 75.55 (C-3), 74.82 (C-2′), 74.57 (C-2), 71.23 (C-4), 67.30 (C-5′), 65.12 (C-5); ESI+ MS [M + Na]+m/z 463.1216 calcd. for C20H24NaO11, observed: 463.1209.Synthesis of MU β-D-xylooligosides with d.p. 3–6
A solution of MUX2 (80 mM) and appropriate amount of the β-D-xylosidase corresponding to 8 U were, incubated in 6 mL of 30 mM sodium phosphate buffer, pH 6.5, for 180 min at 37 °C. Following termination of the reaction by freezing and freeze-drying, individual components of a mixture of transglycosylation products were initially separated on an INERTSIL PREP-ODS column (20.0 × 250 mm) and then purified on a TSK-NH2-60 column (4.6 × 250 mm, Pharmacia) using isocratic elution with 80% MeCN. Finally, the appropriate fractions were freeze-dried and the products obtained were used in further investigations.Acknowledgements
The present work was supported by the grant of the Program for Basic Research in Molecular and Cell Biology from the Presidium of Russian Academy of Sciences (RAS); by the grant no. 03-04-48756 from the Russian Foundation for Basic Research; by the Agreement Rostech-PNPI 585-400-1/2003.References
- H. J. Gilbert and G. P. Hazlewood, J. Gen. Microbiol., 1993, 139, 187–194 CAS.
- A. P. Garg, J. C. Roberts and A. J. McCarthy, Enzyme Microb. Technol., 1998, 22, 594–598 CrossRef CAS.
- A. Gessesse and G. Mamo, Enzyme Microb. Technol., 1999, 25, 68–72 CrossRef CAS.
- Q. K. Beg, M. Kapoor, L. Mahajan and G. S. Hoondal, Appl. Microbiol. Biotechnol., 2001, 56, 326–338 Search PubMed.
- L. Weinstein and P. Albersheim, Plant Physiol., 1979, 63, 425–432 CAS.
- H. Taguchi, T. Hamasaki, T. Akamatsu and H. Okada, Biosci. Biotech. Biochem., 1996, 60, 983–985 CAS.
- Y. Zhao, C. J. Chany II, P. F. G. Sims and M. L. Sinnott, J. Biotechnol., 1997, 57, 181–190 CrossRef CAS.
- B. O. Petersen, M. Krah, J. Ø. Duus and K. K. Thomsen, Eur. J. Biochem., 2000, 267, 361–369 CrossRef CAS.
- L. Kandra, G. Gyémánt, J. Remenyik, G. Hovánszki and A. Lipták, FEBS Lett., 2002, 518, 79–82 CrossRef CAS.
- C. Malet and A. Planas, Biochemistry, 1997, 36, 13838–13848 CrossRef CAS.
- T. Murata, T. Hattori, S. Amarume, A. Koichi and T. Usui, Eur. J. Biochem., 2003, 270, 3709–3719 CrossRef CAS.
- Y. Honda, S. Tanimori, M. Kirihata, S. Kaneko, K. Tokuyasu, M. Hashimoto, T. Watanabe and T. Fukamizo, FEBS Lett., 2000, 476, 194–197 CrossRef CAS.
- V. M. Chernoglazov, A. N. Jafarova and A. A. Klyosov, Anal. Biochem., 1989, 179, 186–189 CrossRef CAS.
- S. Armand, S. Drouillard, M. Schülein, B. Henrissat and H. Driguez, J. Biol. Chem., 1997, 272, 2709–2713 CrossRef CAS.
- S. Kaneko, M. Kitaoka, A. Kuno and K. Hayashi, Biosci. Biotech. Biochem., 2000, 64, 741–745 CAS.
- E. V. Eneyskaya, H. Brumer III, L. V. Backinowsky, D. R. Ivanen, A. A. Kulminskaya, K. A. Shabalin and K. N. Neustroev, Carbohydr. Res., 2003, 338, 313–325 CrossRef CAS.
- F. M. Ibatullin, K. A. Shabalin, J. V. Jänis and A. G. Shavva, Tetrahedron Lett., 2003, 44, 7961–7964 CrossRef CAS.
- R. W. Alder, Chem. Ind. (London), 1973, 983–986 CAS.
- K. Takeo, Y. Ohguchi, R. Hasegawa and S. Kitamura, Carbohydr. Res., 1995, 277, 231–244 CrossRef CAS.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CAS.
- K. G. I. Nilsson, Carbohydr. Res., 1987, 167, 95–103 CrossRef CAS.
- E. V. Eneyskaya, A. M. Golubev, A. M. Kachurin, A. N. Savel'ev and K. N. Neustroev, Carbohydr. Res., 1998, 305, 83–91 CrossRef.
- X. Zeng, R. Yoshimo, T. Murata, K. Ajisaka and T. Usui, Carbohydr. Res., 2000, 325, 120–131 CrossRef CAS.
- H. De Boeck, K. L. Matta, M. Claeyssens, N. Sharon and F. G. Loontiens, Eur. J. Biochem., 1983, 131, 453–460 CAS.
- R. Borriss, M. Krah, H. Brumer III, M. A. Kerzhner, L. A. Elyakova, D. R. Ivanen, E. V. Eneyskaya, S. M. Shishlyannikov, K. A. Shabalin and K. N. Neustroev, Carbohydr. Res., 2003, 338, 1455–1467 CrossRef CAS.
- B. Henrissat, Biochem. J., 1991, 280, 309–316 CAS.
- P. Ntarima, W. Nerinckx, K. Klarskov, B. Devreese, M. K. Bhat, J. Van Beeumen and M. Claeyssens, Biochem. J., 2000, 347, 865–873 CrossRef CAS.
- N. Kitamoto, S. Yoshino, M. Ito, T. Kimura, K. Ohmiya and N. Tsukagoshi, Appl. Microbiol. Biotechnol., 1998, 50, 558–563 CrossRef CAS.
- O. H. Lowry, N. J. Rosenbrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- M. Somogyi, J. Biol. Chem., 1952, 195, 19–23 CAS.
- R. T. Leatherbarrow, Enzifitter., Elsevier Biosoft, Cambridge, 1987 Search PubMed.
- A. Thompson and M. Y. Wolfrom, Methods Carbohydr. Chem., 1963, 2, 215 Search PubMed.
Footnote |
| † In memoriam of Dr Kirill N. Neustroev. |
| This journal is © The Royal Society of Chemistry 2005 |