Bioorthogonal organic chemistry in living cells: novel strategies for labeling biomolecules
Paul F.
van Swieten
a,
Michiel A.
Leeuwenburgh
a,
Benedikt M.
Kessler
b and
Herman S.
Overkleeft
*a
aLeiden Institute of Chemistry, Leiden University, PO Box 9502, 2300, RA Leiden, The Netherlands. E-mail: h.s.overkleeft@chem.leidenuniv.nl; Fax: (+31) 71 5274307; Tel: (+31) 71 5274342
bHenry Wellcome Building of Genomic Medicine, Oxford University, Roosevelt Drive, Oxford, OX3 7BN, UK
First published on 29th November 2004
Abstract
The chemical labeling of biomolecules continues to be an important tool for the study of their function and cellular fate. Attention is increasingly focused on labeling of biomolecules in living cells, since cell lysis introduces many artefacts. In addition, with the advances in biocompatible synthetic organic chemistry, a whole new field of opportunity has opened up, affording high diversity in the nature of the label as well as a choice of ligation reactions. In recent years, several different two-step labeling strategies have emerged. These rely on the introduction of a bioorthogonal attachment site into a biomolecule, then ligation of a reporter molecule to this site using bioorthogonal organic chemistry. This Perspective focuses on these techniques, their implications and future directions.
 Paul F. van Swieten Paul F. van Swieten | Paul van Swieten (1977) is currently in the last year of his PhD research under the guidance of Herman Overkleeft at Leiden University. His thesis elaborates on the development of chemical proteomics techniques for the study of selected protein families, with a special focus on proteolytic activities. |
 Michiel A. Leeuwenburgh Michiel A. Leeuwenburgh | Michiel Leeuwenburgh (1973) obtained his PhD in 2000 under the guidance of Professor Dr J. H. van Boom at Leiden University, the subject of his thesis being organometallic transformations on unsaturated carbohydrate derivatives. Since then, he has shifted attention towards the application of organic synthesis to elucidate biological mechanisms. Currently working as a post-doc in the group of Professor Overkleeft, he is working on the development of novel functional proteomics strategies of proteases. |
 Benedikt M. Kessler Benedikt M. Kessler | Benedikt Kessler graduated from the Swiss Federal Institute of Technology ETH in Zurich, Switzerland in biochemistry in 1992. He received his PhD in immunology at the Ludwig Institute for Cancer Research at the University of Lausanne in 1998. He then joined the laboratory of Hidde L. Ploegh at the Pathology Department at Harvard Medical School in Boston USA to study antigen processing and presentation. After three years, he established a research platform in proteomics at Harvard Medical School. Currently he is University Lecturer at the Department of Clinical Medicine, Oxford University, UK. |
 Herman S. Overkleeft Herman S. Overkleeft | Herman Overkleeft (1969) performed his PhD research in bioorganic chemistry at the University of Amsterdam, where he obtained his PhD degree in 1997 under the guidance of Professor Dr U. K. Pandit. After two years of post-doctoral research at Leiden University, with Professor Dr J. H. van Boom, he moved to the Harvard Medical School (Boston, USA) for two years, where he worked with Professor Dr H. L. Ploegh on chemical biology approaches to study immunological processes. Since July 2001 he has held the chair of bioorganic synthesis at Leiden University. His current research interests are in organic chemistry, glycobiology and chemical biology. |
Introduction
Research in the contemporary life sciences is increasingly directed towards the detailed analysis of biological processes at the molecular level. In order to understand and intervene in processes at the DNA/RNA level, protein–protein interactions and enzymatic action – systems which all involve large biopolymers – a detailed insight into the structural and chemical mode of action is often a prerequisite. At the same time, scientists endeavour to generate such detailed molecular information from macroscopic systems, that is, in whole cells and organisms.The ability to selectively tag a biomolecule in living cells or organisms allows researchers to a) monitor its involvement in a given biological event which progresses in its natural environment and/or b) to, after the fact, retrieve and analyse in detail the tagged biomolecule or its metabolites. The biomolecular toolbox provides a set of useful tags which can be attached to proteins of interest through standard genetic engineering techniques. Widely used tags include short oligopeptide stretches that can be selectively isolated through immunoprecipitation (such as the FLAG tag) or affinity chromatography (for instance the nickel-binding His tag). Of particular interest is the family of fluorescent proteins, the most prominent member of which is green fluorescent protein (GFP).1
The availability of fluorescent proteins emitting at different wavelengths has resulted in the development of fluorescence resonance energy transfer (FRET)2 strategies for the visualisation of the spatial arrangement and interaction dynamics of proteins. In the last decade, a wealth of information on many biological systems has emerged from studies based on the use of fluorescent fusion proteins. An obvious extension of this technique would be to develop systems that express tagged non-proteinogenic entities, or those that report on enzymatic activities. The realisation that this can not be achieved by standard molecular biology techniques alone has led to a shift of attention towards organic chemistry-based approaches. In principle, the use of synthetic tags is not limited to a particular class of biomolecules and, moreover, the variety in the nature of the tag is only limited by the imagination of the researcher. In addition, proper design of the synthetic tag enables not only monitoring of the presence or absence of the tagged biomolecule in a specific system (as with GFP approaches), but will also provide information about enzyme activity or the cellular fate of a metabolite of any given class of biomolecules.
