Organocatalysis with proline derivatives: improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions
Alexander J. A. Cobb, David M. Shaw, Deborah A. Longbottom, Johan B. Gold and Steven V. Ley*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: svl1000@cam.ac.uk; Fax: (+44) 1223-336442; Tel: (+44) 1223-336398
First published on 29th November 2004
Abstract
Tetrazole and acylsulfonamide organocatalysts derived from proline have been synthesised and applied to the asymmetric Mannich, nitro-Michael and aldol reactions to give results that are superior to the proline-catalysed counterpart.
Introduction
The search for asymmetric catalysts that provide high yields and enantioselectivities is an on-going quest for organic chemists. An important area that has been intensively studied over the past few years is that of asymmetric organocatalysis, the primary advantage of which is that it avoids the use of metals, which can be both expensive and toxic. Proline1 is one example of a versatile organocatalyst, which despite having been used effectively in the Hajos–Parrish–Eder–Sauer–Wiechert2 reaction in the 1970s has only recently received full attention in synthetic applications, such as aldol3–6, Mannich7–9 and nitro-Michael10,11 reactions. However, there are a number of drawbacks in the use of proline. First, its limited solvent compatibility; often reactions are performed in very polar solvents such as DMSO, MeOH, or H2O. Secondly a relatively high catalyst loading is usually required to effect the desired reaction in a reasonable timescale; commonly proline is used at levels of around 20 mol%.Catalyst design
The initial aim in our organocatalyst program was to design several organocatalysts with the intention of overcoming some of these problems; in particular we hoped to design a catalyst which could be used in solvents more commonly used in organic synthesis with highly lipophilic substrates.Tetrazoles are used in medicinal chemistry as bioisosteres for carboxylic acids due to the similarity in pKa as well as their increased solubility. It was hoped that replacing the carboxylic acid in proline with a tetrazole unit would give the greater solubility that was desired. This would allow a greater range of solvents to be used with the organocatalyst than it is possible to use with proline. Tetrazole 5 was synthesised according to a modified literature procedure (Scheme 1).8,12 The enantiomer of this compound was also synthesised by the same route from Z-D-proline to give tetrazole 6.
![Reagents and conditions: [a] EDCI, 1-hydroxybenzotriazole, NH3, THF, rt, 24 h; [b]
p-TsCl, pyridine, CH2Cl2, rt, 72 h; [c] NaN3, NH4Cl, DMF, 90–95 °C, 8 h; [d] 10% Pd/C, H2, AcOH–H2O, rt, 4 h. EDCI = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride.](/image/article/2005/OB/b414742a/b414742a-s1.gif) | ||
| Scheme 1 Reagents and conditions: [a] EDCI, 1-hydroxybenzotriazole, NH3, THF, rt, 24 h; [b] p-TsCl, pyridine, CH2Cl2, rt, 72 h; [c] NaN3, NH4Cl, DMF, 90–95 °C, 8 h; [d] 10% Pd/C, H2, AcOH–H2O, rt, 4 h. EDCI = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride. | ||
We also envisaged that acyl sulfonamides 9 and 10 would have increased solubility and act as alternatives to proline in organocatalytic reactions, owing to the acidity of the sulfonamidic proton.13,22 The synthesis of these compounds involved the coupling of Z-L-proline with the relevant sulfonamide (Scheme 2).
![Reagents and conditions: [a] methanesulfonamide, EDCI, DMAP, CH2Cl2, rt, 48 h; [b] benzenesulfonamide, EDCI, DMAP, CH2Cl2, rt, 48 h; [c] 10% Pd/C, H2, MeOH, rt, 20 h. DMAP = 4-dimethylaminopyridine.](/image/article/2005/OB/b414742a/b414742a-s2.gif) | ||
| Scheme 2 Reagents and conditions: [a] methanesulfonamide, EDCI, DMAP, CH2Cl2, rt, 48 h; [b] benzenesulfonamide, EDCI, DMAP, CH2Cl2, rt, 48 h; [c] 10% Pd/C, H2, MeOH, rt, 20 h. DMAP = 4-dimethylaminopyridine. | ||
Results
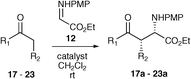
Asymmetric Mannich-type reaction
The asymmetric Mannich-type addition of a ketone directly into an imine has been the subject of much study over recent years. This reaction thus served as an excellent measure by which to compare these new organocatalysts. The reaction of cyclohexanone into the highly reactive electrophile N-PMP-protected α-imino ethyl glyoxalate 12 (synthesised from the condensation of p-anisidine with ethyl glyoxalate)14 was selected as a starting point.15 For the study various solvents were used to ascertain the solvent scope and to ascertain whether the organocatalysts were, indeed, more soluble than proline itself (Table 1).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Cat (mol%) | Solvent | T (h) | Yield (%)a | Dr syn : antib | Ee (%)c |
| a Based on isolated product.b Determined by 1H NMR spectroscopy.c Determined by chiral HPLC. | ||||||
| 1 | 5 (5) | CH2Cl2 | 2 | 65 | >19 : 1 | >99 |
| 2 | L-Pro (5) | CH2Cl2 | 2 | 0 | — | — |
| 3 | 5 (5) | Wet MeCN | 2 | 49 | >19 : 1 | >99 |
| 4 | 5 (5) | Wet THF | 2 | 37 | >19 : 1 | >99 |
| 5 | 5 (1) | CH2Cl2 | 16 | 70 | >19 : 1 | >99 |
| 6 | 9 (20) | MeOH | 24 | 74 | >19 : 1 | 95 |
| 7 | 9 (20) | CH2Cl2 | 24 | 82 | >19 : 1 | 96 |
| 8 | 9 (5) | CH2Cl2 | 24 | 65 | >19 : 1 | 83 |
| 9 | 9 (1) | CH2Cl2 | 24 | 53 | >19 : 1 | 40 |
| 10 | 10 (20) | DMSO | 24 | 87 | >19 : 1 | >99 |
| 11 | 10 (20) | THF | 24 | 87 | >19 : 1 | >99 |
| 12 | 10 (20) | MeOH | 24 | 69 | >19 : 1 | 95 |
| 13 | 10 (20) | CH2Cl2 | 24 | 75 | >19 : 1 | >99 |
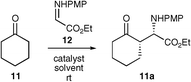
Pleasingly, all three organocatalysts catalysed the reaction in high enantioselectivities with good to excellent yields in all solvents investigated. Significantly, these catalysts worked effectively in dichloromethane, whereas L-proline failed to give any product.
Perhaps the most important observation is that tetrazole 5 gives an expeditious reaction at levels of 1 mol%, with no detrimental effect on enantioselectivity. This represents a vast improvement on L-proline, which is commonly used at levels of 20 mol%. When using organocatalyst 9, levels of 5 mol% maintained enantioselectivity, although a slightly reduced yield was observed. Tetrazole catalyst 5 therefore shows a significant advantage over sulfonamides 9 and 10 in that it can be used in reactions for shorter times, or in lower loading, to achieve similar results.
Also noteworthy is the similarity of the results using the sulfonamide catalysts 9 and 10, suggesting that the functionality appended to this moiety has little effect on the outcome of this reaction.
