New procedure for the preparation of highly sterically hindered alkenes using a hypervalent iodine reagent†
Matthijs K. J.
ter Wiel
,
Javier
Vicario
,
Stephen G.
Davey
,
Auke
Meetsma
and
Ben L.
Feringa
*
Department of Organic and Molecular Inorganic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: Feringa@chem.rug.nl; Fax: + 31 50 363 4296; Tel: +31 50 363 4278
First published on 10th November 2004
Abstract
New methodology is described to construct the olefinic bond in overcrowded alkenes using a hypervalent iodine reagent, and applied in the synthesis of molecular motors.
The design of nanomechanical devices via a bottom-up approach, elegantly illustrated in the recent examples of molecular machines,1 demands for new synthetic methodology to construct such complex molecular systems. Accordingly, we have focused on the development of molecular switches and motors based on sterically overcrowded alkenes.2 Besides the challenge to synthesize these highly overcrowded alkenes, control of steric hindrance at the central olefinic bond is a crucial parameter for the motor function. A key feature of the light-driven molecular motors is that they are capable of repeated unidirectional rotation around their central double bond functioning as the axis of rotation. This rotation takes place in four steps as two photoisomerization steps are each followed by a thermal helix inversion step.3 However, in the so-called first generation of this type of molecules the rate determining thermal isomerization steps are slow at room temperature. The replacement of the two six-membered rings of the original molecular motor by two five-membered rings in 1 led to a significant increase of the speed of rotation due to a decrease in steric hindrance.4 In this paper we present new methodology to prepare sterically overcrowded alkenes 2, with distinct upper and lower halves and which contain in the upper half a five-membered ring bearing the stereocentre needed to control the direction of rotation (Fig. 1).
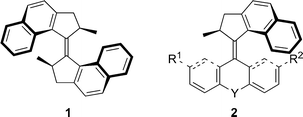 | ||
| Fig. 1 Molecular motors 1 and 2 containing either one or two five-membered rings with a stereocentre. | ||
Where most olefination reactions fail to provide such severely hindered molecules we rely on the diazo–thioketone coupling, more commonly referred to as the Barton–Kellogg5,6 reaction, to prepare sterically overcrowded alkenes. The reaction was introduced in 1920 by Staudinger7 and intensively used in the group of Schönberg.8 The power of this method is that steric strain is gradually introduced into the molecule in a two fold extrusion process.9 In the first step of the reaction, a diazo compound and a thioketone react in a 1,3-dipolar cycloaddition in which a thiadiazoline is formed. In most cases this thiadiazoline is thermally unstable and rapidly eliminates nitrogen to form an episulfide. Extrusion of the remaining sulfur atom is then performed by reaction with copper powder or triphenylphosphine to yield the desired alkene. It was established that the elimination of nitrogen in the thiadiazoline was followed by a conrotatory ring closure to form the corresponding episulfide.6
Apart from the effectiveness in olefin formation due to the gradual build-up of steric hindrance in the molecule, a major advantage of the reaction is the selective coupling of two non-identical halves which is not easily accomplished with, for example, the McMurry reaction. Of major importance for the development of functionalized molecular motors and switches is the tolerance of the olefination reaction towards a variety of functional groups.10