Most biochemical studies are executed in cell lysates, since these are generally easier to exploit experimentally as compared to whole cells or animal models. In line with this, chemical tagging of a biomolecule in a cell lysate obviates the need for the tag to be cell permeable. Reporter entities, such as biotin, fluorescent labels and radiolabels, may therefore be incorporated into the molecular probes liberally, as long as they do not interfere with the process of interest. However, because of the intrinsic disadvantages of working with cell lysates (dilution factor, pH change, loss of subcellular compartmentalisation), live cell systems are preferred where possible.
Tagging biomolecules in living cells does, however, impose some important restrictions on the design of the chemical tags. For instance, the cell-impermeability or the reduced profiling potency3 of many biotinylated constructs essentially precludes the direct incorporation of biotin in a molecular probe. To a lesser extent, the same also holds true for many radiolabels and fluorescent tags. It is therefore not surprising that in recent years a variety of two-step labeling strategies have been developed.4 These strategies are based on a) introduction of a bioorthogonal5 attachment site to a biomolecule in a living cell system, and b) addition of the reporter moiety, through a chemoselective ligation, to the attachment site at a desirable stage in the experiment. The scope and limitations of the recent developments in this exciting area of chemical biology and some future directions are described in this Perspective.
Three different approaches for introducing the bioorthogonal entity into the biomolecule(s) of choice and attaining the chemoselective ligation with a tagging or crosslinking agent can be discerned (see Fig. 1):
A) Treatment of tissue with a chemically modified metabolic precursor will lead to incorporation of a bioorthogonal attachment site in the target biomolecules. Subsequently, the attachment site is addressed in a chemoselective ligation. Examples of this strategy include the introduction of bioorthogonally functionalised amino acids into the cell's protein synthesis machinery6 and the introduction of other modified precursors of biomolecules into post-translational modifications and oligosaccharide clusters.7
B) Creation of mutant cell lines that express fusion proteins with attachment functionalities that specifically react with a complementary synthetic probe of choice.8
C) Tagging of enzymes with small functionalised active site directed probes in living cells, then addressing the functionality in a bioorthogonal chemical ligation step.9
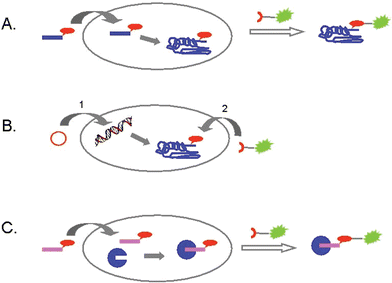 | ||
| Fig. 1 Three different strategies for the labeling of biomolecules in living cells: A) introduction of a chemically modified metabolic precursor, then addressing the modification; B) creation of fusion proteins with a bioorthogonal attachment site, followed by ligation in vivo; C) in vivo tagging of enzymes with activity-based probes, then ligation with a reporter molecule. | ||
A Use of chemically modified metabolic precursors
Methionine tRNA-synthetase is able to accept unnatural methionine isosteres as substrates.10 Using this feature, Van Hest et al. succeeded in introducing amino acids with unsaturated side chains (for instance, 2-amino-5-hexynoic acid) into proteins.11 In a joint effort of the Tirrell and Bertozzi groups, this approach was used for the decoration of E. coli proteins with azide residues.12 Addition of azidohomoalanine (1, see Fig. 2) to a methionine-depleted bacterial culture resulted in the incorporation of azide moieties instead of methionine residues. After cell lysis, the azide functionalities were addressed chemoselectively by a modified Staudinger reaction (these days known as the Staudinger–Bertozzi ligation, see Scheme 1), thereby attaching the antigenic FLAG-peptide to the modified proteins.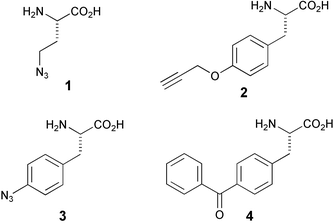 | ||
| Fig. 2 Some bioorthogonally functionalised amino acids that have been built into proteins. | ||
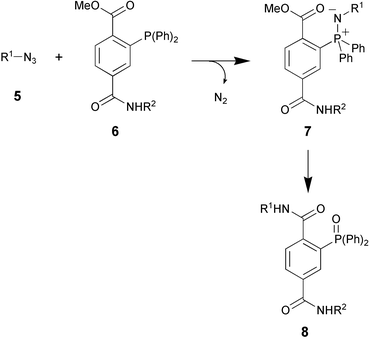 | ||
| Scheme 1 The Staudinger–Bertozzi ligation. | ||
Using the same technique for the construction of modified proteins, Tirrell and coworkers reported that azide-decorated cell surface proteins can be labeled using a copper catalyzed Huisgen-type [3 + 2] cycloaddition (see Scheme 2).13 This reaction, that can be executed under aqueous conditions, and that features two reactive components that are largely inert in physiological environments, was developed independently by the groups of Meldal14 and Sharpless.15 A biotin propargyl amide was used to address the cell surface azides by use of this cycloaddition reaction. After staining with fluorescent avidin, the biotin-modified cells were easily distinguished from the non-modified cells.
