Synthesis and hetero-Michael addition reactions of 2-alkynyl oxazoles and oxazolines†
Peter
Wipf
* and
Thomas H.
Graham
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: pwipf@pitt.edu; Fax: +1-412-624-0787; Tel: +1-412-624-8606
First published on 25th November 2004
Abstract
Stereoselective conjugate additions of alcohols, amines, thiols, and halides to C(2)-alkynyl oxazoles and oxazolines provide a versatile entry to heterocyclic building blocks.
2,4-Disubstituted oxazoles and oxazolines derive biosynthetically from processing of readily available serine residues by a nonribosomal peptide synthetase (NRPS)1 and represent a common structural motif of many biologically active natural products.2 More specifically, 4-formyl-2-(2-oxoalkyl)oxazoles 1 originate from the joint action of NRPS and polyketide synthase (PKS),3 a pathway that results frequently in diverse and very potent secondary metabolites such as phorboxazoles,4 disorazoles,5 and the streptogramin group A antibiotics (virginiamycin,6 madumycin II,7 and griseoviridin)8 (Fig. 1). As part of our program directed towards the synthesis of natural products containing oxazole moieties,9 we have investigated the conversion of 2-alkynyl oxazoles into C2-β-heteroatom-substituted oxazoles. We now report our results involving the conjugate addition of S-, O-, and N-nucleophiles to 2-alkynyl oxazoles.10 Similar additions can be conducted on oxazolines.
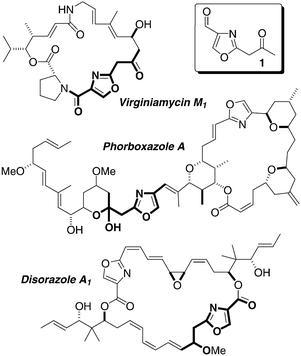 | ||
| Fig. 1 | ||
The most common route toward scaffolds 1 and 2 involves the condensation of an aldehyde with a 2-methyloxazole anion 3 (Fig. 2, path A). Numerous metal counter-ions including lithium,11 zinc,12 chromium,13 samarium,14 and sodium15 have been employed in this transformation. A much less frequently explored disconnection is at the Cβ/Cγ bond, which requires the coupling of the C(2)-β-aldehyde 4 with a suitable nucleophile (path B).16 Consistent with the strategy outlined above, we envisioned the target aldehyde to arise from 2-ethynyl oxazole 5 (path C). Alternatively, an internal alkyne could be converted to C(2)-β-ketone 6via Michael addition of a heteronucleophile followed by hydrolysis (path D).17
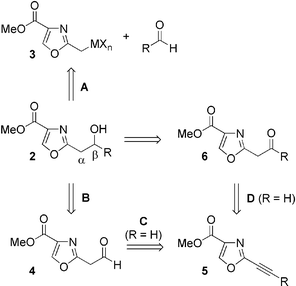 | ||
| Fig. 2 | ||
2-Alkynyl oxazoles 5 can be prepared by (a) the coupling of aminoalcohols with an alkynoic acid followed by cyclodehydration/oxidation,18,19 (b) the transition metal catalyzed cross-coupling of a 2-halo or 2-trifluoromethylsulfonyl oxazole with a terminal alkyne,20 or (c) the condensation of an α-diazoketone with a nitrile.21 We selected a cyclization/oxidation approach since many amino alcohols are commercially available, alkynes can be readily carbonylated to give alkynoic acids and expensive and potentially toxic transition metal catalysts are not required.