However, the diazo–thioketone coupling method for the preparation of target structures 2, starting from hydrazone 4 and thioketones 7, failed to provide any of the desired episulfides 8 (Scheme 1). We found that the approach we commonly use for similar compounds employing silver(I) oxide as the oxidant was not successful.11,12 This prompted an investigation of other oxidizing agents. Based on earlier investigations, the use of hypervalent iodine compounds13 such as 5 seemed to be particularly promising.14 Only recently, similar hypervalent iodine reagents have attracted conderable interest in a number of synthetic applications.15 Indeed, reaction at low temperature (−50 °C) in DMF resulted in formation of the diazo compound 6, as was deduced from the slightly red color of the solution. Upon addition of thioxanthone 7a (Y = O), the evolution of nitrogen gas was observed and after workup episulfide 8a (Y = O) was obtained in good yield. To extend the scope of the reaction and to test the applicability of reagent 5, various thioketones 7a–g were used in the coupling reaction. Moderate to excellent yields were obtained for the various episulfides, as depicted in Table 1.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH
CH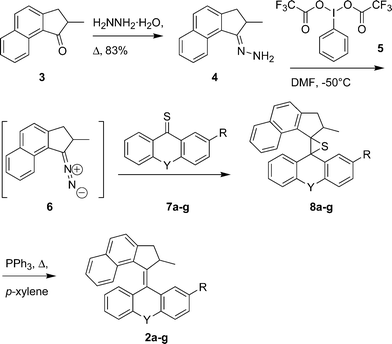 | ||
| Scheme 1 The modified diazo–thioketone coupling for the synthesis of overcrowded alkenes. | ||
Desulfurization of the episulfides 8 was performed preferably by reaction with excess triphenylphosphine in refluxing p-xylene. Although the desulfurization of episulfide 8a could be performed with copper powder, removal of the side products could only be achieved with considerable effort. Much more convenient was the use of triphenylphosphine, which cleanly converted the episulfides 8a–g to the corresponding alkenes. In case of alkenes 2a, 2b, 2c and 2d it was impossible to separate the excess triphenylphosphine by column chromatography. In order to facilitate the purification of these alkenes, this crude reaction mixture was stirred overnight in the presence of excess methyl iodide in p-xylene. Despite the additional reaction with methyl iodide, the reaction sequence was efficient and the conversion of the episulfides to the corresponding alkenes proceeded in all cases in yields exceeding 80%. Surprisingly, the episulfides 8a–8d were obtained as single isomers. To determine the actual structure of the isomer obtained in the stereoselective diazo–thioketone coupling, crystals suitable for X-ray crystallographic analysis were grown by slow diffusion of acetonitrile into a solution of 8a in chloroform (Fig. 1 and Fig. 2).16
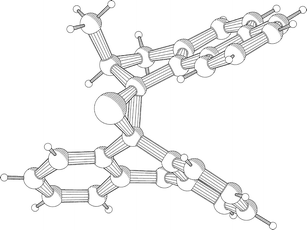 | ||
| Fig. 2 PLUTO drawing of episulfide 8a. | ||
It is evident from the structure shown that the methyl substituent and the naphthalene moiety are oriented in the same direction in order to diminish the steric strain in the molecule. Although the syn-orientation of the sulfur atom with respect to the methyl substituent might be surprising, this is a direct consequence of the reaction mechanism shown in Scheme 2.
In the first step of the reaction sequence (Scheme 2), the diazo compound 6 reacts in a 1,3-dipolar cycloaddition with thioketone 7a. Although there could be a stereochemical preference in this reaction it is anticipated that two isomeric thiadiazolines 9 are formed. These thiadiazolines are thermally unstable and even at low temperatures nitrogen evolution is observed, to form a thiocarbonyl ylide 10. Since the methyl substituent blocks ring closure on one side of the molecule of the essentially flat thiocarbonyl ylide moiety, a single isomer of episulfide 8a is formed.
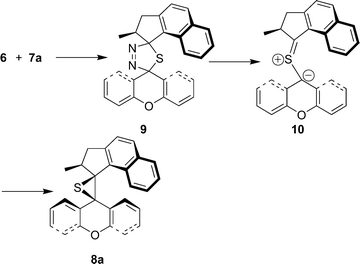 | ||
| Scheme 2 Reaction of diazo compound 6 and thioketone 7a to form episulfide 8avia the intermediacy of thiadiazoline 9 and thiocarbonyl ylide 10. | ||
Although further optimization of the reaction conditions might be achieved, these results represent a major advance in the synthesis of overcrowded alkenes and molecular motors. There are two important advantages of this procedure over the McMurry reaction. First of all, this procedure allows selective coupling of different upper and lower halves without formation of homocoupled products. This is not only more efficient from a synthetic point of view, but also facilitates purification of the desired products. Secondly, many substituted thioketones are available, which allows selective functionalization of the desired motor molecules with substituents that would not have been tolerated by the McMurry reaction. In conclusion, this new method not only allows the synthesis of new molecular motors, but is also highly efficient in the construction of severely hindered non-symmetric alkenes.