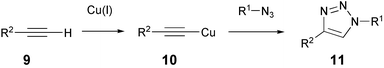 | ||
| Scheme 2 The copper-catalysed Huisgen-type cycloaddition. | ||
Schultz and co-workers have embarked on the development of genetically engineered microorganisms that encode for non-natural amino acids.16 The end goal of this research program is to generate altered microorganisms that endogenously synthesise a 21st amino acid with any desirable functionality. This amino acid residue is recognised by a novel tRNA synthetase that subsequently transfers it to a tRNA equipped with a unique anticodon, allowing site-specific incorporation in selected proteins. The current state of the art dictates that bioorthogonally functionalised amino acids for incorporation still need to be added to the growth medium of altered organisms.17 For example, His-tagged proteins incorporating either O-propargyltyrosine (2, Fig. 2) or p-azidophenylalanine (3),18 depending on the composition of the growth medium, were expressed in yeast. After affinity purification of the target proteins, both modifications were addressed by their fluorescent counterparts (that is, azide- or alkyne-functionalised dansyl or fluorescein derivatives) in a Huisgen-type cycloaddition. This allowed for in-gel visualisation of the modified target proteins. The authors are currently investigating an in vivo labeling strategy based on the same principles.
A major breakthrough in protein engineering is the development of techniques that allow the incorporation, at a predesigned site, of non-natural amino acids that are highly reactive under photolytic conditions.19 After exposure to UV light, the resulting intermediates, normally being carbenes or nitrenes, will react with surrounding functionalities, including amide linkages. Depending on its position, crosslinking may occur with the surrounding proteins, thereby providing information on protein–protein interactions. Until recently, however, the site-specific incorporation of desired amino acids could only be achieved in in vitro translation systems and with low efficiency. Further, the application of the modified proteins obtained through in vitro translation met with considerable technical difficulties (for instance when aiming for the study, through photocrosslinking, of membrane-associated proteins). The in vivo translation system developed by the group of Schultz opens the way to many exciting applications, in which protein–protein interactions can be monitored in the proper environment of the living cell. A relevant example of this strategy entails the following set of experiments.20
After incorporation of azidophenylalanine at the binding surface of glutathione-S-transferase (GST) in the appropriate E. coli mutant, purification of the protein through its fused His-tag and irradiation with UV-light, crosslinking to another GST molecule could be effected. This finding indicates that GST occurs in dimeric form in vitro. In addition, the crosslinking proved to be possible in living cells (see Fig. 3),21 using a different photocrosslinking amino acid, p-benzoyl-L-phenylalanine (4). When incorporated at the same site of GST as azidophenylalanine, similar results were obtained as in the in vitro crosslinking experiment. These results clearly demonstrate the value of the in vivo translation technique for probing protein–protein interactions in living cells.
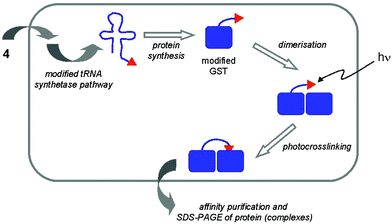 | ||
| Fig. 3 Introduction of a photocrosslinker amino acid (4) specifically into GST of E. coli, followed by irradiation with UV light leads to crosslinked GST molecules, thereby confirming that GST exists as dimers in vivo. | ||
Transforming microorganisms so that they incorporate non-natural amino acids in selected proteins requires considerable efforts in altering their protein synthesis machinery. An alternative strategy for the selective incorporation of bioorthogonal attachment sites is represented by adaptation of specific protein post-translational modification events. In such a strategy, a key intermediate in a given post-translational modification pathway is replaced by a synthetic equivalent containing a ligation handle. Successful incorporation now rests on the ability of the involved enzymes to recognise the chemically altered metabolic precursor and process it like its natural counterpart, without interfering with the incorporated ligation functionality.