Attempts to couple propiolic acid directly to serine methyl ester resulted in decomposition. TMS and TES protective groups at the terminal alkyne, while allowing access to the hydroxyamide, were too labile to survive the reaction conditions of the subsequent oxazole formation. The triisopropylsilyl (TIPS) group proved to be more suitable. Anionic carboxylation of the commercially available acetylene 7 followed by coupling to serine methyl ester hydrochloride with PyBOP22 in the presence of diisopropylethylamine gave hydroxyamide 8 in quantitative yield (Scheme 1). For large scale preparations, a more cost-effective strategy involved converting the triisopropylsilylpropynoic acid to the acid chloride using oxalyl chloride and catalytic DMF. The crude acid chloride was then coupled to serine methyl ester hydrochloride in the presence of diisopropylethylamine to afford 8 in 82% overall yield. Cyclization using diethylaminosulfur trifluoride (DAST) occurred rapidly and afforded crude oxazoline which was oxidized with BrCCl3 and DBU to furnish the desired oxazole in 74% yield.18a Removal of the triisopropylsilyl group with buffered TBAF at ambient temperature gave 9 in low yield. Conducting the reaction at −78 °C increased the yield to 99%. The 2-ethynyl oxazole 9 is a crystalline solid that can be stored for months at ambient temperature with no special precautions.23,24
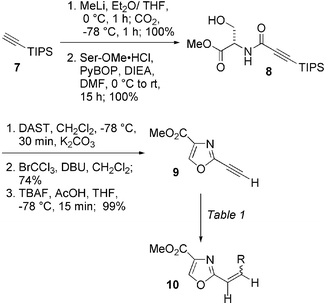 | ||
| Scheme 1 | ||
No examples of Michael additions to 2-alkynyl oxazoles have been reported. However, based on the ease with which the TBAF deprotection of the TIPS-protected alkyne occurred, we expected that this substrate would readily participate in conjugate addition reactions. Indeed, a variety of nucleophiles including ethanethiol and thiophenol added in moderate to good yields and with good (Z)-selectivity (Table 1, entries 1–3).25 2-Mercaptoethanol afforded only the thioenolether and did not cyclize to the thioxolane even after an extended reaction time (entry 4). Benzyl alcohol also furnished an adduct with good kinetic selectivity for the (Z)-isomer, thus allowing access to enol ethers (entry 5).26 Interestingly, secondary amines produced (E)-configured adducts in excellent yields (entries 6 and 7).27 Steric hindrance and a basic reaction medium readily explains the thermodynamic control for the preferred (E)-double bond geometry in the latter case. In agreement with our results for conjugate additions of thiols and alcohols, other studies have determined that the (Z)-selectivity with thiol nucleophiles strongly depends on the nature of the activating group on the alkyne, increasing when the substituent is capable of delocalizing the incipient negative charge.28
| Entry | R–H | Additive | Time/ha | Product | (Z/E) ratio | Yield (%)b |
|---|---|---|---|---|---|---|
| a All reactions were conducted at ambient temperature unless noted otherwise. b Yields were determined after purification on SiO2 and refer to the major isomer, unless stated otherwise. c Purification by recrystallization. d Reaction mixture was heated to 66 °C. e Yield is for the (Z/E) mixture. f Reaction mixture was heated to 60 °C. | ||||||
| 1 | EtSH | n-Bu3P | 36 | 10a | 10.5 : 1.0 | 70 |
| 2 | EtSH | K2CO3/18-C-6 | 1.5 | 10a | 16.4 : 1.0 | 89 |
| 3 | PhSH | NMM | 48 | 10b | 1 : 0 | 97c |
| 4 | HO(CH2)2SH | n-Bu3P | 25d | 10c | 6.4 : 1.0 | 68e |
| 5 | PhCH2OH | n-Bu3P | 0.5 | 10d | 6.4 : 1.0 | 76 |
| 6 | Et2NH | — | 20f | 10e | 0 : 1 | 100 |
| 7 | i-Pr2NH | — | 72 | 10f | 0 : 1 | 100 |
To gain access to aldehyde 4, hydrolysis of 10a–f was attempted under a variety of conditions but only decomposition products were observed. In order to eliminate the conjugation of the donor heteroatom with the electron-deficient oxazole ring and thus facilitate solvolysis, alkyne 9 was exposed to 1,2-ethanedithiol to give the 1,3-dithiolane 11 in 97% yield (Scheme 2).25 In contrast, 1,3-propanedithiol resulted in an inseparable mixture of mono- and bis-addition adducts in low yield. Unfortunately, all attempts to deprotect 11 resulted in decomposition. We reasoned that the apparently highly labile29 aldehyde 4 could be obtained by a mild Fukuyama reduction30 of the corresponding thioester.