Overall, these promising results showed the utility of these organocatalysts and compounds 5 and 9 were used to screen the reaction of the same electrophile with a variety of ketones. Reactions with organocatalyst 5 were conducted at a level of 5 mol% for reasons of practicality and organocatalyst 9 was used at the optimised level of 20 mol% (Table 2).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Carbonyl | Cat (mol%) | T (h) | Yield (%)a | Dr syn : antib | Ee (%)c |
| a Based on isolated product.b Determined by 1H NMR spectroscopy.c Determined by chiral HPLC.d Reaction performed in MeOH.e Reaction performed in neat acetone.f Reaction stopped after 24 h at 55% conversion.g Epimerisation on the silica column led to a deterioration of dr.h Ee measured on corresponding lactone. *Indicates position of enamine formation. | ||||||
| 1 |  | 5 (5) | 16 | 63 | >19 : 1 | >99 |
| 2 | 9 (20)d | 24 | 60 | >19 : 1 | >99 | |
| 3 |  | 5 (5)e | 8 | 99 | — | >99 |
| 4 | 9 (20)d | 24 | 55 | — | 96 | |
| 5 |  | 5 (5) | 8 | 66 | >19 : 1 | >99 |
| 6 | 9 (20)d | 24 | 77 | >19 : 1 | 97 | |
| 7 |  | 5 (5) | 24 | 74 | >19 : 1 | 94 |
| 8 | 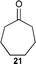 | 5 (5) | 8 | 59 | >19 : 1 | >99 |
| 9 | 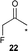 | 5 (5)f | 24 | 31 | — | 14 |
| 10 | 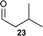 | 5 (5) | 24 | 75 | 7 : 1g | 95h |
The enantioselectivities obtained were excellent with the exception of fluoroacetone as the ketone partner (Table 2, entry 9). With this example a bi-phasic mixture occurred, giving little interaction between the ketone and the catalyst, accounting for the low yield. The low enantioselectivity, however, is attributed to fluorine interfering with the hydrogen bonded transition state thought to be necessary to give a rigid chiral environment, as suggested by Houk (Fig. 1).16
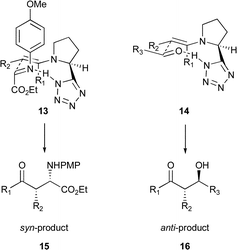 | ||
| Fig. 1 Major pathways in the Mannich and aldol reactions. | ||
In this model, the imine sits in a position where its groups are axial, avoiding any gauche interaction with the tetrazole unit 13 (Fig. 1). This means that although the enamine adopted is the E-isomer, the syn-product results. This is in contrast to the aldol reaction where the major pathway occurs with large substituents in an equatorial position, delivering predominantly the anti-product (Fig. 1).
Tetrazole catalyst 5 was shown to be just as, or more efficient, than L-proline itself. Furthermore, the reaction using this catalyst in dichloromethane appeared to give a more rapid reaction than DL-proline in DMSO as visualised by thin layer chromatography.
In summary, these organocatalysts have been shown to catalyse a Mannich-type reaction in non-polar solvents with either reduced loading or shorter reaction times, thus demonstrating greater versatility than L-proline.
Asymmetric nitro-Michael addition
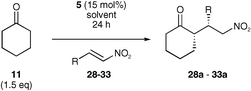
A further use of the tetrazole catalyst 5 was demonstrated with the addition of a ketone to a nitro-olefin (Scheme 3). 17 Recently, there have also been a number of investigations into nitro-Michael additions using various organocatalysts.5,10,11,18,19 Proline was one of the first to be studied and it successfully catalysed the reaction both in DMSO and alcoholic solvents. However, in most cases reported the enantioselectivities obtained were low. It was hoped that one of our organocatalysts would successfully catalyse the reaction in more conventional organic solvents and/or with a greater enantioselectivity. The reaction conditions were investigated using the reaction between cyclohexanone and β-nitrostyrene, using a variety of solvents (Table 3).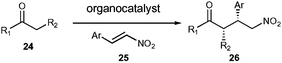 | ||
| Scheme 3 General pyrrolidine mediated nitro-Michael reaction. | ||
| ||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | T (°C) | Yield (%)a | Dr syn : antib | Ee (%)c |
| a Based on isolated product.b Determined by 1H NMR spectroscopy.c Determined by chiral HPLC.d 20 mol% used. | ||||||
| 1 | 5 | DMSO | 20 | 97 | >15 : 1 | 35 |
| 2 | L-Pro | DMSO | 20 | 93 | >15 : 1 | 35 |
| 3 | 5 | MeOH | 20 | 61 | >15 : 1 | 53 |
| 4 | L-Pro | MeOH | 20 | 37 | >15 : 1 | 57 |
| 5 | 5 | MeOH | 50 | 42 | >15 : 1 | 53 |
| 6 | 5 | CH2Cl2 | 20 | 20 | >15 : 1 | 40 |
| 7 | L-Pro | CH2Cl2 | 20 | 0 | — | — |
| 8 | 5 | CH2Cl2 | 40 | 98 | >15 : 1 | 37 |
| 9 | L-Pro | CH2Cl2 | 40 | 0 | — | — |
| 10 | 5 | THF | 20 | 33 | >15 : 1 | 25 |
| 11 | 9d | CH2Cl2 | 20 | 0 | — | — |
| 12 | 10d | CH2Cl2 | 20 | 0 | — | — |
Although the reaction did not proceed using organocatalysts 9 or 10, tetrazole 5 gave good to excellent yields and good enantioselectivities. Again, in dichloromethane, L-proline failed to give any product even at reflux and the tetrazole gave more rapid reaction in methanol than literature reports using L-proline.11 More significantly, reaction with tetrazole 5 in methanol gave the best enantioselectivities and this prompted the screening of the reaction in various mixtures of alcoholic solvents (Table 4).
| Entry | Cat. | Solvent | Cyclohexanone (eq.) | Yield (%)a,b | Ee (%)c |
|---|---|---|---|---|---|
| a Based on isolated product.b All drs were >15 : 1 by 1H NMR spectroscopy.c Determined by chiral HPLC (Daicel Chiralpak AD-H column). | |||||
| 1 | 5 | MeOH | 20 | 61 | 53 |
| 2 | 5 | MeOH–IPA (2 : 1) | 20 | 56 | 53 |
| 3 | 5 | MeOH–IPA (1 : 1) | 20 | 65 | 61 |
| 4 | 5 | MeOH–IPA (1 : 2) | 20 | 76 | 58 |
| 5 | 5 | EtOH | 20 | 65 | 65 |
| 6 | 5 | EtOH–IPA (2 : 1) | 20 | 80 | 59 |
| 7 | L-Pro | EtOH–IPA (1 : 1) | 20 | 78 | 47 |
| 8 | 5 | EtOH–IPA (1 : 1) | 20 | 96 | 62 |
| 9 | 5 | EtOH–IPA (1 : 2) | 20 | 100 | 56 |
| 10 | 5 | IPA | 20 | 80 | 40 |
| 11 | L-Pro | EtOH–IPA (1 : 1) | 1.5 | 52 | 51 |
| 12 | 5 | EtOH–IPA (1 : 1) | 1.5 | 80 | 62 |
The best overall conditions were shown to be those using equal amounts of ethanol and isopropanol. It was these conditions that were used for further optimisation of the system, where variations in the amount of ketone, and organocatalyst were investigated. The reactions with organocatalysts 9 and 10 were also repeated under the optimised ethanol–isopropanol conditions, but no product was observed.