Seminal work by Bertozzi and coworkers on the latent labeling of cell surface glycoproteins comprises one of the hallmarks of contemporary chemical biology research.22 It was demonstrated that a synthetic N-acetylmannosamine derivative equipped with an azide functionality is tolerated by both the sialic acid biosynthetic pathway and the protein glycosylation machinery, resulting in the decoration of the cell surface glycoproteins with azide-containing sialic acid residues. In short, Jurkat cells were grown on medium containing tetra-O-acetyl-N-(azidoacetyl)mannosamine (12, Fig. 4) for three days. After washing, the cells were treated with phosphine-biotin reagent 14 and subsequently stained with fluorescent avidin. In this way, cells fed with 12 could be selectively visualised. This result highlights the metabolic stability of the azide group in a living cell, and thereby its suitability for use in bioorthogonal chemistry. Recently, the scope of the Staudinger–Bertozzi ligation was extended to performing the ligation in living animals. Mice were treated with 12 for 7 days, and the cell surface sialic acid residues were addressed by injection of an analog of 14 where the biotin was replaced with a FLAG-epitope.23
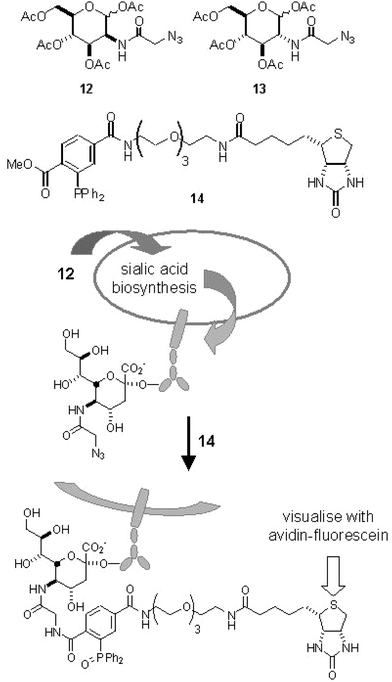 | ||
| Fig. 4 The structures of azide-modified carbohydrates and the use of modified ManNAc 12 in cell surface labeling. | ||
The modification of specific serine and threonine residues in a wide array of cytosolic and nuclear proteins with a single N-acetylglucosamine residue (Ser/Thr O-GlcNAc modification) has attracted considerable interest in recent years. In many cases, the modification appears to be reversible and of a highly dynamic nature. Further, the finding that this modification often occurs at sites also prone to phosphorylation has led to speculations that protein O-GlcNAcylation, as is the case with protein phosphorylation, is involved in the control of many biological events. These considerations prompted the Bertozzi group24 to establish whether protein O-GlcNAcylation could be addressed through selective chemical ligation in a similar fashion as outlined above for sialic acid containing glycoproteins. A cell line having a defect in its de novo hexosamine synthesis pathway was cultured in the presence of azide-modified GlcNAc (compound 13). After cell lysis and isolation of nuclear material, the modified GlcNAc moieties were equipped with an affinity label using the Staudinger–Bertozzi ligation. As a result, the authors detected a large number of nuclear proteins that bear the azido-GlcNAc modification.
B Tagging of fusion proteins with synthetic probes
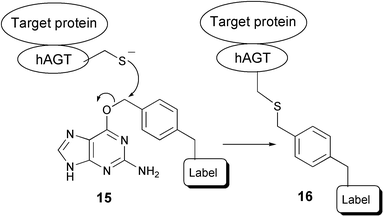
The ability to site-selectively label a protein of choice, in order to track its cellular localisation and fate, has been the objective of many research efforts over recent decades. Fusion proteins containing GFP or its analogues remain the tools of choice for this purpose. However, the fact that these proteins are bulky and thereby potentially perturbative, has spurred the search for alternatives. Some examples of recent results are described in the following section.Johnsson and co-workers have developed a labeling strategy that exploits the activity of the human DNA repair protein 6-O-alkylguanine-DNA-alkyltransferase (hAGT).25 The biological function of hAGT resides in its ability to recognise 6-O-alkylated guanine and transfer the alkyl group to one of its cysteine thiols, thereby restoring the structural integrity of the DNA. It appears that hAGT possesses broad substrate specificity with respect to the alkyl moiety and, especially, that functionalised benzyl moieties are well tolerated. This observation paved the way for the following strategy. A fusion is created between the protein of interest and hAGT. In the next step, ligation between the fusion protein and the label of choice (biotin, fluorescent or bifunctional tag26) is accomplished by nucleophilic attack of hAGT's activated sulfur on the benzylic position of modified guanine derivatives (see Scheme 3). It should be noted that special care needs to be taken with mammalian cells, since these also express AGT which can react with the 6-O-alkylated guanine, thereby causing high background labeling.27 This problem can be avoided by the use of an AGT knock-out cell line. Furthermore, the higher activity of AGT-mutants relative to wild-type AGT allows selective substoichiometric labeling of AGT fusion proteins.28 The same group developed an extension of this approach, in which a chemical inducer of dimerization (methotrexate) was linked to a hAGT-fusion protein, thereby constructing heterodimers of proteins that regulate DNA transcription in yeast.29 This method can be viewed as a more flexible anologue of the GFP method, in the sense that the tag that is transferred to the fusion protein of interest can be chosen liberally. But even though hAGT is 31 amino acid residues smaller than GFP and it is claimed that perturbation of the protein of interest should therefore be less, this approach does not take away the main objection against the GFP method.
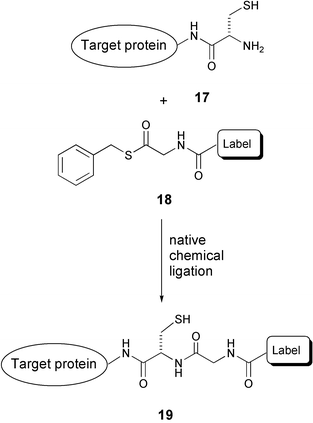
The problem of unwanted cross-reactivity with native proteins, as noted in the example above, is circumvented in an elegant approach by the group of Yao,30 who make use of the highly selective and biocompatible native chemical ligation reaction of an N-terminal cysteine and a thioester (Scheme 4). Thus, cell permeable probes consisting of a reporter moiety (label) and a benzyl thioester (18) are reacted with an N-terminal cysteine modified target protein overexpressed in E. coli cells. In this way, a highly site-selective labeling strategy is attained, without having to resort to additional knock-out cell lines. It should be noted that labeling of internal cysteine residues or other thiols is highly likely. However, this non-specific labeling will not be observed since under the conditions of sample preparation for gel electrophoresis all thioesters will be hydrolysed. The advantage of this technique, as compared to the above example, is that the necessary modification (only one cysteine residue) is less perturbing in terms of size. However, protein engineering still remains a necessity and side reactions on the N-terminal cysteine modified proteins may be introduced.