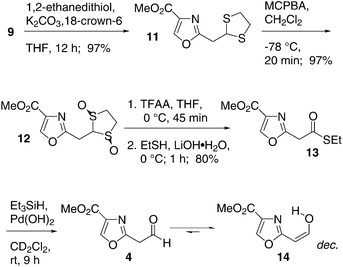 | ||
| Scheme 2 | ||
Oxidation of 11 under Trost's conditions31 gave the 1,3-dithiolane dioxide 12 in 97% yield. The hydrophilic properties of 12 required an anhydrous workup that involved concentrating the reaction mixture onto a mixture of Florisil and Celite. The crude mixture was then directly subjected to chromatography on silica gel. Using a variation of a method developed by Aggarwal and co-workers,32 rearrangement of the 1,3-dithiolanedioxide 12 under Pummerer conditions and quenching with ethanethiol gave thioester 13 in 80% yield. Reduction with Pd(OH)233 in dichloromethane gave a new compound by TLC analysis; however, preparative isolation failed. When the experiment was conducted in CD2Cl2 and monitored by 1H NMR, no aldehyde signal was observed. Instead, a pair of doublets with J = 6.4 Hz was observed, at 6.76 and 5.47 ppm. These data suggest that compound 4 exists predominantly in the tautomeric form 14, which is possibly stabilized by hydrogen bonding to the oxazole nitrogen.34 However, 14 decomposes in CD2Cl2 at room temperature with a half-life of several hours, and we were unable to obtain preparative samples of this compound.
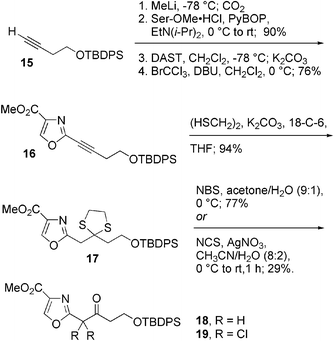
The transient nature of 4 and the shift of the equilibrium to enol 14 discouraged any further attempts to use this compound as a synthetic intermediate. However, based on the ease of preparation of 13, we expected the corresponding ketones to have greater chemical stability. The 2-alkynyl oxazole 16 was readily obtained from 15 (Scheme 3). Deprotonation with methyl lithium followed by the addition of CO2 gave the alkynoic acid. Coupling with serine methyl ester hydrochloride in the presence of PyBOP22 and diisopropylethylamine led to the hydroxyamide in 90% yield over 2 steps. Cyclodehydration with DAST and oxidation of the crude oxazoline with BrCCl3 and DBU18a gave 16 in 76% yield. 2-Alkynyl oxazole 16 was found to readily accept ethanedithiol at the β-position to give the 1,3-dithiolane 17 in 94% yield. Further conversion with N-bromosuccinimide in 10% aqueous acetone35 provided ketone 18 in 77% yield. In contrast, deprotection with a combination of N-chlorosuccinimide and silver nitrate35 gave the corresponding dichloroketone 19 in low yield, suggesting that the α-methylene group readily enolizes under the reaction conditions. Both ketones are markedly more stable than aldehyde 4.
 | ||
| Scheme 3 | ||
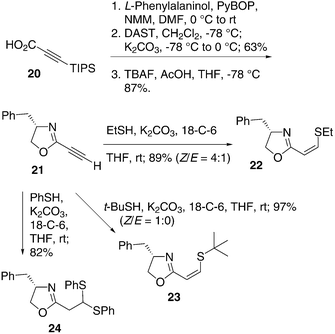
Oxazolines with additional chelating ligands have found exceptional use in asymmetric catalysis and are among few “privileged” metal ligands.36–38 Since a range of heteroatoms could be introduced in the conjugate addition to 2-ethynyl oxazoles 9, we extended the scope of this process to the corresponding oxazolines (Scheme 4). Oxazoline 2139 was obtained in 3 steps from L-phenylalaninol and the TIPS-protected alkynoic acid 20. As expected, 21 was found to participate readily in the conjugate addition reaction with thiols. Ethanethiol and even the sterically hindered t-butyl thiol formed adducts 22 and 23, respectively, in good to excellent yields and (Z)-selectivity. Interestingly, similar conditions using thiophenol gave the dithioacetal 24. These results indicate that the conjugate addition methodology is equally effective for 2-ethynyl oxazolines as for the corresponding oxazoles. Along with varying the thiol or the C4-substituent of the oxazoline, the conjugate addition methodology affords products that offer the potential for further modications. The mild reaction conditions, good yields and (Z/E)-selectivity offer significant promise for accessing novel bidentate ligands for asymmetric catalysis.