It is clear that in the case of the alcoholic solvents, reducing the amount of catalyst lowers the yield. In dichloromethane this is not the case. Nevertheless, the decrease in catalyst loading does not seem to effect the enantioselectivities in either case (Table 5, entries 1 and 9). Decreasing the amount of ketone in the reaction lowers the yield of product in the case of the alcoholic solvent system (Table 5, entry 4), but again, this reduction seems to have little effect in dichloromethane (Table 5, entry 11).
| Entry | Cat. | mol% | Cyclohexanone (eq.) | Solvent | T (°C) | Yield (%)a,b | Ee (%)c |
|---|---|---|---|---|---|---|---|
| a Based on isolated product.b All drs were >15 : 1 by 1H NMR spectroscopy.c Determined by chiral HPLC (Daicel Chiralpak AD-H column). | |||||||
| 1 | 5 | 1 | 20 | EtOH–IPA (1 : 1) | 20 | 10 | 59 |
| 2 | 5 | 5 | 20 | EtOH–IPA (1 : 1) | 20 | 57 | 60 |
| 3 | L-Pro | 5 | 20 | EtOH–IPA (1 : 1) | 20 | 25 | 51 |
| 4 | 5 | 15 | 1.1 | EtOH–IPA (1 : 1) | 20 | 69 | 68 |
| 5 | 5 | 15 | 1.5 | EtOH–IPA (1 : 1) | 20 | 80 | 62 |
| 6 | 5 | 15 | 5 | EtOH–IPA (1 : 1) | 20 | 84 | 65 |
| 7 | 5 | 15 | 20 | EtOH–IPA (1 : 1) | 20 | 96 | 62 |
| 8 | L-Pro | 15 | 20 | EtOH–IPA (1 : 1) | 20 | 78 | 47 |
| 9 | 5 | 1 | 20 | CH2Cl2 | Reflux | 62 | 46 |
| 10 | 5 | 5 | 20 | CH2Cl2 | Reflux | 100 | 44 |
| 11 | 5 | 15 | 1.1 | CH2Cl2 | Reflux | 86 | 49 |
| 12 | 5 | 15 | 5 | CH2Cl2 | Reflux | 68 | 51 |
| 13 | 5 | 15 | 10 | CH2Cl2 | Reflux | 64 | 37 |
In alcoholic solvents, tetrazole outperforms proline, both in terms of product yield and enantioselectivity (5 provides almost a 20% better yield than L-proline and a 15% improvement in ee. Table 5, entries 7 and 8).
Despite the obvious superiority of yields in dichloromethane, the better enantioselectivities obtained in the alcoholic solvent led to this being the system of choice for further investigations into the scope of this reaction. The best system was that using ethanol–isopropanol (1 : 1) and 1.5 equivalents of ketone. Several nitro-olefins were screened under these conditions using cyclohexanone as the ketone and the results are shown (Table 6). Reactions in dichloromethane are also shown in comparison.
| ||||
|---|---|---|---|---|
| Entry | Product | Solvent (T/°C) | Yield (%)a,b | Ee (%)c |
| a Based on isolated product.b All drs were>15 : 1 by 1H NMR spectroscopy.c Determined by chiral HPLC (Daicel Chiralpak AD-H column). | ||||
| 1 | 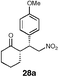 | EtOH–IPA (1 : 1), (20) | 83 | 58 |
| 2 | CH2Cl2, (reflux) | 96 | 37 | |
| 3 | 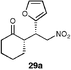 | EtOH–IPA (1 : 1) (20) | 59 | 65 |
| 4 | CH2Cl2, (reflux) | 100 | 23 | |
| 5 | 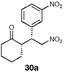 | EtOH–IPA (1 : 1), (20) | 92 | 65 |
| 6 | 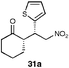 | EtOH–IPA (1 : 1), (20) | 74 | 57 |
| 7 | 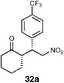 | EtOH–IPA (1 : 1), (20) | 58 | 55 |
| 8 |  | EtOH–IPA (1 : 1), (20) | 47 | 60 |
It was found that under these conditions, the yields were generally good and ranged between 47% and 92%. However, the substituent of the nitro-olefin appears to have little effect on the enantioselectivities of the reaction, which range from 55% to 65%. The relative configuration of compound 27a has been confirmed by X-ray crystallographic methods (Fig. 2).†
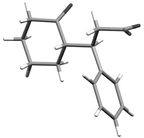 | ||
| Fig. 2 X-Ray structure of adduct 27a. | ||
β-3-Dinitrostyrene was found to produce the best result providing an excellent yield and good enantioselectivity (entry 5). It was this Michael acceptor that was used to investigate the scope of ketones within the reaction (Table 7).
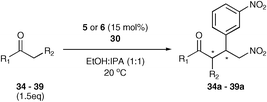
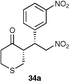
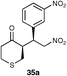
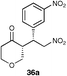
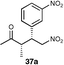
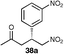
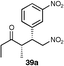
The results of this study showed that in general, cyclic ketones performed best, with the exception of 3-pentanone (entry 7) which gave good enantioselectivity. The best example was that using tetrahydrothiopyran-4-one as the ketone which gave the corresponding Michael adduct 34a in 62% yield and 70% enantiomeric excess (Table 7, entry 2). This is in stark contrast with L-proline which falls short of the tetrazole-induced enantioselectivity by 30% (Table 7, entry 1). The enantiomeric tetrazole 6 was also applied to this example to give the product of the opposite stereochemistry, in a comparable yield and enantioselectivity (Table 7, entry 3).
The improvement in enantioselectivity of the tetrazole catalysts 5 and 6 over proline, suggests that there is an inherent difference between the two organocatalysts that alters the transition state. On one hand, if the tetrazole participates in a hydrogen bonded framework as is suggested by Enders for proline (Fig. 3),11 then it would be expected for them to give similar enantioselectivities unless there is an inherent difference in the hydrogen bonding strengths between tetrazole catalyst 5 and L-proline, resulting in a tighter transition state. The hydrogen bonding strength is, of course, affected by the solvent and this would be consistent with the observed range of enantioselectivities in the various solvents investigated.
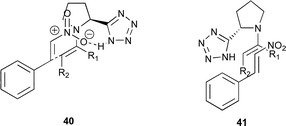 | ||
| Fig. 3 Potential transition states. | ||
A second explanation is that the slightly larger tetrazole moiety occupies a larger region of space than a carboxylic acid, thereby providing more of a facial preference for an approaching substrate. This explanation has been used recently to explain the performance of some other organocatalysts in the same reaction (Fig. 3).18
In conclusion, tetrazole organocatalysts 5 and 6 have been shown to catalyse the asymmetric addition of a ketone to a nitro-olefin to better yields and enantioselectivities than L-proline itself. This demonstrates the advantages of having an organocatalyst that can be used under a diversity of conditions and in a wider solvent scope than the previously limited L-proline reactions.