An exquisite combination of organic chemistry and biochemical engineering is represented by the fluorescent labeling strategy developed by Tsien and coworkers.31 A report that the toxicity of trivalent arsenic compounds originates from strong binding to thiol pairs of adjacent cysteine residues spurred the design of novel protein labels. Fluorescent biarsenides (exemplified by the so-called FlAsH derivative 21, see Scheme 5) were prepared and studied with respect to their binding affinity with synthetic cysteine-containing oligopeptides. It was found that peptides containing the CCXXCC motive bind strongly and selectively to FlAsH. Fusion of the tetracysteine motif to target proteins now allows their labeling with FlAsH or its analogues32 at the desired time. The validity of this approach was illustrated by the Tsien group in the following experiment.33 Cells were generated that express recombinant connexin fused with the tetracysteine tag. Connexins are cell surface proteins that establish channels between two aligned cells for the exchange of subcellular material. Treatment of the culture with the cell-permeable FlAsH compound resulted in selective labeling of clusters of cell surface connexin molecules. At this point, an interesting feature of the two-step labeling presented itself. Whereas FlAsH molecules bound to recombinant connexins appeared to be relatively stable, excess non-bound FlAsH could be readily washed from the culture. Then, treatment with another biarsenic label, ReAsH (which colours red as opposed to the green FlAsH), rendered any newly synthesised connexin red, while the earlier synthesised connexins remained green. In this way, important information on the development of connexin patches was obtained. It is unlikely that similar results can be obtained by making use of GFP fusion type constructs.
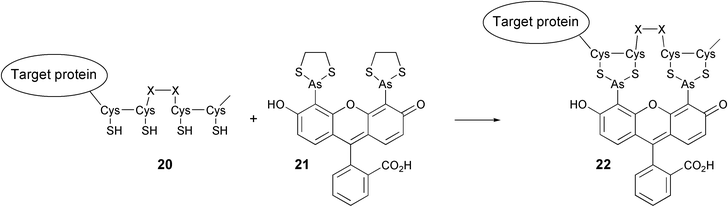 | ||
| Scheme 5 The reaction of a fusion protein containing a tetracysteine motif with the FlAsH reagent (21). | ||
The toxicity and the high background signals arising from FlAsH derivatives as well as the cross reactivity as observed in the hAGT-approach are addressed in the approach of Nolan.34 The high-affinity interaction between a FKBP12 mutant protein and the synthetic ligand, SLF′, is exploited to label and inactivate proteins in living cells by fluorophore assisted laser inactivation (FALI). Fusion proteins of β-galactosidase and mutant FKBP12 were expressed in NIH 3T3 cells. The cells were incubated with a cell permeable fluorescein derivative of SLF′, allowing detection of labeled fusion proteins with confocal microscopy. In a subsequent FALI-experiment, irradiation of the fluorescein group generated oxygen radicals near the galactosidase fusion protein, thereby inactivating the enzyme. This method can be used to create spatially and temporally defined enzyme inactivation.
C Tagging of enzymes with activity-based probes
The examples described thus far entail the development of techniques that enable the labeling of protein families through the incorporation of selective attachment sites in either the polypeptide backbone or in post-translationally added entities. After chemoselective ligation of the reporter group, information on both expression levels and the localisation of the targeted protein (family) is obtained. Families of enzymatic activities provide an alternative strategy for chemoselective attachment of reporter molecules. Chemical probes that are designed to bind specifically and irreversibly within the active site of a given enzymatic activity, and that are equipped with a reporter molecule, may in principle report on both localisation and expression level. The fact that binding is dependent on actual enzymatic activity introduces a third functional read-out, namely that of proper functioning of the targeted enzyme(s). The value of this strategy has been widely recognised in recent years, especially in the field of proteolysis.35 In general, short oligopeptides that serve as recognition elements for a given protease (family) are equipped with both an electrophilic trap (a chemical entity with high reactivity towards the enzyme active site) and a reporter group (for instance biotin, fluorescent group, radioisotope). The resulting construct is referred to as an activity-based probe (ABP). Treatment of cell lysates with an ABP will result in irreversible labeling of the targeted proteases, which can be visualised and analysed using standard gel electrophoresis. A distinct advantage of the ABP strategy over related strategies that are based on the use of fluorogenic oligopeptide substrates is the covalent and irreversible nature. Modified proteases can thereby be visualised and, optionally, isolated