 | ||
| Scheme 4 | ||
Electron-deficient alkynes are known to hydrohalogenate when exposed to HI or HBr.40 Taniguchi as well as Lu and co-workers have developed a convenient protocol involving a lithium halide salt in the presence of acetic or trifluoroacetic acid.41–43 Indeed, heating 9 in acetic acid in the presence of sodium or lithium halides afforded moderate to good yields of the corresponding vinyl halides (Table 2). For the preparation of vinyl bromide 10h, NaBr gave a slightly better (Z/E)-ratio than LiBr (entry 2 vs. entry 6). For the formation of vinyl iodide 10i, the (Z/E)-ratio was found to decrease with increasing quantities of NaI (entries 8, 10, 12) or increasing temperature (entry 10 vs. entry 13). The presence of lithium or sodium acetate provided remarkably increased (Z)-selectivities.
| Entry | H–R | Additive (equiv.) | Time/h | Product | (Z/E) ratioa | Yield (%)b |
|---|---|---|---|---|---|---|
| a The (Z/E)-ratio was determined by 1H NMR analysis of the crude reaction mixture after aqueous workup. b Yield refers to material isolated by chromatography on SiO2. c The crude reaction mixture contained approximately 10% starting material (alkyne). d The crude reaction mixture contained approximately 3% starting material (alkyne). e The reaction was conducted in a sealed tube at 150 °C. | ||||||
| 1 | HCl | NaCl (1.5) | 16 | 10g | 1.3 : 1.0 | 62 |
| 2 | HBr | LiBr (1.5) | 16 | 10h | 4.0 : 1.0 | 71 (Z) |
| 3 | HBr | LiBr/LiOAc (1.0 : 3.0) | 16 | 10h | 9.1 : 1.0c | 70 (Z) |
| 4 | HBr | LiBr/LiOAc (1.5 : 4.5) | 16 | 10h | 20 : 1.0d | 87 (Z) |
| 5 | HBr | NaBr (1.0) | 20 | 10h | 8.0 : 1.0c | 70 (Z) |
| 6 | HBr | NaBr (1.5) | 16 | 10h | 9.0 : 1.0c | 68 (Z) |
| 7 | HI | LiI (1.5) | 12 | 10i | 5.6 : 1.0 | 77 (Z) |
| 8 | HI | NaI (1.0) | 12 | 10i | 10.7 : 1.0 | 90 (Z) |
| 9 | HI | NaI/NaOAc (1.0 : 3.0) | 12 | 10i | >50 : 1.0 | 92 (Z) |
| 10 | HI | NaI (1.5) | 12 | 10i | 4.2 : 1.0 | 75 (Z) |
| 11 | HI | NaI/NaOAc (1.5 : 1.5) | 12 | 10i | 9.5 : 1.0 | 84 (Z) |
| 12 | HI | NaI (3.0) | 12 | 10i | 1.1 : 1.0 | 70 |
| 13e | HI | NaI (1.5) | 12 | 10i | 1.0 : 1.2 | 50 |
Some of the results shown in Table 2 can be explained by an alkene isomerization, and therefore the pure (Z)-isomers were re-subjected to the reaction conditions. In the absence of NaOAc, 1.5 to 3.0 equiv. of NaBr and NaI led to a thermodynamic ratio of 1 : 6–8 for 10h and 10i, in preference for (E)-isomers.44 If NaOAc was added to the reaction mixture, the rate of (Z)- to (E)-isomerization decreased dramatically. In addition, NaOAc also inhibited the isomerization of (E)-10i to (Z)-10i.
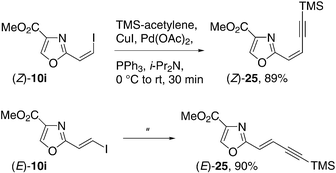
Vinyl halides are suitable for a wide range of metal-catalyzed cross-coupling reactions leading to efficient strategies for C–C bond formation.45 Iodide (Z)-10i readily participated in the cross-coupling reaction with trimethylsilylacetylene under typical Sonogashira conditions to afford (Z)-25 in 89% yield (Scheme 5). In an analogous fashion, (E)-10i was converted stereospecifically to (E)-25 in 90% yield.