Financial support from the Netherlands Foundation for Scientific Research (NWO-CW, MKJtW), the Basque Country Government (JV), and the EU Project on Molecular Level Devices and Machines HPRN-CT-2000-00029 (SGD) is gratefully acknowledged.
References and notes
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS.
- B. L. Feringa, N. Koumura, R. A. van Delden and M. K. J. ter Wiel, Appl. Phys. A., 2002, 75, 301 CrossRef CAS.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS.
- M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 15076 CrossRef CAS.
- (a) D. H. R. Barton and B. J. Willis, J. Chem. Soc., Chem. Commun., 1970, 1225 RSC; (b) D. H. R. Barton, E. H. Smith and B. J. Willis, J. Chem. Soc., Chem. Commun., 1970, 1226 RSC; (c) D. H. R. Barton and B. J. Willis, J. Chem. Soc., Perkin Trans. 1, 1972, 305 RSC.
- (a) R. M. Kellogg, Tetrahedron, 1976, 32, 2165 CrossRef CAS; (b) J. Buter, S. Wassenaar and R. M. Kellogg, J. Org. Chem., 1972, 37, 4045–4060 CrossRef.
- H. Staudinger and J. Siegwart, Helv. Chim. Acta, 1920, 3, 833 CrossRef CAS.
- For example: (a) A. Schönberg, D. Cernik and W. Urban, Ber. Dtsch. Chem. Gess. B, 1931, 64, 2577 Search PubMed; (b) A. Schönberg and E. Frese, Chem. Ber., 1968, 101, 701.
- For a review concerning organic extrusion reactions, see: F. S. Guziec and L. J. Sanfilippo, Tetrahedron, 1988, 44, 6241 Search PubMed.
- For example, the strong reducing conditions in the McMurry reaction are not compatible with a nitro-group.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037 CrossRef CAS.
- Presumably due to the low stability of 6 and the low rate of oxidation by Ag2O, this approach is not effective..
- Encyclopedia of Reagents for Organic Synthesis, ed. L. A. Paquette, Wiley, New York, 1995, vol. 6, p. 3982. Search PubMed.
- (a) P. A. S. Smith and E. M. Bruckmann, J. Org. Chem., 1974, 39, 1047 CrossRef CAS; (b) L. Lapatsanis, G. Milias and S. Paraskewas, Synthesis, 1985, 513 CrossRef CAS.
- (a) M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222 CrossRef CAS; (b) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192 CrossRef CAS.
- Crystal data for 8a, C27H20OS, Mr = 392.52, orthorhombic, P212121, a = 6.4273(3), b = 15.6636(7), c = 18.9920(9) Å, V = 1912.01(16) Å3, Z = 4, Dc = 1.363 g cm−3, λ (Mo–Kα) = 0.71073 Å, µ = 1.86 cm−1, F(000) = 824, T = 100 K, GoF = 0.956, wR(F2) = 0.773 for 4735 reflections with F0 ≥ 4δ(F0) criterion of observability. CCDC reference number 251068. See http://www.rsc.org/suppdata/ob/b4/b414959a/ for crystallographic data in .cif format.
- The compounds showed spectroscopic and analytical data in accordance with the structures. See the electronic supplementary information† for typical procedures.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures for compounds 4, 7f, 8a–g, and 2a–g; crystal data for 8a. See http://www.rsc.org/suppdata/ob/b4/b414959a/ |
| This journal is © The Royal Society of Chemistry 2005 |