and identified in a later stage of the experiment, facilitating profiling of the enzymatic activities in complex mixtures. ABP labeling of many protease families, including serine proteases,36 cysteine proteases (cathepsins,37 caspases38), metalloproteases39 and the proteasome,3 have been reported in recent years. A drawback encountered in these procedures, however, is the finding that the presence of the reporter entity generally diminishes or even precludes cellular uptake3 of the probes, necessitating cellular lysis prior to the labeling experiment. To overcome this shortcoming, several groups have endeavoured to introduce small latent ligation sites in ABPs, with the idea that such constructs would retain cell permeability. After labeling in living cells, attachment of the reporter entity may then be executed ex vivo through a chemoselective ligation reaction.This strategy is well illustrated by the recent work of Cravatt and coworkers on the profiling of a range of enzyme activities in mammalian tissue cultures (for instance human breast cancer glutathione-S-transferase and mouse liver aldehyde dehydrogenase).40 Making use of the finding that a diverse range of enzymes are irreversibly disabled by simple benzenesulfonyl derivatives, a set of azide containing ABPs were prepared (such as compound 23, Fig. 5). Cells were incubated with the sulfonate, washed, lysed and the azide moiety was addressed in the cell lysate by an alkyne-functionalysed rhodamine (24) in a stepwise Huisgen reaction. In a more recent report,41 it was shown that the alkyne moiety is not fully bioorthogonal. In a reversed experiment, the ABP was equipped with an acetylene moiety and the reporter entity with an azide. It appeared that application of the alkyne-functionalysed rhodamine (and therefore an azide-ABP) led to higher background labeling than an azide-functionalised rhodamine (with an alkyne ABP). Also, the Cravatt group demonstrated the first labeling of proteins in live organisms (mice), and subsequent in-gel visualisation of retrieved proteins.40
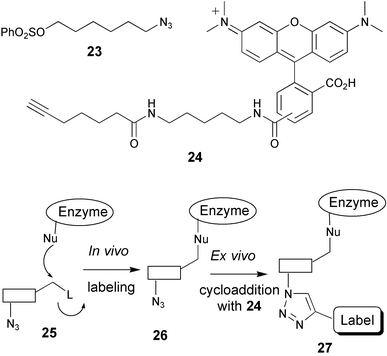 | ||
| Fig. 5 Two-step activity-based enzyme profiling using Huisgen-type cyclisation. | ||
In a related approach, we equipped a cell permeable ABP directed against the proteasome with an azide moiety.42 Cells were incubated with the cell permeable, irreversible inhibitor 28 (see Fig. 6) and after cell lysis the inhibitor–proteasome construct was decorated with a biotin moiety by Staudinger–Bertozzi ligation with a biotinylated phosphine reagent. Western blotting showed that labeling of all catalytic subunits was effected and, importantly, the in vivo labeling proceeded with higher efficiency than the labeling in cell lysates with the biotinylated proteasome inhibitor 29. This underlines the value of labeling the enzymes in their natural environment.
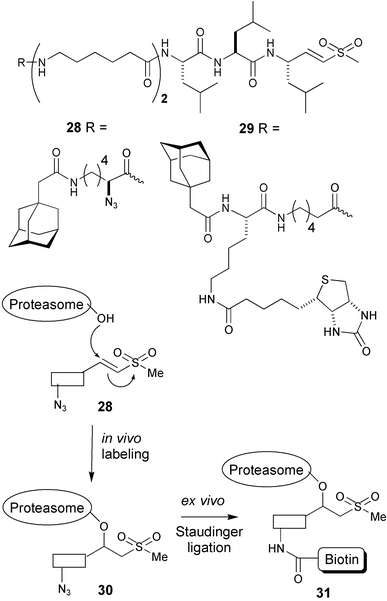 | ||
| Fig. 6 Two-step labeling of active proteasomes using Staudinger–Bertozzi ligation. | ||
Conclusion and perspectives
The approaches shown in this Perspective all rely on the organic-chemical modification of biomolecules (in most cases proteins) in living cells, with the aim of visualising and/or retrieving the biomolecules or their metabolites. Application of organic chemistry allows, in theory, the introduction of a multitude of modifications into selected biomolecules. The philosophy behind executing the labeling in living cells rather than in lysates stems from the desire to trap the targets as much as possible in their natural environment, so that artefacts created during cell lysis are reduced to a minimum.In this sense, it may seem counterintuitive to use molecular biological techniques to engineer proteins in such a way as to accommodate a modification. In the examples shown in category B (see above), fusion proteins are created, containing an attachment site (ranging from a single amino acid – N-terminal Cys – to an entire protein – hAGT). Although these modifications are all smaller than GFP, the target protein that is studied is considerably altered compared to its natural counterpart. Further, in the case of the hAGT examples, an additional artefact is introduced by the need to knock out the native AGT mechanism (in the case of mammalian tissue) in order to diminish background labeling. Finally, the creation of fusion proteins for each separate process that one would like to study constitutes a tremendous amount of biotechnological effort. However, the collective strongpoint of the approaches described in this category is that these are, up to now, the only truly in vivo chemical labeling strategies, in the sense that both steps (the tagging and ligation) are executed in live cells.