 | ||
| Scheme 5 | ||
In conclusion, 2-alkynyl oxazoles and oxazolines are synthesized from readily available starting materials and are sufficiently polarized to participate in nucleophilic conjugate addition reactions. Although the C2-β-aldehyde 4 proved to be unstable, alkyne 9 could be converted to 1,3-dithiolane 11 and thioester 13. The 1,3-dithiolane 17, derived from internal alkyne 16, was readily converted to the C2-β-keto oxazole scaffold found in numerous biologically active natural products. Internal alkynes can therefore serve as a masked form of the C2-β-carbonyl function which can be unveiled under mild conditions. Thioenol ethers such as 10a and 22–24 represent readily accessible metal-binding motifs. In addition, hydrohalogenation of 9 affords predominantly (Z)-configured vinyl bromides and iodides under kinetic control. The addition of an acetate salt to the reaction mixture improves the (Z/E)-ratio and the overall yield. The corresponding (E)-vinyl halides are obtained under thermodynamic control. Both (Z)- and (E)-vinyl iodides readily participate in cross-coupling reactions. Accordingly, the conjugate addition of heteronucleophiles to oxazole and oxazoline alkynes offers a unique, mild and flexible approach to C(2)-functionalized heterocyclic building blocks for the synthesis of biologically active products.
Acknowledgements
This work has been supported by a grant from the National Institutes of Health (GM-55433).References and notes
- R. S. Roy, A. M. Gehring, J. C. Milne, P. J. Belshaw and C. T. Walsh, Nat. Prod. Rep., 1999, 16, 249 RSC.
- (a) P. Wipf, Chem. Rev., 1995, 95, 2115 CrossRef CAS; (b) Z. Jin, Nat. Prod. Rep., 2003, 20, 584 RSC; (c) Oxazoles: Synthesis, reactions, and spectroscopy, Part A, D. C. Palmer, ed., J. Wiley & Sons, Inc., Hoboken, NJ, 2003, vol. 60 Search PubMed.
- C. T. Walsh, ChemBioChem, 2002, 3, 124 CrossRef CAS.
- T. F. Molinski, Tetrahedron Lett., 1996, 37, 7879 CrossRef CAS.
- (a) R. Jansen, H. Irschik, H. Reichenbach, V. Wray and G. Höfle, Liebigs Ann. Chem., 1994, 759 CrossRef CAS; (b) H. Irschik, R. Jansen, K. Gerth, G. Höfle and H. Reichenbach, J. Antibiot., 1995, 48, 31 CAS; (c) P. Wipf and T. H. Graham, J. Am. Chem. Soc., 2004, 126, 15346 CrossRef CAS.
- G. R. Delpierre, F. W. Eastwood, G. E. Gream, D. G. I. Kingston, P. S. Sarin, A. R. Todd and D. H. Williams, J. Chem. Soc. C, 1966, 1653 RSC.
- J. W. Chamberlin and S. Chen, J. Antibiot., 1977, 30, 197 CAS.
- G. I. Birnbaum and S. R. Hall, J. Am. Chem. Soc., 1976, 98, 1926 CrossRef CAS.
- (a) P. Wipf, Y. Aoyama and T. E. Benedum, Org. Lett., 2004, 6, 3593 CrossRef CAS; (b) L. Scarone, D. Sellanes, E. Manta, P. Wipf and G. Serra, Heterocycles, 2004, 63, 773 CAS; (c) P. Wipf and J.-L. Methot, Org. Lett., 2001, 3, 1261 CrossRef CAS; (d) P. Wipf, C. P. Miller and C. M. Grant, Tetrahedron, 2000, 56, 9143 CrossRef CAS.
- To the best of our knowledge, this transformation has never been reported with oxazoles or oxazolines. However, HCl has been added to a 2-alkynyl thioazoline: R. C. Cambie, D. Chambers, P. S. Rutledge and P. D. Woodgate, J. Chem. Soc., Perkin Trans. 1, 1981, 40 Search PubMed.
- D. A. Evans, V. J. Cee, T. E. Smith and K. J. Santiago, Org. Lett., 1999, 1, 87 CrossRef CAS.
- A. R. Gangloff, B. Åkermark and P. Helquist, J. Org. Chem., 1992, 57, 4797 CrossRef CAS.
- P. Breuilles and D. Uguen, Tetrahedron Lett., 1998, 39, 3149 CrossRef CAS.
- D. R. Williams, M. P. Clark, U. Emde and M. A. Berliner, Org. Lett., 2000, 2, 3023 CrossRef CAS.
- Y. Nagao, S. Yamada and E. Fujita, Tetrahedron Lett., 1983, 24, 2287 CrossRef CAS.