Asymmetric aldol reaction
Following our initial publication of organocatalyst 58, its application in the asymmetric aldol reaction has been studied thoroughly.19,20,21 Here we report only our investigations with organocatalysts 9 and 10 using a range of ketones with p-nitrobenzaldehyde as a test to measure their utility (Table 8).22
| |||||
|---|---|---|---|---|---|
| Entry | Product | Cat. | Solvent | Yield (%)asyn ( anti) | Ee (%)csyn (anti) |
| a Based on isolated product.b Determined by chiral HPLC.c Reaction stirred for 7 days. | |||||
| 1 |  | 9 | DMSO | 52 | 87 |
| 2 | 9 | CH2Cl2 | 78 | 79 | |
| 3 | 9 | MeOH | 49 | 49 | |
| 4 | 9 | CHCl3 | 67 | 78 | |
| 5 | 9 | IPA–EtOH | 57 | 44 | |
| 6 |  | 10 | CH2Cl2 | 49 | 84 |
| 7 | 10 | MeOH | 42 | 61 | |
| 8 | 10 | CHCl3 | 62 | 65 | |
| 9 | 10 | IPA–EtOH | 39 | 75 | |
| 10 | 10 | Acetone | 100 | 92 | |
| 11 |  | 9 | CH2Cl2 | 42c | 76 |
| 12 | 10 | CH2Cl2 | 48c | 77 | |
| 13 |  | 9 | CH2Cl2 | 24 (46) | 78 (84) |
| 14 | 10 | CH2Cl2 | 21 (43) | 86 (94) | |
| 15 |  | 9 | CH2Cl2 | 30 (55) | 41 (36) |
| 16 | 10 | CH2Cl2 | 30 (54) | 33 (23) | |
| 17 |  | 9 | CH2Cl2 | 29 (51) | 74 (78) |
| 18 | 10 | CH2Cl2 | 35 (53) | 63 (90) | |
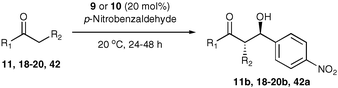
Firstly, a solvent screen was investigated with 9 and 10 and optimum results were observed in methylene chloride, providing excellent enantioselectivity and practical reaction times (Table 8). Alcoholic solvents were observed to give reduced enantioselectivities, consistent with the interruption of a hydrogen bonded transition state (Fig. 1) and also promoted the elimination of the product. Aprotic solvents gave comparably high enantioselectivities, although dichloromethane was shown to give the highest selectivity and reaction rate of the apolar solvents studied, with the sulfonamide catalysts observed to have even greater solubility than the tetrazole catalyst 5. The excellent enantioselectivity observed in DMSO shows that it is indeed the sulfonamide catalyst that is superior to proline in this asymmetric aldol reaction, not just in solubility, but also in enantioselectivity, (87% with catalyst 9 compared to 76% with L-proline).
A range of ketones were then explored and the results summarised in Table 8. Excellent enantioselectivities were observed for straight chain and cyclic ketones, although the diastereomeric ratios observed with cyclic ketones remain an unsolved problem. Noteworthy examples include that of 19b with an observed enantioselectivity of 77% compared to that of L-proline 59% and 11b where an enantioselectivities of 90% and 68% (entries 17 and 28) are observed for the anti and syn products respectively with catalyst 10, compared with 63% and 36% for L-proline. Excellent yields and enantioselectivities were also produced for the syn and anti aldol products from the reaction of cyclobutanone 20 (entries 13 and 14). Only cyclopentanone 42 proved a disappointing substrate in this reaction, providing moderate enantioselectivities.
A possible rationale of these marked increases in enantioselectivity through a hydrogen bonded Houk transition state (Fig. 1) is through the increased pKa of the sulfonamide proton giving a stronger hydrogen bond to the carbonyl moiety and subsequently a more tightly bound transition state, leading to greater selectivity. It is hard to envisage a steric argument for the differences between proline and the sulfonamide catalysts 9 and 10, as well as between the sulfonamide catalysts themselves, as the relevant groups are too distant from the point of chiral induction. The differences between the sulfonamide catalysts 9 and 10 can also be rationalised by the opposing electron donating and withdrawing nature of the methyl and phenyl substituents, consistent with 10 having the lowest pKa and giving predominantly the best enantioselectivities.
The effect of changing DMSO for methylene chloride could also be a factor in the strength of hydrogen bonding in the transition state (supported by the difference in enantioselectivity shown for example 18a, Table 8) as DMSO is likely to alter the pKa of the sulfonamidic proton, effecting the enantioselectivity as previously discussed.
Conclusions
New tetrazole and sulfonamide derivatised proline catalysts have been shown to give good to excellent yields and enantioselectivities in a range of important transformations. In all cases studied these new catalysts gave superior or equivalent results in terms of enantioselectivity, catalyst loading, solvent tolerance and reaction times, when compared with proline itself.We are currently investigating rational design of new catalysts for enamine derived reactions and the application to combinatorial and multi-step synthesis, the results of which will be published in due course.
Experimental
All reactions were carried out in freshly distilled solvent under an atmosphere of Argon unless otherwise stated. Dichloromethane, toluene, methanol and tetrahydrofuran were distilled from calcium hydride. All other solvents were anhydrous grade and used as received. All other reagents were used as received. Flash column chromatography was carried out using Merck 60 Kieselgel (230–400 mesh) under pressure. Analytical thin layer chromatography (TLC) was performed on glass plates pre-coated with Merck Kieselgel 60 F254, and visualised by ultra-violet irradiation (254 nm) or by staining with aqueous acidic ammonium hexamolybdate, or aqueous acidic potassium permanganate solutions as appropriate. Melting points were performed on a Reichert hot-stage apparatus, and are uncorrected. Optical rotations were recorded on a Perkin-Elmer 343 digital polarimeter using a sodium lamp (589 nm) as the light source. Infra-red spectra were obtained on a Spectrum One FT-IR ATR (Attenuated Total Reflectance) spectrometer, from a thin film deposited on the ATR. Mass spectra and accurate mass data were obtained on a Micromass Platform LC-MS, Kratos MS890MS, Kratos Concept IH, Micromass Q-TOF, or Bruker BIOAPEX 4.7 T FTICR spectrometer, by electron ionisation, chemical ionisation or fast atom/ion bombardment techniques at the Department of Chemistry, Lensfield Road, Cambridge. 1H NMR spectra were recorded at ambient temperature on Bruker DPX-400, Bruker DRX-500 or Bruker DRX-600 spectrometers at 400, 500 or 600 MHz with residual protic solvent CHCl3 as the internal reference (δH = 7.26 ppm); Chemical shifts (δ) are given in parts per million (ppm) and coupling constants (J) are given in Hertz (Hz). The proton spectra are reported as follows δ/ppm (number of protons, multiplicity, coupling constant J/Hz, assignment). 13C NMR spectra were recorded at ambient temperatures on the same spectrometers at 100, 125 or 150 MHz, with the central peak of CHCl3 as the internal reference (δC = 77.0 ppm). Where rotamers are apparent, peaks for major and minor rotamers are reported, when resolved. DEPT135 and two dimensional (COSY, HMQC, HMBC) NMR spectroscopy were used where appropriate, to aid the assignment of signals in the 1H and 13C NMR spectra. Where a compound has been characterised as an inseparable mixture of diastereoisomers, the NMR data for the major isomer has been reported as far as was discernable from the spectrum of the mixture. Where coincident coupling constants have been observed in the 1H NMR spectrum, the apparent multiplicity of the proton resonance concerned has been reported. Evaporation refers to the removal of solvent under reduced pressure.(2S)-2-Carbamoyl-pyrrolidine-1-carboxylic acid benzyl ester212
To a solution of Z-L-proline (4.00 g, 16.1 mmol, 1 eq.) in THF (80 mL) were added 1-hydroxybenzotriazole (3.26 g, 24.1 mmol, 1.5 eq.) and EDCI (3.08 g, 16.1 mmol, 1 eq.). The resulting mixture was stirred at room temperature for 30 min whereupon aqueous ammonia (11 mL) was added slowly by syringe. The resulting mixture was allowed to stir for 24 h. After this time, saturated aqueous ammonium chloride (100 mL) was added and the aqueous phase extracted with ethyl acetate (3 × 50 mL). The combined organic layers were dried (MgSO4), filtered and evaporated to give a pale yellow oil, which was purified by flash column chromatography (EtOAc–petroleum ether 40/60, 1 : 4) to give the title compound as a clear colourless oil (3.98 g, 100%). [α]D = −82.8° (c = 0.50, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.28 (5H, m, ArH), 6.71 (1H, s, NHH′), 5.80 (1H, s, NHH′), 5.19 (1H, d, J = 12 Hz, ArCHH′), 5.15 (1H, d, J = 12 Hz, ArCHH′), 4.39 (1H, m, NCHC(O)), 3.59–3.45 (2H, m, NCH2), 2.40–1.90 (4H, m, CH2CH2).(2S)-2-Cyano-pyrrolidine-1-carboxylic acid benzyl ester123
To a solution of (2S)-2-carbamoyl-pyrrolidine-1-carboxylic acid benzyl ester 2 (3.98 g, 16.1 mmol, 1 eq.) in dichloromethane (40 mL) at room temperature was added pyridine (6.49 mL, 80.2 mmol, 5 eq.) followed by neat tosyl chloride (6.13 g, 32.1 mmol, 2 eq.). The resulting mixture was allowed to stir for 72 h after which time, saturated aqueous ammonium chloride (30 mL) was added. The aqueous layer was extracted with ethyl acetate (3 × 50 mL) and the combined organic layers were dried (MgSO4), filtered and evaporated in vacuo to give a yellow oil which was purified by flash column chromatography (EtOAc–petroleum ether 40/60, 7 : 3) to give the title compound as a pale yellow oil (2.80 g, 75%). [α]D = −89.0° (c = 1.00, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.42–7.31 (5H, m, ArH), 5.22–5.14 (2H, m, ArCH2), 4.61–4.54 (1H, dd, J = 26.4, 5.5 Hz, NCHCO), 3.60–3.57 (1H, m, NCHH′), 3.46–3.39 (1H, m, NCHH′), 2.39–2.04 (4H, m, CH2CH2).(2S)-2-(1H-Tetrazol-5-yl)-pyrrolidine-1-carboxylic acid benzyl ester124
To a solution of (2S)-2-cyano-pyrrolidine-1-carboxylic acid benzyl ester 3 (1.50 g, 6.52 mmol, 1 eq.) in DMF (15 mL) were added sodium azide (440 mg, 6.78 mmol, 1.04 eq.) and ammonium chloride (380 mg, 7.17 mmol, 1.1 eq.). The resulting mixture was heated to 90 °C for 8 h. After this time, the reaction was allowed to cool to room temperature and acidified to pH 2 with 1 M aqueous HCl. The aqueous layer was extracted with chloroform (3 × 25 mL) and the combined organic layers were washed with saturated aqueous lithium chloride (50 mL). The organic layer was then dried (MgSO4), filtered and evaporated in vacuo to give the title compound analytically pure as a clear colourless residue (1.39 g, 78%). [α]D = −85.7° (c = 1.18, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.35 (5H, m, ArH), 7.06 (1H, s, NH), 5.40–4.98 (3H, m, ArCH2 and NCHCN), 3.66–3.45 (2H, m, NCH2), 2.62–1.86 (4H, m, CH2CH2).(2S)-5-Pyrrolidin-2-yl-1H-tetrazole125
(2S)-2-(1H-Tetrazol-5-yl)-pyrrolidine-1-carboxylic acid benzyl ester 4 (1.40 g, 5.08 mmol, 1 eq.) and 10% Pd/C (279 mg) in acetic acid–water (9 : 1, 75 mL) were stirred under an atmosphere of hydrogen at room temperature for 4 h. After this time the mixture was filtered through Celite® and the filtrate evaporated in vacuo. The residue was azeotroped using toluene to aid removal of acetic acid. The resulting solid was recrystallised with a mixture of toluene and methanol to give the title compound as an off-white solid (590 mg, 89%). [α]D = +1.2° (c = 0.5, H2O). 1H NMR (400 MHz, CD3COOD) δ = 4.95 (1H, t, J = 8.1 Hz, NHCHCN), 3.45 (2H, m, NHCH2), 2.60–2.16 (4H, m, CH2CH2).(2R)-2-Carbamoyl-pyrrolidine-1-carboxylic acid benzyl ester23
Synthesised in the same way as described above for 2 from Z-D-proline (8.00 g, 32.1 mmol). Purified by flash column chromatography (EtOAc–petroleum ether 40/60, 1 : 4) to give the title compound as a white solid (5.43 g, 68%). Mp = 289–291 °C, [α]D = −82.8° (c = 0.50, CHCl3). 1H NMR (600 MHz, CDCl3) δ = 7.34 (5H, m, ArH), 6.70 (1H, s, NH, major rot), 6.04 (1H, broad, s, NH, minor rot), 5.87 (1H, broad, s, NH, minor rot) 5.80 (1H, broad, s, NH, major rot), 5.15 (1H, d, J = 12.3 Hz, ArCHH′), 5.15 (1H, d, J = 12.3 Hz, ArCHH′), 4.35 (1H, m, NCHC(O)), 3.53–3.44 (2H, m, NCH2), 2.31–1.89 (4H, m, CH2CH2).(2R)-2-Cyano-pyrrolidine-1-carboxylic acid benzyl ester. (ent-3)
Synthesised in the same way as described above for 3 from (2R)-2-carbamoyl-pyrrolidine-1-carboxylic acid (5.43 g, 21.8 mmol) to yield the title compound as a pale yellow oil (3.22 g, 64%). νmax (film)/cm−1 2959, 2883, 1701, 1405, 1355, 1118, 1091. [α]D = +89.2° (c = 0.47, CHCl3). 1H NMR (600 MHz, CDCl3) δ = 7.42–7.31 (5H, m, ArH), 5.22–5.14 (2H, m, ArCH2), 4.61–4.54 (1H, m, NCHCN), 3.60–3.57 (1H, m, NCHH′), 3.46–3.39 (1H, m, NCHH′), 2.39–2.04 (4H, m, CH2CH2). 13C NMR (150 MHz, CHCl3) (major and minor rotamers) δ = 154.3, 153.6, 136.1, 136.0, 128.5, 128.2, 128.1, 118.9, 118.7, 67.8, 67.6, 47.5, 47.0, 46.3, 45.9, 31.7, 30.8, 30.3, 24.6, 23.7. m/z (ES) found 231.1134 ([MH]+ C13H15N2O2 requires 231.1128).(2R)-2-(1H-Tetrazol-5-yl)-pyrrolidine-1-carboxylic acid benzyl ester. (ent-4)
Synthesised in the same way as described above for 4 from (2R)-2-cyano-pyrrolidine-1-carboxylic acid benzyl ester (3.22 g, 14.0 mmol) to yield the title compound analytically pure (3.12 g, 82%). νmax (film)/cm−1 2957, 2919, 2851, 1663, 1411, 1355, 1120, 1099, 696. [α]D = +89.0° (c = 0.96, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.35 (5H, m, ArH), 7.06 (1H, s, NH), 5.40–4.98 (3H, m, ArCH2 and NCHCN), 3.66–3.45 (2H, m, NCH2), 2.62–1.86 (4H, m, CH2CH2). 13C NMR (100 MHz, CHCl3) δ = 156.3, 135.7, 128.6, 128.3, 127.8, 68.0, 52.6, 51.4, 47.0, 33.0, 24.6. m/z (ES) found 274.1304 ([MH]+ C13H16N5O2 requires 274.1299).(2R)-5-Pyrrolidin-2-yl-1H-tetrazole 6
Synthesised in the same way as described above for 5 from (2R)-2-(1H-tetrazol-5-yl)-pyrrolidine-1-carboxylic acid benzyl ester (3.12 g, 11.4 mmol) to yield the title compound as an off white solid (1.37 g, 84%). Mp = 272–274 °C. νmax (film)/cm−1 2940, 2580, 2460, 1627, 1456, 1420, 1396, 1046, 1012, 954. [α]D = −0.8° (c = 1.00, H2O). 1H NMR (400 MHz, CD3COOD) δ = 4.95 (1H, t, J = 8.1 Hz, NHCH), 3.45 (2H, m, NHCH2), 2.60–2.16 (4H, m, CH2CH2). 13C NMR (100 MHz, CD3COOD) δ = 156.5, 54.7, 46.6, 30.3, 23.6. m/z (EI) found 139.0856 ([M]+ C5H9N5 requires 139.0858).(2S)-2-Methanesulfonylaminocarbonyl-pyrrolidine-1-carboxylic acid benzyl ester 7
To a stirred solution of Z-L-proline (5.00 g, 20.1 mmol, 1 eq.) in dichloromethane (150 mL) were added methanesulfonamide (2.10 g, 22.1 mmol, 1.1 eq.), DMAP (380 mg, 3.11 mmol, 0.15 eq.) and EDCI (3.85 g, 20.1 mmol, 1 eq.) respectively. The resulting mixture was stirred at room temperature for 2 days. The reaction was concentrated to half the volume in vacuo and the resulting mixture was partitioned between EtOAc (250 mL) and 1 M aqueous HCl (100 mL). The organic layer was washed with half-saturated brine (50 mL), dried (NaSO4) and concentrated in vacuo. The crude product was purified by flash column chromatography (dichloromethane–EtOAc, 7 : 3) to give the title compound as a clear colourless residue (3.92 g, 60%). The crude product may be used directly in the next step. νmax (film)/cm−1 3207, 2963, 1674, 1416, 1336, 1121, 969, 697. [α]D = −86.4° (c = 2.10, CHCl3). 1H NMR (500 MHz CDCl3, major rotamer) δ = 10.1 (1H, broad, s, NH), 7.36 (5H, m, ArH), 5.21 (1H, d, J = 12.2 Hz, CHH′Ar), 5.15 (1H, d, J = 12.2 Hz, CHH′Ar), 4.36 (1H, m, NHCH), 3.46 (2H, m, NHCH2), 3.25 (3H, s, CH3), 2.46 (1H, s, CHCHH′), 1.94 (3H, m, CH2CHH′). 13C NMR (125 MHz, CDCl3) δ = 170.2, 157.2, 135.7, 128.6, 128.5, 128.2, 68.2, 61.1, 47.3, 41.3, 26.9, 24.5. m/z (ES) found 327.1018 ([MH]+ C14H19N2O5S requires 327.1015).(2S)-2-Benzenesulfonylaminocarbonyl-pyrrolidine-1-carboxylic acid benzyl ester 8
To a stirred solution of Z-L-proline (5.00 g, 20.1 mmol, 1 eq.) in dichloromethane (150 mL) were added benzenesulfonamide (3.16 g, 20.1 mmol, 1 eq.), DMAP (400 mg, 3.28 mmol, 0.16 eq.) and EDCI (3.85 g, 20.1 mmol, 1 eq.) respectively. The resulting mixture was stirred at room temperature for 2 days before being partitioned between EtOAc (250 mL) and 1 M aqueous HCl (100 mL). The organic layer was washed with half-saturated brine, dried (NaSO4) and concentrated in vacuo. The residue was treated with dichloromethane and the resulting white solid was filtered-off. Following evaporation in vacuo the crude product was purified by flash column chromatography (dichloromethane–EtOAc 7 : 3) to give the title compound as an off white solid (4.92 g, 63%). The crude product may be used directly in the next step. Mp = 196–197 °C, νmax (film)/cm−1 3063, 2955, 2882, 1673, 1448, 1416 1351, 1185, 1174, 1123, 1084, 686. [α]D = −87.5° (c = 0.24, CHCl3). 1H NMR (500 MHz, CDCl3, major rotamer) δ = 10.5 (1H, broad, s, NH), 8.06 (2H, d, J = 7.4 Hz, SO2ArH), 7.65 (1H, t, J = 7.4 Hz, SO2ArH), 7.54 (2H, t, J = 7.4 Hz, SO2ArH), 7.42 (5H, m, CH2ArH), 5.24 (2H, s, CH2Ar), 4.32 (1H, d, J = 6.8 Hz, NCH), 3.42 (2H, m, NCH2), 2.45 (1H, d, J = 8.2 Hz, CH2CH2CHH′), 1.90 (3H, m, CH2CH2CHH′). 13C NMR (125 MHz, CDCl3) δ = 168.7, 157.5, 138.7, 135.8, 133.8, 128.9, 128.7, 128.5, 128.4, 128.3, 68.3, 60.8, 47.2, 26.8, 24.4. m/z (ES) found 389.1171 ([MH]+ C19H21N2O5S requires 389.1171).(2S)-N-(2-Pyrrolidine-2-carbonyl)-methanesulfonamide 9
To a solution of (S)-2-methanesulfonylaminocarbonyl-pyrrolidine-1-carboxylic acid benzyl ester 7 (1.00 g, 3.06 mmol, 1 eq.) in MeOH (100 mL) was added 10% Pd/C (180 mg). The mixture was stirred at room temperature for 20 hours under an atmosphere of hydrogen. The reaction was filtered through Celite® and 1 cm of silica gel and the filtrate concentrated in vacuo. The crude product was purified by flash column chromatography (dichloromethane–MeOH, 8 : 2) to give the title compound as a white solid (576 mg, 98%). Mp 214–216 °C; νmax (film)/cm−1 3096, 1575, 1253, 1109, 834. [α]D = −42.6° (c = 1.03, DMSO). 1H NMR (500 MHz, DMSO) δ = 8.55 (2H, broad, s, CH2NH, SO2NH), 3.82 (1H, dd, J = 7.0, 8.3 Hz, CHC(O)), 3.21 (1H, m, NHCHH′), 3.06 (1H, m, NHCHH′), 2.79 (3H, s, CH3), 2.12–1.74 (4H, m, CH2CH2). 13C NMR (125 MHz, DMSO) δ = 23.5, 29.3, 40.2, 45.4, 62.0, 172.0. m/z (ES) found 193.0643 ([MH]+ C6H13N2O3S requires 193.0647).(2S)-N-(2-Pyrrolidine-2-carbonyl)-benzenesulfonamide 10
To a solution of (S)-2-benzenesulfonylaminocarbonyl-pyrrolidine-1-carboxylic acid benzyl ester 8 (5.85 g, 15.0 mmol) in MeOH (300 mL) was added 10% Pd/C (900 mg). The mixture was stirred at room temperature for 20 h under an atmosphere of hydrogen. The reaction was filtered through Celite® and 1 cm of silica gel, and the filtrate was concentrated in vacuo to give a white solid. The crude product was purified by flash column chromatography (dichloromethane–MeOH 8 : 2) to give the title compound as a white solid (2.01 g, 91%). Mp 237–239 °C; [α]D = −21.1° (c = 1.00, DMSO). νmax (film)/cm−1 3066, 1623, 1576, 1256, 1130, 1083, 831, 690. 1H NMR (500 MHz, DMSO) δ = 8.51 (2H, broad, s, NH), 7.78 (2H, dd, J = 10.4, 2.7 Hz, ArH), 7.41 (3H, m, ArH), 3.82 (1H, dd, J = 6.8, 8.3 Hz, CHC(O)), 3.14 (1H, m, NHCHH′), 3.03 (1H, m, NHCHH′), 2.10–1.67 (4H, m, CH2CH2). 13C NMR (125 MHz, DMSO) δ = 23.4, 29.1, 45.3, 62.0, 126.9, 127.8, 130.3, 145.3, 171.4. m/z (ES) found 255.0815 ([MH]+ C11H15N2O3S requires 255.0803).N-p-Methoxybenzyl-α-iminoglyoxalate1412
Methyl glyoxalate (8.14 mL, 50% sol in toluene, 40 mmol) was dissolved in dichloromethane (150 mL) and a solution of p-anisidine (4.92 g, 40 mmol) in dichloromethane (50 mL) was added slowly. The reaction mixture was stirred at room temperature for 30 min and pre-activated 4 Å molecular sieves were added. After stirring for an additional 1 h, the mixture was filtered and the filtrate evaporated in vacuo to give the title compound, analytically pure, as a yellow oil (8.20 g, 99%). 1H NMR (400 MHz, CDCl3) δ = 7.95 (1H, s, HC(N)), 7.38 (2H, d, J = 8.8 Hz, ArH), 6.92 (2H, d, J = 8.8 Hz, ArH), 4.42 (2H, q, J = 6.9 Hz, CH2CH3), 3.84 (3H, s, OCH3), 1.41 (3H, t, J = 6.9 Hz, CH2CH3).General procedure for the addition of a carbonyl-containing compound to N-p-methoxybenzyl-α-iminoglyoxalate 12
N-p-Methoxybenzyl-α-iminoglyoxalate 12 (93.5 mg, 0.5 mmol) was dissolved in dichloromethane (4 mL). Carbonyl-containing compound (1 mL, 20 vol%) was added to this solution followed by 5-pyrrolidin-2-(S)-yl-1H-tetrazole 5 (3.5 mg, 5 mol%) or N-((S)-pyrrolidine-2-carbonyl)-benzenesulfonamide (25.5 mg, 20 mol%) and the resulting mixture stirred for 2–24 h. After this time, the mixture was quenched with saturated aqueous ammonium chloride (10 mL) and extracted with ethyl acetate (2 × 25 mL). The combined organic layers were dried (MgSO4), filtered and evaporated in vacuo to give a residue, which was purified by flash column chromatography using varying mixtures of ethyl acetate and petroleum ether 40/60 as eluent.General procedure for the conjugate addition of a ketone to a nitro-olefin
To a suspension of 5-pyrrolidin-2-yl-1H-tetrazole 5 or 6 (10.5 mg, 15 mol%) and nitro-olefin (0.5 mmol) in isopropanol–ethanol (1 : 1, 4 mL) was added the relevant ketone (0.75 mmol). The resulting mixture was allowed to stir at room temperature for 24 h, whereupon the reaction was quenched with saturated aqueous ammonium chloride (25 mL) and the aqueous layer extracted with ethyl acetate (2 × 25 mL). The combined organic layers were dried over MgSO4, filtered and evaporated in vacuo to give an oil which was purified by flash column chromatography using varying amounts of ethyl acetate and petroleum ether 40/60 as eluent.General procedure for the sulfonamide catalysed aldol reaction
To a solution of p-nitrobenzaldehyde (151 mg, 1 mmol, 1 eq.) in dichloromethane (8 mL) was added the relevant ketone (2 mL, 20 vol%) and N-(pyrrolidin-2-(S)-carbonyl)-methylsulfonamide 9 (38.6 mg, 0.2 mmol, 20 mol%) or N-(pyrrolidin-2-(S)-carbonyl)-benzenesulfonamide 10 (51.0 mg, 0.2 mmol, 20 mol%) respectively. The solution was stirred at room temperature and evaporated in vacuo after the time reported. The resulting yellow/orange residue was purified by flash column chromatography using varying amounts of ethyl acetate and petroleum ether 40/60 as eluent.Acknowledgements
We thank Pentraxin Therapeutics for funding (to A. J. A. C.), AstraZeneca (to D. M. S.), EPSRC (to D. A. L.) and Dr J. Davies for X-ray crystallographic analysis.References
- (a) M. Movassaghi and E. N. Jacobsen, Science, 2002, 298, 1904 CrossRef CAS; (b) B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS; (c) B. List, Synlett, 2001, 1675 CrossRef CAS; (d) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS.
- (a) Z. G. Hajos and D. R. Parrish, Angew. Chem., Int. Ed. Engl., 1974, 39, 1615 CAS; (b) U. Eder, R. Wiechert and G. Sauer: German Patent DE 2014757, 1971; (c) U. Eder, G. Sauer and R. Wiechert, J. Org. Chem., 1971, 10, 496 CAS; (d) Z. G. Hajos and D. R. Parrish: German Patent DE 2102623, 1971.
- (a) S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475 CrossRef CAS; (b) A. Bogevig, N. Kumaragurubaran and K. A. Jorgensen, Chem. Commun., 2002, 620 RSC; (c) V. Chan, J. G. Kim, C. Jimeno, P. J. Carroll and P. J. Walsh, Org. Lett., 2004, 6, 2051 CrossRef CAS; (d) A. Cordova, W. Notz and C. F. Barbas III, Chem. Commun., 2002, 3024 RSC; (e) Y. Hayashi, W. Tsuboi, M. Shoji and N. Suzuki, Tetrahedron Lett., 2004, 45, 4353 CrossRef CAS; (f) L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16 CrossRef CAS; (g) J. Kofoed, J. Nielsen and J. L. Reymond, Bioorg. Med. Chem. Lett., 2003, 13, 2445 CrossRef CAS; (h) P. Kotrusz, I. Kmentova, B. Gotov, S. Toma and E. Solcaniova, Chem. Commun., 2002, 2510 RSC; (i) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (j) B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573 CrossRef CAS; (k) B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839 CrossRef CAS; (l) T. P. Loh, L. C. Feng, H. Y. Yang and J. Y. Yang, Tetrahedron Lett., 2002, 43, 8741 CrossRef CAS; (m) H. J. Martin and B. List, Synlett, 2003, 1901 CAS; (n) M. Nakadai, S. Saito and H. Yamamoto, Tetrahedron, 2002, 58, 8167 CrossRef CAS; (o) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798 CrossRef CAS; (p) A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152 CrossRef CAS; (q) Y. Y. Peng, Q. P. Ding, Z. C. Li, P. G. Wang and J. P. Cheng, Tetrahedron Lett., 2003, 44, 3871 CrossRef CAS; (r) C. Pidathala, L. Hoang, N. Vignola and B. List, Angew. Chem., Int. Ed., 2003, 42, 2785 CrossRef CAS; (s) S. Saito, M. Nakadai and H. Yamamoto, Synlett, 2001, 1245 CrossRef CAS; (t) Y. Sekiguchi, A. Sasaoka, A. Shimomoto, S. Fujioka and H. Kotsuki, Synlett, 2003, 1655; (u) Z. Tang, F. Jiang, L. T. Yu, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, J. Am. Chem. Soc., 2003, 125, 5262 CrossRef CAS; (v) Z. Tang, F. Jiang, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5755 CrossRef CAS.
- A. Cordova and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 7749 CrossRef CAS.
- N. Mase, F. Tanaka and C. F. Barbas III, Angew. Chem., Int. Ed., 2004, 43, 2420 CrossRef CAS.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS.
- (a) B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS; (b) A. Cordova, Acc. Chem. Res., 2004, 37, 102 CrossRef CAS; (c) W. Notz, F. Tanaka, S. Watanabe, N. S. Chowdari, J. M. Turner, R. Thayumanavan and C. F. Barbas III, J. Org. Chem., 2003, 68, 9624 CrossRef CAS; (d) P. Pojarliev, W. T. Biller, H. J. Martin and B. List, Synlett, 2003, 1903 CAS; (e) N. S. Chowdari, D. B. Ramachary and C. Barbas III, Synlett, 2003, 1906 CAS; (f) A. Cordova, Synlett, 2003, 1651 CrossRef CAS; (g) Y. Hayashi, W. Tsuboi, M. Shoji and N. Suzuki, J. Am. Chem. Soc., 2003, 125, 11208 CrossRef CAS; (h) Y. Hayashi, W. Tsuboi, I. Ashimine, T. Urushima, M. Shoji and K. Sakai, Angew. Chem., Int. Ed., 2003, 42, 3677 CrossRef CAS; (i) A. Cordova and C. F. Barbas III, Tetrahedron Lett., 2003, 44, 1923 CrossRef CAS; (j) S. Watanabe, A. Cordova, F. Tanaka and C. F. Barbas III, Org. Lett., 2002, 4, 4519 CrossRef CAS; (k) A. Cordova and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 7749 CrossRef CAS; (l) A. Cordova, S. Watanabe, F. Tanaka, W. Notz and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1866 CrossRef CAS; (m) B. List, P. Pojarliev, W. T. Biller and H. J. Martin, J. Am. Chem. Soc., 2002, 124, 827 CrossRef CAS; (n) W. Notz, K. Sakthivel, T. Bui, G. F. Zhong and C. F. Barbas III, Tetrahedron Lett., 2001, 42, 199 CrossRef.
- A. J. A. Cobb, D. M. Shaw and S. V. Ley, Synlett, 2004, 558 CAS.
- A. Cordova, W. Notz, G. F. Zhong, J. M. Betancort and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1842 CrossRef CAS.
- B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423 CrossRef CAS.
- D. Enders and A. Seki, Synlett, 2002, 26 CrossRef CAS.
- R. G. Almquist, C. Jenningswhite, W. R. Chao, T. Steeger, K. Wheeler, J. Rogers and C. Mitoma, J. Med. Chem., 1985, 28, 1062 CrossRef CAS.
- We thank Steve Challenger (Pfizer Inc) for the suggestion of the acyl sulfonamide functionality as an acid surrogate.
- M. S. Manhas, M. Ghosh and A. K. Bose, J. Org. Chem., 1990, 55, 575 CrossRef CAS.
- W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS.
- (a) L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16 CrossRef CAS; (b) S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 12911 CrossRef CAS; (c) S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475 CrossRef CAS.
- A. J. A. Cobb, D. A. Longbottom, D. M. Shaw and S. V. Ley, Chem. Commun., 2004, 1808 RSC.
- J. M. Betancort, K. Sakthivel, R. Thayumanavan, F. Tanaka and C. F. Barbas III, Synthesis, 2004, 1509 CAS.
- (a) T. Ishii, S. Fujioka, Y. Sekiguchi and H. Kotsuki, J. Am. Chem. Soc., 2004, 126, 9558 CrossRef CAS; (b) H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS.
- H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983 CrossRef CAS.
- A. Hartikka and P. I. Arvidsson, Tetrahedron: Asymmetry, 2004, 15, 1831 CrossRef CAS.
- Note added in proof:since submission an aldol reaction has been reported with related acyl sulfonamides; (a) A. Berkessel, B. Koch and J. Lex, Adv. Synth. Catal., 2004, 1141 CrossRef CAS ; a sulfonamide catalyst has been employed in Mannich and a-aminoxylation reactions; (b) W. Wang, J. Wang, H. Li and L. X. Liao, Tetrahedron Lett., 2004, 45, 7235 CrossRef CAS; (c) W. Wang, J. Wang and H. Li, Tetrahedron Lett., 2004, 45, 7243 CrossRef CAS.
- K. Morikawa, M. Honda, K. I. Endoh, T. Matsumoto, K. I. Akamatsu, H. Mitsui and M. Koizumi, J. Pharm. Sci., 1991, 80, 837 CAS.
- S. J. Blarer, W. B. Schweizer and D. Seebach, Helv. Chim. Acta., 1982, 65, 1637 CAS.
- D. Seebach and M. A. Brook, Helv. Chim. Acta., 1985, 68, 319 CAS.
- D. Seebach, I. M. Lyapkalo and R. Dahinden, Helv. Chim Acta., 1999, 82, 1829 CrossRef CAS.
- D. Enders and T. Otten, Synlett, 1999, 747 CAS.
Footnote |
| † CCDC reference number 256659. See http://www.rsc.org/suppdata/ob/b4/b414742a/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2005 |