A less invasive strategy is to incorporate, at a designated site of a biomolecule, an inconspicuous bioorthogonal modification that can be addressed after the fact (as described under category A). Techniques required for altering the protein synthesis machinery in such a way that it accepts a non-natural amino acid with a bioorthogonal handle are by no means standardised yet. Furthermore, all potential target proteins have to be engineered separately. In this respect, the other approach described under A (to feed the cell certain modified metabolic precursors that are later expressed in sites of interest) seems less laborious, in the sense that non-engineered (wild type) tissues can be studied. Of course, this strategy only works if the modified precursors are accepted by the natural metabolic pathway. But with the groundbreaking work described in section A, and some careful consideration of the modified precursor to be introduced (in terms of site of modification, its size and thereby conspicuousness), it is possible to envisage applications in the area of, for instance, lipid metabolism and post-translational modifications other than glycanation or glycosidation. In fact, the metabolic incorporation of a synthetic azido-farnesyl analog into farnesylated proteins has been shown recently.43
The third category, the activity-based enzyme profiling approach, is also independent of protein engineering. The intrinsic reactivity of native enzymes is exploited to a) visualise their activity and b) retrieve the biomolecules from a biological sample. It may seem that this strategy can only be applied to enzymatic activities that actually partake, through the involvement of an amino acid side chain functionality inherent to the enzyme, in the chemical transformation at hand. However, it may well be that, in the near future, hydrolytic activities that involve water as the nucleophilic species (rather than a serine- or cysteine residue), may be addressed in an ABP fashion, for instance through the application of well-designed suicide substrates.
As for any technique that is only just past its infancy, there are many areas still to be developed. Firstly, all studies done so far in the area of activity-based profiling have been on the qualitative level (whether a labeled protein can be detected or not). Not much effort has been devoted to efficacy studies of both the tagging and the subsequent ligation step, in terms of side reactions, molar yield and kinetics. Of course, this requires robust analytical methods for the characterisation of the tagging and ligation products. With detailed information in this area in hand, it will be possible to advance the in vivo labeling strategy to a truly quantitative approach, in which the levels of certain enzymatic activities can be compared between, for instance, diseased and control tissues. One can readily imagine a merging of an ABP based profiling of enzymatic activities with the isotope-encoded affinity probe techniques developed by Aebersold and coworkers.44,45 In such a strategy, tissue from different sources would be treated with a pair of ABPs differing only in hydrogen/deuterium content.
Furthermore, there is much to be gained from studies on enzymatic activities other than proteases, which up to now have been the subject of in vivo ABP-based techniques. With the advances made in the design and synthesis of irreversible inhibitors for other enzymes (for instance glycosidases and phosphatases), it is now possible to embark on similar in vivo strategies with these enzymes. The first step toward in vivo activity based profiling of glycosidases has been made by the Bertozzi group. A known mechanism-based glycosidase inhibitor was equipped with an azide moiety and used to profile enzyme activity in cell lysates.46
The last, but certainly not least, perspective of strategy C entails the approach best described as forward chemical genomics.47 This entails the incubation of a certain tissue with a label of choice, followed by the ligation reaction, retrieval and identification of the products. This may lead to the discovery of enzymatic activities or metabolites that may not have been envisaged or have been overlooked, because they are, for instance, not present in cell lysates.
It can be concluded from the examples described in this Perspective that the labeling of proteins in living cells using bioorthogonal organic chemistry is an effective and reliable technique with high potential for further application. Although the described approaches which employ organic chemistry, rather than biotechnology, are only just past the stage of proof-of-principle, it is obvious that there is a considerable interest from both the organic chemistry and the biology research communities. It is therefore only a matter of time before these techniques will find acceptance and applicability in fundamental biochemistry, pharmaceutics and medicine alike.
References
- R. Y. Tsien, Ann. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS.
- (a) J. Neefjes and N. P. Dantuma, Nat. Rev. Drug Discovery, 2004, 3, 58–69 CrossRef CAS; (b) J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS.
- B. M. Kessler, D. Tortorella, M. Altun, A. F. Kisselev, E. Fiebinger, B. G. Hekking, H. L. Ploegh and H. S. Overkleeft, Chem. Biol., 2001, 8, 913–929 CrossRef CAS.
- S. W. Michnick, Drug Discovery Today, 2004, 9, 262–267 CrossRef CAS.
- Bioorthogonal is defined in this paper as: not associated with – and chemically inert to – the conditions in the particular physiological system.
- (a) T. Hohsaka and M. Sisido, Curr. Opin. Chem. Biol., 2002, 6, 809–815 CrossRef CAS; (b) A. Strømgaard, A. A. Jensen and K. Strømgaard, ChemBioChem, 2004, 5, 909–916 CrossRef CAS.
- D. H. Dube and C. R. Bertozzi, Curr. Opin. Chem. Biol., 2003, 7, 616–625 CrossRef CAS.
- N. Johnsson and K. Johnsson, ChemBioChem, 2003, 4, 803–810 CrossRef CAS.
- N. Jessani and B. F. Cravatt, Curr. Opin. Chem. Biol., 2004, 8, 54–59 CrossRef CAS.
- (a) W. A. Hendrickson, J. R. Horton and D. M. Lemaster, EMBO J., 1990, 9, 1665–1672 CAS; (b) N. Budisa, B. Steipe, P. Demange, C. Eckerskorn, J. Kellerman and R. Huber, Eur. J. Biochem., 1995, 230, 788–796 CAS; (c) H. Duewel, E. Daub, V. Robinson and J. F. Honek, Biochemistry, 1997, 36, 3404–3416 CrossRef CAS.