- For an example of a nucleophilic addition to a non-enolizable β-carbonyl group, see: A. Yanagisawa, Y. Matsuzaki and H. Yamamoto, Synlett, 2001, 1855 Search PubMed.
- A. Nishida, M. Shibasaki and S. Ikegami, S. Chem. Pharm. Bull., 1986, 34, 1434 Search PubMed.
- For examples of cyclization–oxidation sequences, see: (a) A. J. Phillips, Y. Uto, P. Wipf, M. J. Reno and D. R. Williams, Org. Lett., 2000, 2, 1165 CrossRef CAS; (b) A. I. Meyers and F. X. Tavares, J. Org. Chem., 1996, 61, 8207 CrossRef CAS; (c) F. Tavares and A. I. Meyers, Tetrahedron Lett., 1994, 35, 6803 CrossRef CAS; (d) F. W. Eastwood, P. Perlmutter and Q. Yang, Tetrahedron Lett., 1994, 35, 2039 CrossRef CAS; (e) J. C. Barrish, J. Singh, S. H. Spergel, W.-C. Han, T. P. Kissick, D. R. Kronenthal and R. H. Mueller, J. Org. Chem., 1993, 58, 4494 CrossRef CAS; (f) F. Yokokawa, Y. Hamada and T. Shioiri, Synlett, 1992, 153 CrossRef CAS; (g) C. Kashima and H. Arao, Synthesis, 1989, 873 CrossRef CAS; (h) D. L. Evans, D. K. Minster, U. Jordis, S. M. Hecht, A. L. Mazzu and A. I. Meyers, J. Org. Chem., 1979, 44, 497 CrossRef.
- For an oxidation–cyclization sequence, see: (a) P. Wipf and T. H. Graham, J. Org. Chem., 2001, 66, 3242 CrossRef CAS; (b) P. Wipf and S. Lim, Chimia, 1996, 50, 157 CAS; (c) P. Wipf and S. Lim, J. Am. Chem. Soc., 1995, 117, 558 CrossRef CAS; (d) P. Wipf and C. P. Miller, J. Org. Chem., 1993, 58, 3604 CrossRef CAS.
- (a) L. A. Dakin, N. F. Langille and J. S. Panek, J. Org. Chem., 2002, 67, 6812 CrossRef CAS; (b) K. J. Hodgetts and M. T. Kershaw, Org. Lett., 2002, 4, 2905 CrossRef CAS; (c) N. F. Langille, L. A. Dakin and J. S. Panek, Org. Lett., 2002, 4, 2485 CrossRef CAS.
- (a) Y. Wang, J. Janjic and S. A. Kozmin, J. Am. Chem. Soc., 2002, 124, 13670 CrossRef CAS; (b) R. D. Connell, F. Scavo, P. Helquist and B. Åkermark, Tetrahedron Lett., 1986, 27, 5559 CrossRef CAS.
- J. Coste, D. Le-Nguyen and B. Castro, Tetrahedron Lett., 1990, 31, 205 CrossRef.
- While this work was in progress, Hoffmann and co-workers reported the synthesis of the ethyl ester of 9 by a different route: (a) I. V. Hartung, U. Eggert, L. O. Haustedt, B. Niess, P. M. Schäfer and H. M. R. Hoffmann, Synthesis, 2003, 1844 CAS; (b) L. O. Haustedt, S. B. Panicker, M. Kleinert, I. V. Hartung, U. Eggert, B. Niess and H. M. R. Hoffmann, Tetrahedron, 2003, 59, 6967 CrossRef CAS.
- In contrast, 2-vinyloxazole-4-carboxylic acid ethyl ester is reported to be unstable to storage: W. A. Donaldson and F. Ahmed, Synth. Commun., 2003, 33, 2685 Search PubMed.
- H. Kuroda, I. Tomita and T. Endo, Synth. Commun., 1996, 26, 1539 CAS.
- J. Inanaga, Y. Baba and T. Hanamoto, Chem. Lett., 1993, 241.
- S. Cossu, O. De Lucchi and R. Durr, Synth. Commun., 1996, 26, 4597.
- W. E. Truce and G. J. W. Tichenor, J. Org. Chem., 1972, 37, 2391 CrossRef CAS.
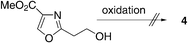
- Attempts to oxidize 2-(2-hydroxyethyl)oxazole-4-carboxylic acid methyl ester also resulted in decomposition:
. - (a) H. Tokuyama, S. Yokoshima, S.-C. Lin, L. Li and T. Fukuyama, Synthesis, 2002, 1121 CrossRef CAS; (b) T. Fukuyama, S.-C. Lin and L. Li, J. Am. Chem. Soc., 1990, 112, 7050 CrossRef CAS.
- B. M. Trost, T. N. Salzmann and K. Hiroi, J. Am. Chem. Soc., 1976, 98, 4887 CrossRef CAS.
- (a) V. K. Aggarwal and B. N. Esquivel-Zamora, J. Org. Chem., 2002, 67, 8618 CrossRef CAS; (b) V. K. Aggarwal, A. Thomas and S. Schade, Tetrahedron, 1997, 53, 16213 CrossRef CAS; (c) V. K. Aggarwal, A. Thomas and R. J. Franklin, J. Chem. Soc., Chem. Commun., 1994, 1653 RSC.
- Palladium on carbon (Pd/C) failed to catalyze the reaction. For the use of Pd(OH)2/C, see: Y. Mori and M. Seki, J. Org. Chem., 2003, 68, 1571 Search PubMed.
- For examples of enols that display intramolecular hydrogen bonding to nitrogen heterocycles, see: (a) L. W. Deady and T. Rodemann, J. Heterocycl. Chem., 2001, 38, 1185 CAS; (b) O. L. Acevedo, R. S. Andrews, M. Dunkel and P. D. Cook, J. Heterocycl. Chem., 1994, 31, 989 CAS.
- E. J. Corey and B. W. Erickson, J. Org. Chem., 1971, 36, 3553 CrossRef CAS.
- 1H NMR studies in CDCl3 revealed strong chelation of thioether 10a to Ag(I) and Zn(II) salts. This scaffold may therefore be of broad utility in applications where rapid and straightforward access to metal-binding ligands is required. For a review of thioether ligands, see: A. M. Masdeu-Bultó, M. Diéguez, E. Martin and M. Gómez, Coord. Chem. Rev., 2003, 242, 159 Search PubMed.
- A. Pfaltz, Chimia, 2001, 55, 708 CAS.
- P. Wipf and X. Wang, Org. Lett., 2002, 4, 1197 CrossRef CAS.
- A. Cevallos, R. Rios, A. Moyano, M. A. Pericàs and A. Riera, Tetrahedron: Asymmetry, 2000, 11, 4407 CrossRef CAS.
- K. Bowden and M. J. Price, J. Chem. Soc. B, 1970, 1466 RSC.
- M. Taniguchi, S. Kobayashi, M. Nakagawa, T. Hino and Y. Kishi, Tetrahedron Lett., 1986, 27, 4763 CrossRef CAS.
- (a) S. Ma and X. Lu, J. Chem. Soc., Chem. Commun., 1990, 1643 RSC; (b) S. Ma and X. Lu, Tetrahedron Lett., 1990, 31, 7653 CrossRef CAS; (c) X. Lu, X. Huang and S. Ma, Tetrahedron Lett., 1992, 33, 2535 CrossRef CAS; (d) S. Ma, X. Lu and Z. Li, J. Org. Chem., 1992, 57, 709 CrossRef CAS.
- For additional studies of the hydrohalogenation of electron-deficient alkynes, see: (a) I. Marek, A. Alexakis and J.-F. Normant, Tetrahedron Lett., 1991, 32, 5329 CrossRef CAS; (b) E. Piers, T. Wong, P. D. Coish and C. Rogers, Can. J. Chem., 1994, 72, 1816 CAS; (c) T. Zoller and D. Uguen, Tetrahedron Lett, 1998, 39, 6719 CrossRef CAS; (d) R. Takeuchi, K. Tanabe and S. Tanaka, J. Org. Chem., 2000, 65, 1558 CrossRef CAS.
- According to our DFT calculations (RB3LYP/6-31G(D); Spartan 02), (Z)-10h is 1.8 kcal mol−1 less stable than (E)-10h.
- See, for example: (a) S.-I. Murahashi, J. Organomet. Chem., 2002, 653, 27 CrossRef CAS; (b) E. Negishi, J. Organomet. Chem., 2002, 653, 34 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures, 1H and 13C NMR spectra for all new compounds. See http://www.rsc.org/suppdata/ob/b4/b413604g/ |
| This journal is © The Royal Society of Chemistry 2005 |