- (a) J. C. M. van Hest, K. L. Kiick and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 1282–1288 CrossRef CAS; (b) K. L. Kiick, J. C. M. van Hest and D. A. Tirell, Angew. Chem., Int. Ed., 2000, 39, 2148–2152 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirell and C. R. Bertozzi, Proc. Natl. Acad. Sci. USA, 2002, 99, 19–24 CrossRef CAS.
- A. J. Link and D. A. Tirell, J. Am. Chem. Soc., 2003, 125, 11164–11165 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- V. V. Rostovsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS.
- R. A. Mehl, J. C. Anderson, S. W. Santoro, L. Wang, A. B. Martin, D. S. King, D. M. Horn and P. G. Schulz, J. Am. Chem. Soc., 2003, 125, 935–939 CrossRef CAS.
- A. Deiters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 11782–11783 CrossRef.
- J. Brunner, Chem. Soc. Rev., 1993, 22, 183–189 RSC.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- J. W. Chin and P. G. Schultz, ChemBioChem, 2002, 3, 1135–1137 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS.
- D. J. Vocadlo, H. C. Hang, E.-J. Kim, J. H. Hanover and C. R. Bertozzi, Proc. Natl. Acad. Sci. USA, 2003, 100, 9116–9121 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS.
- M. Kindermann, I. Sielaff and K. Johnsson, Bioorg. Med. Chem. Lett., 2004, 14, 2725–2728 CrossRef CAS.
- A. Keppler, M. Kindermann, S. Gendreizig, H. Pick, H. Vogel and K. Johnsson, Methods, 2004, 32, 437–444 Search PubMed.
- A. Keppler, H. Pick, C. Arrivoli, H. Vogel and K. Johnsson, Proc. Natl. Acad. Sci. USA, 2004, 101, 9955–9959 CrossRef CAS.
- S. Gendreizig, M. Kindermann and K. Johnsson, J. Am. Chem. Soc., 2003, 125, 14970–14971 CrossRef CAS.
- D. S. Y. Yeo, R. Srinivasan, M. Uttamchandani, G. Y. J. Chen, Q. Zhu and S. Q. Yao, Chem. Commun., 2003, 2870–2871 RSC.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS.
- G. Gaietta, T. J. Deerinck, S. R. Adams, J. Bouwer, O. Tour, D. W. Laird, G. E. Sosinsky, R. Y. Tsien and M. H. Ellisman, Science, 2002, 296, 503–507 CrossRef CAS.
- K. M. Marks, P. D. Braun and G. P. Nolan, Proc. Natl. Acad. Sci. USA, 2004, 101, 9982–9987 CrossRef CAS.
- A. E. Speers and B. F. Cravatt, ChemBioChem, 2004, 5, 41–47 CrossRef CAS.
- D. Kidd, Y. Liu and B. F. Cravatt, Biochemistry, 2001, 40, 4005–4015 CrossRef CAS.
- D. Greenbaum, K. F. Medzihradszky, A. Burlingame and M. Bogyo, Chem. Biol., 2000, 7, 569–581 CrossRef.
- M.-L. Liau, R. C. Panicker and S. Q. Yao, Tetrahedron Lett., 2003, 44, 1043–1046 CrossRef CAS.
- A. Saghatelian, N. Jessani, A. Joseph, M. Humphrey and B. F. Cravatt, Proc. Natl. Acad. Sci. USA, 2004, 101, 10000–10005 CrossRef CAS.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS.
- A. E. Speers and B. F. Cravatt, Chem. Biol., 2004, 11, 535–546 CrossRef CAS.
- H. Ovaa, P. F. van Swieten, B. M. Kessler, M. A. Leeuwenburgh, E. Fiebinger, A. M. C. H. van den Nieuwendijk, P. J. Galardy, G. A. van der Marel, H. L. Ploegh and H. S. Overkleeft, Angew. Chem., Int. Ed., 2003, 42, 3626–3629 CrossRef CAS.
- Y. Kho, S. C. Kim, C. Jiang, D. Barma, S. W. Kwon, J. Cheng, J. Jaunbergs, C. Weinbaum, F. Tamanoi, J. Falck and Y. Zhao, Proc. Natl. Acad. Sci. USA, 2004, 101, 12479–12484 CrossRef CAS.
- S. P. Gygi, B. Rist, S. A. Gerber, F. Turecek, M. H. Gelb and R. Aebersold, Nat. Biotechnol., 1999, 17, 994–999 CrossRef CAS.
- An example of an isotope-coded activity-based affinity tag has been published: P. F. van Swieten, R. Maehr, A. M. C. H. van den Nieuwendijk, B. M. Kessler, M. Reich, C.-S. Wong, H. Kalbacher, M. A. Leeuwenburgh, C. Driessen, G. A. van der Marel, H. L. Ploegh and H. S. Overkleeft, Bioorg. Med. Chem. Lett., 2004, 14, 3131–3134 Search PubMed.
- D. J. Vocadlo and C. R. Bertozzi, Angew. Chem., Int. Ed., 2004, 43, 5338–5342 CrossRef CAS.
- S. M. Khersonsky and Y.-T. Chang, ChemBioChem, 2004, 5, 903–908 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |