Combining two-directional synthesis and tandem reactions:† an efficient strategy for the total syntheses of (±)-hippodamine and (±)-epi-hippodamine‡
Martin
Rejzek
a,
Robert A.
Stockman
*a and
David L.
Hughes
b
aSchool of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: r.stockman@uea.ac.uk; Fax: +44 (0)1603 592003; Tel: +44 (0)1603 593890
bX-Ray Crystallography Unit, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ
First published on 8th November 2004
Abstract
Two-directional total stereoselective syntheses of (±)-hippodamine and (±)-epi-hippodamine, utilising a tandem deprotection/intramolecular double Michael addition sequence as the key step, are presented.
Introduction
Ladybird beetles (Coleoptera: Coccinellidae) are important predators contributing to the natural control of pest aphid populations and are therefore of considerable interest for insect pest management. However, ladybirds themselves are attacked by a range of natural enemies. General predation on ladybirds by vertebrates such as birds is largely prevented by highly toxic, bitter tasting, defence alkaloids contained in a reflex bleed released when the ladybird is attacked. To date, eight alkaloids of this type have been isolated from coccinellid beetles,1 all of them being formally derivatives of perhydro-9b-azaphenalene (or dodecahydro-pyrido[2,1,6-de]quinolizine) (Fig. 1).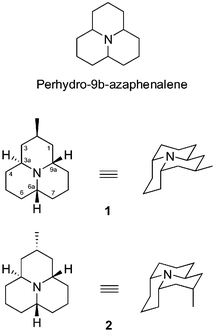 | ||
| Fig. 1 Structures of hippodamine (1) and epi-hippodamine (2), derivatives of perhydro-9b-azaphenalene. | ||
Another group of natural enemies, parasitic insects, can cause substantial reductions in populations of ladybird species. Recent research2 has shown that the parasites locate the ladybirds through perception of certain defence alkaloids that they emit. If ladybirds are to be used effectively in insect pest control then their parasites must be controlled as well. The significant attraction of parasitic insects to the ladybird alkaloids suggests that there is potential for development of control strategies for this particular natural enemy.
In addition to their bitter taste, the coccinellid alkaloids also seem to interfere with the nervous systems of higher animals. Certain derivatives of coccinellid alkaloids have recently been shown3 to be potent receptor antagonists of the neurotransmitter serotonin. This pharmacological property makes these compounds attractive for the treatment of a number of gastrointestinal and brain disorders. One of the most intriguing properties is the ability of these compounds to prevent behavioural consequences of the withdrawal of drugs such as nicotine. For these reasons much attention has been paid to developing syntheses of the coccinellid defence alkaloids.1
Hippodamine (1) is a naturally occurring alkaloid isolated from a ladybird beetle Hippodamia convergens by Tursch and co-workers in 1972.4 The structure of hippodamine (1) was established two years later by the same group5 on the basis of a single-crystal X-ray diffraction experiment (Fig. 1). epi-Hippodamine (2) is a non-natural isomer with an axial C(2) methyl group. Both hippodamine (1)6,7,8 and epi-hippodamine (2)9 have been the subjects of total synthesis.
In our previous short communication10 we reported on the formation of quinolizidine 8. Herein, we give a full account of a concise synthetic strategy to the title azaphenalene alkaloids 1 and 2 using a two-directional approach to a common key intermediate, with a tandem deprotection/intramolecular double Michael addition (IDM) sequence as the key step.
Results and discussion
Retrosynthesis
Assembling the tricyclic skeleton and controlling the stereochemistry of the title azaphenalene alkaloids in a minimum of synthetic steps represents a formidable synthetic challenge. Our plan was to establish the axial or equatorial C(2) methyl substituents of the title compounds by manipulation of the exocyclic double bond in 3 (Fig. 2). The olefin 3 could be formed by de-alkoxycarbonylation of A and a subsequent Wittig olefination.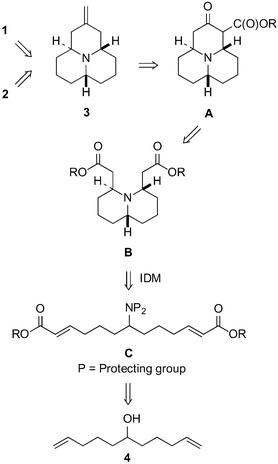 | ||
| Fig. 2 Retrosynthesis for (±)-hippodamine (1) and (±)-epi-hippodamine (2). | ||
Dieckmann condensation of diester B would in turn furnish the third ring of the tricyclic structure. The formation of trans-4,6-disubstituted quinolizidine derivative B represents the key step in the synthetic plan. It was assumed that the quinolizidine derivative B could be accessed via an intramolecular double Michael addition of the acyclic intermediate C and the desired trans-relative configuration of the substituents in B could be installed at the same time. The acyclic intermediate C is a symmetric molecule opening up the possibility of two-directional synthesis. It was thought that C could be obtained from undeca-1,10-dien-6-ol (4) by firstly installing and protecting the required amino group, and secondly by oxidative cleavage of both double bonds and by a subsequent olefination. Finally, undeca-1,10-dien-6-ol (4) can be obtained from 5-bromo-1-pentene by a known procedure.
Synthesis of common intermediate 3
Diester 7 (Scheme 1) was prepared using the principles of two-directional synthesis. The symmetrical alcohol (4) was prepared in 89% yield following the method of Kitching et al.11 Mitsunobu substitution12 of alcohol 4 with phthalimide was then carried out to give diene 5 in 97% yield after column chromatography.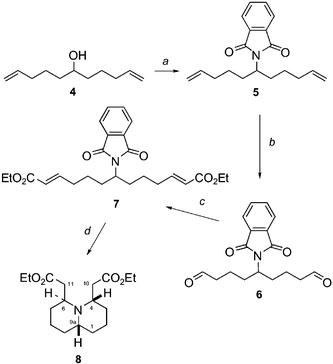 | ||
| Scheme 1 (a) Phthalimide, PPh3, DIAD, THF (97%); (b) Method A: OsO4, NaIO4, Method B: RuCl3, NaIO4, Method C: (1) O3, CH2Cl2, −78 °C, (2) Me2S, PPh3, rt; (c) Et2O(O)PCH2CO2Et, NaH, THF (over 2 steps Method A: 75%, Method B: 71%, Method C: 45%); (d) (1) NaBH4 (1.4 eq.), i-PrOH/H2O (6.2:1), (2) AcOH glac. (71%). | ||
Oxidative cleavage of the terminal alkenes in 5 was evaluated using three different methods: treatment of 5 with sodium periodate (6 eq.) and a catalytic amount of osmium tetroxide (2 mol%) in 3 : 1 THF–water13 (Method A); ruthenium(III) chloride (4.1 mol%) and sodium periodate (4 eq.) in 6 : 1 CH3CN–water14 (Method B); and ozone in dichloromethane and subsequent treatment with Me2S and PPh315 (Method C). The resulting crude dialdehyde 6 was doubly homologated under standard Wadsworth–Emmons conditions16 to yield acyclic diester 7 in 75% (Method A), 71% (Method B) or 45% (Method C) yield respectively over two steps from diene 5 as a single E,E-stereoisomer. The ruthenium(III) chloride catalysed oxidation (Method B) gave varying results which was later put down to the varying quality of the catalyst batches. Ozonolysis (Method C) proved to be difficult to perform on a larger scale. The osmium tetroxide catalysed oxidative cleavage (Method A) gave excellent yields on a small scale (up to 92%).10 Although the yield dropped to some extent (75%) when performed on a large scale (>10 g), Method A was the method of choice for preparation of 7 as the results were easily reproducible.
With the successful formation of protected amine 7, the stage was now set for the key tandem deprotection/double intramolecular Michael addition, which we surmised should give our desired trans-4,6-disubstituted quinolizidine 8. Following Ganem's protocol,17 the phthalimide group was removed by selective hydride reduction (Scheme 2) of the alkyl succinimide 7 to furnish an ortho-hydroxymethyl benzamide 10 (probably via hemiaminal 9). Addition of acetic acid to the reaction solution (c in Scheme 2) promotes lactonisation of the ortho-hydroxymethyl benzamide 10 to form phthalide 11 with concomitant release of the primary amine, or its hydroacetate 15 respectively. The hydroacetate must however, at least to some extent, equilibrate with the free amine representing the reactive species in the desired Michael addition. Under the acidic conditions the amine undergoes an intramolecular Michael addition to give the product of the first cyclisation 12, which could be isolated from the reaction mixture with 13. The second cyclisation can follow to give the desired quinolizidine derivative 8. However, under the initial unoptimised conditions (c in Scheme 2) other side-reactions were noted: the side chain double bond of 12 was partially reduced (to give 13) and so were both double bonds in 15 (to give 14). As a consequence the Michael addition could not occur and the yield of 8 was low. We suspected that sodium triacetoxyborohydride18 formed in situ from sodium borohydride and acetic acid might be responsible for this undesired over-reduction.
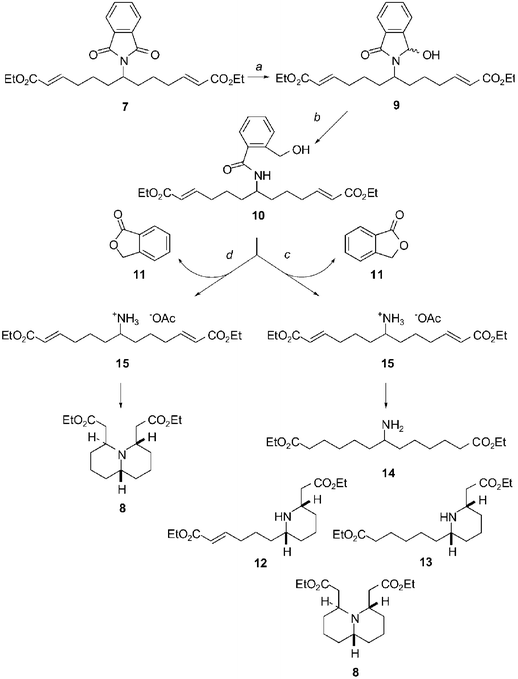 | ||
| Scheme 2 Proposed mechanism of the tandem deprotection/intramolecular double Michael addition sequence: (a) NaBH4 (0.25 eq.), i-PrOH–H2O (6.2 : 1) (product not isolated); (b) NaBH4 (0.25 eq.), i-PrOH–H2O (6.2 : 1) (67%); (c) (1) AcOH glac., (2) K2CO3, benzene (mixture of products 8, 11, 12, 13 and 14); (d) (1) acetone, 12 h, (2) AcOH glac., (3) K2CO3, benzene (71%). | ||
To overcome this problem any surplus of the hydride was quenched by addition of an aliquot of acetone prior to addition of the acetic acid (d in Scheme 2). When all remaining hydride was removed acetic acid was added promoting lactonisation as in the previous case and liberating the amine hydroacetate 15. Under the acidic conditions the acyclic unsaturated amine undergoes the intramolecular double Michael addition to give the desired quinolizidine derivative 8 in high yield.
The 2,6-disubstituted piperidine 13 showed a cis-relative configuration of the ring substituents. The cis-configuration was assigned on the basis of NMR spectra and from analogy with spectra of a cis/trans pair of another 2,6-dialkyl piperidine compound.19 The structure of 12 was assigned from analogy with 13. From this observation it is possible to conclude that the first Michael addition of 15 leads to a thermodynamically more stable cis-2,6-dialkyl piperidine 12. The stereochemistry of the subsequent Michael addition of 12 is then controlled by the stereochemistry of the first ring. As a result the formation of trans-4,6-disubstituted quinolizidine 8 as a single isomer is observed.
Optimal conditions for the tandem deprotection/double cyclisation were as follows: N-alkyl phthalimide 7 (Scheme 1) was subjected to sodium borohydride (1.4 eq.) in 6.2 : 1 isopropanol–water mixture at 25 °C for 24 hours.17 After this time acetone (3.6 eq.) was added and stirring was continued overnight to quench any remaining hydride. Then glacial acetic acid (30 eq.) was added dropwise at 0 °C, and the reaction mixture was heated to 80 °C (bath temperature) for a further 24 hours. An aqueous work-up gave a crude hydroacetate of quinolizidine 8. The free base 8 was liberated by treating the hydroacetate with K2CO3 in benzene. Final purification on activity IV neutral alumina produced pure quinolizidine 8 as a single (±)-trans-isomer (according to NMR) in 71% yield.
The trans relative configuration of substituents in positions C(4) and C(6) of the quinolizidine 8 was determined by NMR. Protons C(4)-H and C(6)-H (Scheme 1) situated on the α-carbon atoms to the nitrogen atom show different behaviour in the 1H NMR spectrum. Proton C(6)-H (2.78–2.72, m) is more shielded than C(4)-H (3.83–3.80, m), the deshielding being about 1 ppm. This can only be explained by an antiperiplanar spatial arrangement assumed by C(6)-H and the nitrogen lone pair. Consequently C(6)-H proton must be in an axial and C(4)-H in an equatorial position with respect to the trans-quinolizidine moiety. Moreover, in the 13C NMR spectrum, the axial methylene group C(4)-CH2 (26.3, t) of the side chain, is more shielded than the respective equatorial C(6)-CH2 (38.5, t), the deshielding being 12.2 ppm. This can be explained by a double γ-gauche effect20 experienced by the axial C(4)-CH2 methylene group from C(2) and C(9a). No such effect is suffered by the respective equatorial C(6)-CH2 group. In addition, in the 13C NMR, 16 signals were detected rather than the 9 that would be expected for the more symmetric cis-isomer of the quinolizidine 8.
We had planned that the formation of the third ring of the perhydro-9b-azaphenalene skeleton would be achieved using a Dieckmann condensation.9,21 To this effect, the 4,6-disubstituted quinolizidine derivative 8 was subjected to a variety of bases and reaction conditions. Two methods were found to give the desired cyclisation product. When 8 was treated with LDA in THF at −78 °C (Scheme 3) a mixture of β-ketoester 16 and its corresponding enol form 17 was obtained. Initially when the reaction mixture was directly subjected to an aqueous work-up the resulting yields were very low. This was later put down to the low stability of the cyclic products 16 and 17 towards aqueous basic solutions. This problem was circumvented by removal of THF and by taking the residue into benzene prior to quenching of the reaction by an aqueous solution. This method eventually gave a reasonable yield (48%) of the desired products 16 and 17. To further improve the yield we explored the alternative of performing the Dieckmann condensation in a lipophilic solvent. When 8 was treated with tBuOK in refluxing benzene (Scheme 3) under continuous azeotropic removal of EtOH using a Dean–Stark trap with lithium in the trap to remove EtOH, a mixture of β-ketoester 16 and its corresponding enol form 17 was obtained in a very good yield (84%). The Dieckmann condensation gave a mixture of regioisomeric β-ketoesters 16 differing in the position of the ester moiety on either C(1) or C(3). Moreover, both regioisomers of 16 are in equilibrium with their enol forms 17. However, the following step, a de-ethoxycarbonylation of all the possible isomers, would lead to the ketone 18 only and consequently we did not purify and identify the components of the isomeric mixture any further.
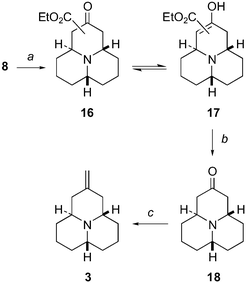 | ||
| Scheme 3 (a) Method A: tBuOK, Li, benzene, azeotropic removal of EtOH (84%), Method B: LDA, THF, −78 °C (48%); (b) (1) HClg, Et2O, (2) LiCl, wet DMF, reflux (66%); (c) CH3PPh3Br, nBuLi, THF (87%). | ||
The de-ethoxycarbonylation of the isomeric mixture of 16 and 17 gave initially only limited yields of 18 when the mixture was directly subjected to LiCl in wet refluxing DMF. It appeared that only the β-ketoester 16 was able to undergo the desired decarboxylation. The enol form 17 was either recovered or it decomposed after a prolonged reaction time. This problem was eventually solved by exposing the isomeric mixture 16 and 17 to HCl in Et2O prior to the treatment with LiCl in wet refluxing DMF. This method gave the desired ketone 18 in 66% yield after purification.
Wittig olefination7 of the ketone 18 using methyltriphenylphosphonium bromide in THF and nBuLi gave low yields of the desired product 3 when the olefination was performed between −78 °C and room temperature. However, a short reflux of the reaction mixture gave the desired product 3 in a good yield (87%) after purification.
(±)-Hippodamine (1)
In order to achieve our objective the exocyclic double bond of the olefin 3 had to be hydrogenated in a stereoselective way. In fact the olefin 3 would have to approach the catalyst surface via its more hindered face in order to obtain the required equatorial methyl on C(2) (Scheme 4) which of course is less likely than the opposite case. Indeed, when a catalytic hydrogenation using 10% palladium on carbon in absolute methanol was performed (Table 1, Entry 1) a mixture of (±)-hippodamine (1) and (±)-epi-hippodamine (2) was obtained in a high combined yield (99%), but in a ratio 1 : 2 of 1 : 1.4, thus supporting the theory that the olefin 3 is much more likely to approach the catalyst surface via its less hindered face, with the hydrogenation resulting in an axial methyl substituent.| Entry | Additive | Solvent | Time/days | T/°C; solventb | Combined yield (%) | Ratio 1 : 2a | dsc (%) |
|---|---|---|---|---|---|---|---|
| a The ratio was determined from relative intensities of the C(2) methyl substituent signals in 400 MHz 1H NMR spectra. b Temperature and solvent (if other than the hydrogenation solvent) of the salt formation. c Diastereoselectivity in favour of either (1) or (2). | |||||||
| 1 | — | MeOH | 1 | rt | 99 | 1 : 1.4 | 17.4 (2) |
| 2 | Oxalic acid (0.5 eq.) | MeOH | 1 | rt | 98 | 1.2 : 1 | 10.0 (1) |
| 3 | Diphenylacetic acid | Et2O | 1 | −20 | 92 | 2.0 : 1 | 34.0 (1) |
| 4 | (1S)-(+)-10-Camphorsulfonic acid | MeOH | 1 | rt; CH2Cl2 | 99 | 1.8 : 1 | 28.0 (1) |
| 5 | (1S)-(+)-10-Camphorsulfonic acid | AcOEt | 2 | rt; CH2Cl2 | 97 | 2.7 : 1 | 46.0 (1) |
| 6 | (1S)-(+)-10-Camphorsulfonic acid | THF | 3 | rt; CH2Cl2 | 97 | 3.3 : 1 | 54.0 (1) |
| 7 | Mesitylenesulfonic acid | THF | 2 | rt | 93 | 3.5 : 1 | 56.0 (1) |
| 8 | Mesitylenesulfonic acid | THF | 2 at −20 °C | −20 | 95 | 2.8 : 1 | 48.0 (1) |
| 9 | Mesitylenesulfonic acid | Et2O–THF 4 : 1 | 2 | rt; Et2O | 99 | 2.0 : 1 | 34.0 (1) |
| 10 | 2,4,6-Triisopropylbenzenesulfonic acid | Et2O | 1 | rt | 98 | 1.8 : 1 | 28.0 (1) |
| 11 | 9-Cyclohexyl 9-borabicyclo[3.3.1]nonane | MeOH | 1 | rt | 79 | 1 : 1.6 | 24.0 (2) |
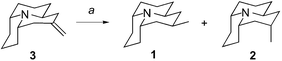
In order to prepare the target compound (±)-hippodamine (1) the selectivity of the hydrogenation had to be reversed. In order to achieve this we decided to make use of the nitrogen lone pair in 3. Reacting the nitrogen lone pair with a convenient reagent would hopefully block one face of the rigid olefin 3 thus allowing the resulting product (a salt or a complex) to approach the catalyst via the desired opposite face only.
When the free base of olefin 3 was reacted at room temperature with 0.5 equivalent of oxalic acid in MeOH a dimeric salt was formed immediately (Table 1, Entry 2). This salt was subjected to catalytic hydrogenation at room temperature and atmospheric pressure of hydrogen. The hydrogenation was monitored using TLC. When the conversion was complete, the mixture was filtered, evaporated and the residue treated with K2CO3 in benzene to liberate the free base. After purification on activity IV neutral alumina a mixture of (±)-hippodamine (1) and (±)-epi-hippodamine (2) was obtained in a high combined yield (98%). This time the ratio 1 : 2 amounted to 1.2 : 1 in favour of the desired (±)-hippodamine (1). Although not a significant advance, this experiment did show that it was possible to reverse the stereoselectivity of the hydrogenation.
To further increase the selectivity of the hydrogenation for (±)-hippodamine (1) a range of salts and boron complexes were screened for their behaviour in hydrogenation (Table 1). The best results were obtained when a salt of 3 with mesitylenesulfonic acid (Table 1, entries 7–9) was hydrogenated. A variety of solvents and salt formation conditions were tested to show that THF at room temperature gave a 1 : 2 ratio of 3.5 : 1 (56% ds) in a very high combined yield (93%). This mixture was then subjected to multiple column chromatography on neutral aluminium oxide activity IV affording isomerically pure hippodamine in 37% yield.
In summary, we have achieved the synthesis of (±)-hippodamine (1) in 8 synthetic steps in an overall yield of 7.2%, using the concept of combining two-directional synthesis and tandem reactions.
In an attempt to further improve the control over the formation of the equatorial methyl substituent in 1 we planned to perform a deoxygenation of tertiary alcohol 25 (Scheme 5) via its xanthate 26. The Barton–McCombie deoxygenation of xanthate ester 26 should in theory proceed via a planar radical species. We assumed that the nitrogen lone pair might donate into the half-filled p-orbital and force the unpaired electron into the opposite face. As a consequence, the hydrogen radical should only react from the required face which would then lead to the desired equatorial methyl substituent of 1.
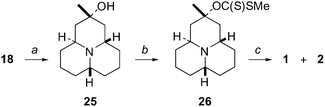 | ||
| Scheme 5 (a) (1) MeLi, toluene, 0 °C, (2) crystallization from Et2O (61%); (b) (1) nBuLi, THF, −78 °C, (2) CS2, rt, (3) MeI (42%); (c) Bu3SnH, AIBN, toluene, reflux (46%). | ||
The tricyclic ketone 18 was transformed into tertiary alcohol 25 (Scheme 5). Interestingly, when the ketone was treated with MeMgBr in either polar or lipophilic aprotic solvents, no or only very limited formation of the desired alcohol was observed. This was most likely due to competing enol formation. When the ketone 18 was treated with MeLi in THF the outcome was very similar. When however toluene was used as a solvent and the reaction was performed at 0 °C the desired tertiary alcohol was formed and isolated in 61% yield after crystallization from diethyl ether.
A single crystal X-ray diffraction experiment of 2522 (Fig. 3) confirmed isolation of a single isomer only with axial hydroxyl and equatorial methyl substituents. It also confirmed the expected trans,cis,trans-stereochemistry of the tricyclic system. The ability of this alcohol to crystallize was greatly enhanced by formation of a hydrogen bond between the hydroxy group and the nitrogen lone pair of a neighbouring molecule.
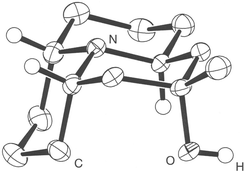 | ||
| Fig. 3 Crystal structure of alcohol 25. | ||
The alcohol 25 was then reacted with 1,1′-thiocarbonyldiimidazole in THF in an attempt to prepare the corresponding xanthate. This method failed, probably due to steric hindrance. For this reason the alcohol 25 was first deprotonated using nBuLi in THF at −78 °C. The corresponding alkoxide was then treated with CS2 and the reaction mixture was allowed to reach room temperature. The reaction was then quenched using MeI to give the desired xanthate 26. This compound was not very stable and consequently it was used after a short filtration through a column of neutral alumina without further purification. Finally, xanthate 26 was subjected to the Barton–McCombie deoxygenation conditions giving 46% combined yield of a mixture of 1 and 2 in a ratio of 2.3 : 1.
The predicted donation of the nitrogen lone pair in the planar radical intermediate of the Barton–McCombie deoxygenation is very likely operative but the control over the formation of the desired equatorial methyl substituent was only limited, most likely due to the high reaction temperature employed. The desired (±)-hippodamine (1) was formed in 39.4% ds.
(±)-epi-Hippodamine (2)
With the successful completion of our synthesis of (±)-hippodamine (1) we also wished to prepare the non-natural epimer (±)-epi-hippodamine (2) as it was thought that it might provide insight into the influence of stereochemistry on the biological activity of these compounds.We surmised that we could use the nitrogen lone pair to deliver a borohydride reagent selectively to one face of the exocyclic olefin 3. Hydroboration of the exocyclic double bond in 3 (Scheme 6) should lead to an anti-Markovnikov primary alcohol. The hydroboration however can proceed from two possible faces leading to either the equatorial 21 or the axial alcohol 22. The tertiary amine moiety in 3 makes this molecule a good Lewis base. Such a base should be capable of quickly replacing ligands of ether type (weaker Lewis bases) in borane etherate complexes and form complexes such as 19 and 20 at low temperatures. At elevated temperatures the hydroboration should hopefully then occur in an intramolecular fashion.
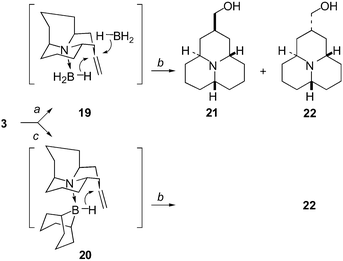 | ||
| Scheme 6 (a) BH3·THF, THF, −78 °C; (b) (1) BH3·THF, THF, rt to reflux, (2) H2O2, NaOH, H2O (21 + 22 90%); (c) 9-BBN·THF, THF, −78 °C; (d) (1) 9-BBN·THF, THF, rt to reflux, (2) H2O2, NaOH, H2O (22 only 84%). | ||
Olefin 3 was treated with borane tetrahydrofuran complex in absolute THF at −78 °C for a short time period. The mixture was then allowed to warm up to room temperature and finally the mixture was heated at reflux. Oxidative work-up under basic conditions furnished a mixture of alcohols 21 and 22 in a ratio 1 : 4.3, with the axial alcohol being favoured, to our gratification. In an attempt to further increase the selectivity olefin 3 was treated with 9-borabicyclo[3.3.1]nonane tetrahydrofuran complex. The hydroboration furnished exclusively the axial alcohol 22 in a high yield (84%) after crystallization from Et2O.
A single crystal X-ray diffraction experiment of the alcohol 2223 (Fig. 4) confirmed the axial position of the hydroxymethylene moiety. Similarly to the alcohol 25 the ability of this compound to crystallize was greatly enhanced by formation of hydrogen bonds between the hydroxy group and the nitrogen lone pair of a neighbouring molecule.
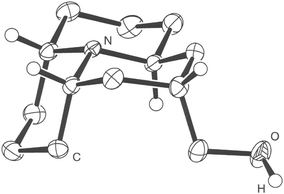 | ||
| Fig. 4 Crystal structure of alcohol 22. | ||
Removal of the oxygen functionality (Scheme 7) by conversion to the iodide 24via tosylate 2324 followed by reduction with zinc25 in refluxing acetic acid gave the desired (±)-epi-hippodamine (2) in a good yield (83% from 23) after purification. Attempts to directly reduce (LiAlH4, LiAlH4–AlCl3 to give AlH3, super-hydride LiB(C2H5)3H) the tosylate 23 to the corresponding alkane 2 failed. Similarly all attempts to proto-deborylate the putative alkyl borane intermediate formed on hydroboration of 3 also failed.
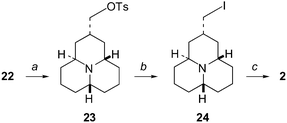 | ||
| Scheme 7 (a) Method A: TsCl, Et3N, Me3N·HCl, CH2Cl2 (25%), Method B: (1) nBuLi, THF, (2) TsCl, THF (95%); (b) NaI, 2-butanone (usually not isolated); (c) (1) Zn activated, AcOH glac., (2) filtration, evaporation, (3) K2CO3, benzene (83%). | ||
In summary, we have managed to synthesise isomerically pure (±)-epi-hippodamine (2) in 11 synthetic steps in an overall yield of 17.0%.
Studies on syntheses of the other members of the perhydro-9b-azaphenalene class of alkaloids and their application in insect pest management are ongoing in these laboratories, and will be reported in due course.
Experimental
General methods
All chemicals were purchased as reagent grade and used without further purification. Tetrahydrofuran was freshly distilled from sodium diphenyl ketyl just prior to use. Dichloromethane was freshly distilled over CaH2. Other solvents for experiments were purified by the usual methods or purchased from a commercial source. If not stated otherwise TLC was performed on precoated silica plates containing a fluorescence indicator. Silica gel (63–200 μm) was used for analytical and preparative TLC (PLC) and for column chromatography, unless otherwise noted. Compounds were visualised under UV (254 nm), by heating after dipping in a solution of alkaline KMnO4, or by exposing to iodine vapours. NMR spectra were recorded on 300 or 400 MHz instruments. Chemical shifts are reported in ppm with respect to TMS. Coupling constants are reported in Hz. Multiplicity of signals in 13C NMR spectra was determined from DEPT or gHSQC spectra. IR spectra of neat (unless otherwise noted) compounds were expressed as wave numbers (cm−1). The Mass Spectrometry Service Centre at the University of Wales, Swansea performed the low- and high-resolution mass spectra. Melting points were taken on a micro melting point apparatus and are uncorrected. Water- and air-sensitive reactions were routinely carried out under argon atmosphere. Compound 4 was synthesized according to literature procedure.112-(1′-Pent-4″-enyl-hex-5′-enyl)-isoindole-1,3-dione (5)
A 2-necked 1000 mL flask equipped with addition funnel and magnetic stirrer was charged with triphenyl phosphine (42.9 g, 163.4 mmol), phthalimide (24.0 g, 163.4 mmol), anhydrous THF (700 mL) and undeca-1,10-dien-6-ol (4) (25 g, 148.6 mmol). The mixture was cooled to −20 °C under stirring and argon atmosphere. Diisopropyl azodicarboxylate (33.0 g, 32.4 mL, 163.4 mmol) in anhydrous THF (100 mL) was added dropwise over 1 h. When the addition was finished the mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure and the oily residue was purified by three subsequent crystallizations from first EtOAc, then Et2O and finally petroleum ether–Et2O (1 : 1) in order to remove triphenylphosphine oxide. The resulting oily mixture was purified by column chromatography on silica gel (stepwise gradient of petroleum ether–Et2O 20 : 3 to pure Et2O) to afford 5 (42.4 g, 96%) as a yellow oil: Rf 0.54 (petroleum ether–EtOAc 10 : 1); IR (cm−1) 3076, 3040, 2927, 2860, 1772, 1708, 1641, 1614, 1468, 1459, 1441, 1396, 1372, 1334; 1H NMR (400 MHz, CDCl3) 7.85–7.82 (2H, m), 7.73–7.70 (2H, m), 5.74 (2H, ddt, J = 16.8, 10.0 and 6.8), 5.01–4.90 (4H, m), 4.26–4.18 (1H, m), 2.16–1.99 (6H, m), 1.78–1.69 (2H, m), 1.51–1.23 (4H, m); 13C NMR (75 MHz, CDCl3) 169.0 (2C, s), 138.6 (2C, d), 134.1 (2C, d), 132.1 (2C, s), 123.3 (2C, d), 115.0 (2C, t), 52.0 (d), 33.4 (2C, t), 31.9 (2C, t), 26.0 (2C, t); MS (EI) m/z 297 (43, M+), 256 (47), 242 (25), 228 (29), 159 (40), 147 (100), 129 (63), 81 (65), 41 (76); HRMS calcd for C19H27N2O2 (M + NH4) 315.2073, found 315.2072.7-(1′,3′-Dioxo-1′,3′-dihydro-isoindol-2′-yl)-trideca-2,11-dienedioic acid diethyl ester (7)
Method A: Alkyl phthalimide 5 (8.751 g, 29.43 mmol) was dissolved in THF (437.6 mL) and distilled water (145.9 mL). A solution (2.5% wt in 2-methyl-2-propanol) of osmium tetroxide (5.984 g, 0.59 mmol, 2 mol%) was added in one portion. After 5 min stirring at room temperature the mixture was cooled by water bath to about 15 °C and sodium periodate (37.769 g, 176.58 mmol) was added in portions over 20 min. The mixture was stirred at room temperature for 20 h. Volatiles were evaporated under reduced pressure (bath temperature did not exceed 30 °C) to give a white solid. The solid was extracted/decanted with dichloromethane (3 × 100 mL). Combined extracts were dried over Na2SO4 and concentrated to give a crude dialdehyde 6 (10.7 g) that was used for the subsequent reaction without purification. To an oven dried flask a 60% dispersion of NaH in mineral oil (3.95 g, 98.8 mmol) was added and washed with n-pentane (3 × 20 mL) under argon atmosphere. The remaining solvent was evaporated under reduced pressure and freshly distilled THF (400 mL) was added. Triethyl phosphonoacetate (17.52 mL, 19.80 g, 88.3 mmol) was added dropwise over 40 min at room temperature. Stirring was continued for 2 h. The mixture was cooled to 0 °C. A solution of the crude dialdehyde 6 in freshly distilled THF (100 mL) was added dropwise over 2 h and the mixture was stirred for 48 h. A saturated solution of NH4Cl (100 mL) and ethyl acetate (100 mL) were added, the organic layer was separated and the aqueous one was extracted with ethyl acetate (3 × 200 mL). Combined organic extracts were washed with sodium bicarbonate (20 mL), brine (10 mL) and dried over Na2SO4. Evaporation gave a crude product (13 g) that was purified using column chromatography on silica gel (stepwise gradient of petroleum ether–ethyl acetate 9 : 1 to 1 : 1) to afford 7 (9.76 g, 75.1% over 2 steps). Method B: A stock solution of ruthenium(III) chloride was prepared by dissolving RuCl3·(H2O)2 (842 mg, 3.5 mmol) in distilled water (100 mL) to give a concentration of 0.035 M. To a stirred solution of alkyl phthalimide 5 (9.432 g, 31.7 mmol) and RuCl3 stock solution (31.7 mL, 1.11 mmol, 3.5 mol%) in CH3CN (158.6 mL) and distilled water (26.4 mL) was added in portions NaIO4 (27.2 g, 127.2 mmol) over a period of 5 min at room temperature. After completion (TLC monitoring) in 3 h, the reaction was quenched with a saturated aqueous solution of Na2S2O3 (100 mL). The resulting mixture was filtered through sintered glass and the aqueous layer was extracted with AcOEt (3 × 100 mL). The combined organic layers were washed with water (50 mL), brine (50 mL) and dried over Na2SO4. The solvents were evaporated under reduced pressure to give a crude dialdehyde 6 (10.9 g) that was used for the subsequent reaction without purification. To an oven dried flask a 60% dispersion of NaH in mineral oil (7.75 g, 194.0 mmol) was added and washed with n-pentane (3 × 20 mL) under argon atmosphere. The remaining solvent was evaporated under reduced pressure and freshly distilled THF (700 mL) was added. Triethyl phosphonoacetate (31.5 mL, 35.60 g, 159.1 mmol) was added dropwise over 40 min at room temperature. Stirring was continued for 2 h. The mixture was cooled to 0 °C. A solution of the crude dialdehyde 6 in freshly distilled THF (100 mL) was added dropwise over 2 h and the mixture was stirred for 48 h. A saturated solution of NH4Cl (100 mL) and ethyl acetate (100 mL) were added, the organic layer was separated and the aqueous one was extracted with ethyl acetate (3 × 200 mL). Combined organic extracts were washed with sodium bicarbonate (20 mL), brine (10 mL) and dried over Na2SO4. Evaporation gave a crude product (15 g) that was purified using column chromatography on silica gel (stepwise gradient of petroleum ether–ethyl acetate 9 : 1 to 1 : 1) to afford 7 (9.9 g, 70.7% over 2 steps). Method C: Alkyl phthalimide 5 (1.03 g, 3.477 mmol) was dissolved in dichloromethane (20 mL). Ozone was passed through the mixture at −78 °C until a blue colour persisted. The excess of O3 gas was removed by blowing argon through the mixture for 15 min. Me2S (5 mL, large excess) was added and the mixture was stirred 0.5 h at −78 °C. The cold bath was removed and stirring was continued for another 2 h. Triphenylphosphine (600 mg, 2.29 mmol) in dichloromethane (5 mL) was added and stirring was continued until no more ozonide could be detected (TLC). The mixture was concentrated under reduced pressure. The residue was triturated with Et2O (3 × 10 mL) and the combined extracts were washed with water (5 mL), brine (5 mL) and dried over Na2SO4. Evaporation gave a crude dialdehyde 6 (1.2 g) that was used for the subsequent reaction without purification. To an oven dried flask a 60% dispersion of NaH in mineral oil (0.51 g, 12.8 mmol) was added and washed with n-pentane (3 × 5 mL) under argon atmosphere. The remaining solvent was evaporated under reduced pressure and freshly distilled THF (40 mL) was added. Triethyl phosphonoacetate (2.07 mL, 2.339 g, 10.4 mmol) was added dropwise over 20 min at room temperature. Stirring was continued for 1 h. The mixture was cooled to 0 °C. A solution of the crude dialdehyde 6 in freshly distilled THF (10 mL) was added dropwise over 1 h and the mixture was stirred for 48 h. A saturated solution of NH4Cl (10 mL) and ethyl acetate (10 mL) were added, the organic layer was separated and the aqueous one was extracted with ethyl acetate (3 × 20 mL). Combined organic extracts were washed with sodium bicarbonate (5 mL), brine (5 mL) and dried over Na2SO4. Evaporation gave a crude product (1.7 g) that was purified using column chromatography on silica gel (stepwise gradient of petroleum ether–ethyl acetate 9 : 1 to 1 : 1) to afford 7 (0.688 g, 44.8% over 2 steps) as a yellow oil: Rf 0.56 (petroleum ether–EtOAc 3 : 1); IR (cm−1) 3462, 3411, 2980, 2924, 2852, 1767, 1705, 1643, 1607, 1464, 1444, 1398, 1367; 1H NMR (400 MHz, CDCl3) 7.86–7.84 (2H, m), 7.78–7.75 (2H, m), 6.90 (2H, dt, J = 15.6 and 6.8), 5.79 (2H, d, J = 15.6), 4.25–4.20 (1H, m), 4.18 (4H, q, J = 7.2), 2.30–2.12 (6H, m), 1.80–1.72 (2H, m), 1.48–1.43 (4H, m), 1.27 (6H, t, J = 7.2); 13C NMR (100 MHz, CDCl3) 168.1 (2C, s), 165.9 (2C, s), 147.8 (2C, d), 133.6 (2C, d), 131.2 (2C, s), 122.8 (2C, d), 121.3 (2C, d), 59.6 (2C, t), 50.9 (d), 31.4 (2C, t), 31.2 (2C, t), 24.6 (2C, t), 13.8 (2C, q); MS (EI) m/z 441 (7, M+), 254 (13), 185 (40), 147 (100), 129 (59); HRMS calcd for C25H32NO6 (M + H) 442.2230, found 442.2227.7-(2′-Hydroxymethyl-benzoylamino)-trideca-2,11-dienedioic acid diethyl ester (10)
To a flask containing NaBH4 (0.166 g, 4.39 mmol) a solution of diester 7 (1.292 g, 2.93 mmol) in isopropanol (47.4 mL) and water (7.6 mL) was added. The mixture was stirred for 24 h at room temperature. The formation of 10 (Rf 0.63, petroleum ether–EtOAc 1 : 1) was monitored by TLC. Brine (10 mL) was added and the mixture was extracted with Et2O (4 × 20 mL). Combined organic layers were dried over Na2SO4. Evaporation gave a crude product (1.771 g) that was purified using column chromatography on silica gel (stepwise gradient of petroleum ether–EtOAc 3 : 1 to 1 : 1) to afford 10 (0.873 g, 66.9%) as a yellow oil: IR (cm−1) 3344, 3288, 3062, 2980, 2934, 2852, 1716, 1629, 1536, 1454, 1362, 1311, 1260, 1178; 1H NMR (400 MHz, CDCl3) 7.54–7.34 (4H, m), 6.96–6.88 (2H, m), 6.51–6.39 (1H, m), 5.82 (2H, dd, J = 15.6 and 1.2), 4.57 (2H, s), 4.20–4.10 (5H, m), 2.31–2.22 (4H, m), 2.0 (1H, bs), 1.63–1.31 (8H, m), 1.30–1.23 (6H, m); 13C NMR (100 MHz, CDCl3) 173.8 (s), 169.7 (s), 166.7 (s), 148.5 (2C, d), 139.6 (s), 136.0 (s), 131.1 (d), 130.7 (d), 128.2 (d), 127.7 (d), 121.8 (2C, d), 64.6 (t), 60.3 (2C, t), 49.4 (d), 34.8 (2C, t), 31.8 (2C, t), 24.5 (2C, t), 14.3 (2C, q); MS (EI) m/z 445 (21, M+), 428 (100), 427 (48), 416 (25), 135 (75), 132 (99); HRMS calcd for C25H36NO6 (M + H) 446.2542, found 446.2543.(4S*, 6S*)-(6-Ethoxycarbonylmethyl-octahydro-quinolizin-4-yl)-acetic acid ethyl ester (8)
To a flask containing NaBH4 (1.046 g, 27.65 mmol) a solution of diester 7 (8.721 g, 19.75 mmol) in isopropanol (320.0 mL) and water (51.6 mL) was added. The mixture was stirred for 24 h at room temperature. The formation of 10 was monitored by TLC. Acetone (5.23 mL, 71.23 mmol) was added and stirring was continued overnight. The mixture was cooled to 0 °C and glacial AcOH (33.9 mL, 592.76 mmol) was added dropwise. The mixture was heated to 80 °C (bath temperature) under stirring for 24 h. The mixture was allowed to cool and brine (20 mL) was added. Solids were filtered off and petroleum ether (100 mL) was added to the filtrate to initiate separation of organic and aqueous layers. The aqueous layer was extracted with ethyl acetate (3 × 100 mL). Combined organic layers were washed with brine (20 mL) and dried over Na2SO4. Evaporation gave a crude hydroacetate of quinolizidine 8 that was taken into benzene (200 mL) and K2CO3 (5.6 g) was added. The mixture was stirred overnight. Solids were filtered off and the filtrate was evaporated. The residue was crystallized from first Et2O and then from Et2O–petroleum ether 1 : 1 to separate phthalide 11 (Rf 0.48, alumina, petroleum ether–EtOAc 5 : 1). Crystals of phthalide 11 were filtered off, washed with Et2O–petroleum ether 1 : 1 (3 × 2 mL) and the combined filtrates were evaporated to give crude free base 8 (5.3 g) that was purified using column chromatography on activity IV neutral alumina (40 g, petroleum ether–EtOAc 30 : 1) to afford 8 (4.38 g, 71.2%) as a yellow oil: Rf 0.79 (alumina, petroleum ether–EtOAc 5 : 1); IR (cm−1) 2980, 2929, 2853, 1726, 1465, 1445, 1368, 1337, 1301, 1276, 1163, 1096, 1030; 1H NMR (400 MHz, benzene-d6) 4.04–3.93 (4H, m), 3.83–3.80 (1H, m), 2.81 (1H, dd, J = 14.2 and 4.4), 2.78–2.72 (1H, m), 2.65 (1H, dd, J = 14.6 and 3.2), 2.41 (1H, dd, J = 14.6 and 10.4), 2.29 (1H, dd, J = 14.2 and 6.8), 1.98–1.92 (1H, m), 1.72–1.66 (3H, m), 1.47–1.36 (2H, m), 1.34–1.29 (4H, m), 1.24–1.04 (3H, m), 1.02–0.98 (6H, m); 13C NMR (75 MHz, benzene-d6) 171.9 (s), 170.9 (s), 59.0 (t), 58.9 (t), 53.9 (d), 52.9 (d), 50.0 (d), 38.5 (t), 33.7 (t), 33.6 (t), 32.0 (t), 28.6 (t), 26.3 (t), 22.9 (t), 17.7 (t), 12.9 (2C, q); MS (EI) m/z 311 (60, M+), 224 (100), 167 (57), 149 (95), 136 (62); HRMS calcd for C17H30NO4 (M + H) 312.2175, found 312.2171.(4S*, 6S*)-(6-Ethoxycarbonylmethyl-octahydro-quinolizin-4-yl)-acetic acid ethyl ester (8), (2′S*, 6′S*)-6-(6′-ethoxycarbonylmethyl-piperidin-2′-yl)-hexanoic acid ethyl ester (13) and 7-amino-tridecanedioic acid diethyl ester (14)
To a flask containing NaBH4 (261 g, 6.9 mmol) a solution of diester 7 (2.437 g, 5.52 mmol) in isopropanol (89.4 mL) and water (14.4 mL) was added. The mixture was stirred for 24 h at room temperature. The formation of 10 was monitored by TLC. The mixture was cooled to 0 °C and glacial AcOH (9.46 mL, 165.4 mmol) was added dropwise. The mixture was heated to 80 °C (bath temperature) under stirring for 24 h. The mixture was allowed to cool down and brine (10 mL) was added. Solids were filtered off and petroleum ether (10 mL) was added to the filtrate to initiate separation of organic and aqueous layers. The aqueous layer was extracted with ethyl acetate (3 × 100 mL). Combined organic layers were washed with brine (5 mL) and dried over Na2SO4. Evaporation gave a mixture of crude amine hydroacetates that was taken into benzene (50 mL) and K2CO3 (3 g) was added. The mixture was stirred overnight. Solids were filtered off and the filtrate was evaporated. The residue was crystallized from first Et2O and then from Et2O–petroleum ether 1 : 1 to separate phthalide 11. Crystals of phthalide 11 were filtered off, washed with Et2O–petroleum ether 1 : 1 (3 × 2 mL) and the combined filtrates were evaporated to give a mixture of crude free amines (2.5 g) that was purified using column chromatography on activity IV neutral alumina (40 g, stepwise gradient petroleum ether–EtOAc 30 : 1 to MeOH pure) to afford 8 (0.539 g, 31.4%), 13 (0.36 g, 21.0%) and 14 (0.275 g, 15.8%) as yellow oils.For 13: Rf 0.30 (alumina, petroleum ether–EtOAc 5 : 1); IR (cm−1) 3385, 2929, 2852, 1731, 1454, 1372, 1183; 1H NMR (400 MHz, CDCl3) 4.17–4.10 (4H, m), 2.98–2.90 (1H, m), 2.56–2.50 (1H, m), 2.41 (2H, d, J = 6.0), 2.29 (2H, t, J = 7.6), 2.20 (1H, bs), 1.81–1.76 (2H, m), 1.66–1.58 (6H, m), 1.50–1.30 (6H, m), 1.28–1.24 (6H, m); 13C NMR (100 MHz, CDCl3) 173.8 (s), 172.6 (s), 60.4 (t), 60.2 (t), 56.7 (d), 53.4 (d), 41.5 (t), 37.1 (t), 34.3 (t), 32.5 (t), 32.1 (t), 29.3 (t), 25.6 (t), 24.9 (t), 24.6 (t), 14.2 (2C, q); MS (EI) m/z 313 (2, M+), 300 (99), 290 (20), 276 (26), 226 (15), 156 (100), 124 (29), 82 (44); HRMS calcd for C17H32NO4 (M + H) 314.2326, found 314.2324.
For 14: Rf 0.07 (alumina, petroleum ether–EtOAc 5 : 1); IR (nujol, cm−1) 3426, 2955, 2852, 1726, 1577; 1H NMR (300 MHz, CDCl3) 4.12 (4H, q, J = 7.2), 3.02–3.19 (1H, m), 2.30 (4H, t, J = 7.1), 1.98–1.32 (16H, m), 1.25 (6H, t, J = 7.2); 13C NMR (100 MHz, CDCl3) 173.6 (2C, s), 60.3 (2C, t), 52.2 (d), 34.2 (2C, t), 33.0 (2C, t), 28.8 (2C, t), 25.1 (2C, t), 24.7 (2C, t), 14.3 (2C, q); MS (EI) m/z 315 (1, M+), 184 (25), 170 (100), 82 (38); HRMS calcd for C17H34NO4 (M + H) 316.2488, found 316.2492.
(3aS*, 9aS*)-2-Oxo-dodecahydro-pyrido[2,1,6-de]quinolizine (18)
Method A: An oven dried double-neck flask was equipped with magnetic stirring bar and a Dean–Stark trap. The flask was charged with tBuOK (3.899 g, 34.74 mmol) and absolute benzene (180 mL). The Dean–Stark trap was filled with absolute benzene and charged with a lithium stick (1 g). Quinolizidine 8 (5.41 g, 17.37 mmol) was added in absolute benzene (20 mL) via a septum. The mixture was refluxed (bath temperature 120 °C) under continuous removal of EtOH for 2 h. The septum was replaced by a glass stopper and reflux was continued for another 17 h. The formation of 16 (Rf 0.46, MeOH–EtOAc 1 : 10) and 17 (Rf 0.33, MeOH–EtOAc 1 : 10) was monitored by TLC. The mixture was allowed to cool down and water (50 mL) was added. The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 100 mL). Combined organic layers were washed with brine (25 mL) and finally dried over Na2SO4. Evaporation gave a crude mixture of 16 and 17 (4.1 g) that was purified by filtration through a pad of activity IV neutral alumina (2 g, EtOAc) to afford a regioisomeric mixture of keto and enol tautomers 16 and 17 (3.85 g, 83.5%) as a yellow oil. The mixture of 16 and 17 (1.6 g, 6.03 mmol) was dissolved in CHCl3 (10 mL) and 2 M HClg in Et2O (40 mL) was added. The mixture was stirred for 1 h at room temperature. The volatiles were evaporated under reduced pressure and the residue was taken into DMF (50 mL). Water (540 μL, 30.15 mmol) and LiCl (1.278, 30.15 mmol) were added. The mixture was heated at reflux (oil bath 140 °C) for 4 h. The volatiles were evaporated at 60 °C under reduced pressure. The residue was taken into dichloromethane (20 mL) and filtered through a pad of activity IV neutral alumina (2 g, dichloromethane). Evaporation gave a crude product (0.97 g) that was purified using column chromatography on activity IV neutral alumina (10 g, stepwise gradient of hexane–Et2O 20 : 1 to 1 : 1) to afford 18 (0.764 g, 65.6%) as a yellow oil. Method B: To a stirred solution of quinolizidine 8 (108.2 mg, 0.347 mmol) in absolute THF (10 mL) was added LDA (74.3 mg, 0.694 mmol) via septum at −78 °C and under argon atmosphere. The formation of 16 and 17 was monitored by TLC. After 8 h the mixture was allowed to reach room temperature. THF was evaporated and the residue was taken into benzene (10 mL). Water (10 mL) was added. The organic layer was separated and the aqueous one was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over Na2SO4. Evaporation gave a crude mixture of 16 and 17 (100 mg) that was purified by filtration through a pad of activity IV neutral alumina (2 g, EtOAc) to afford a mixture of pure 16 and 17 (44.3 mg, 48.1%) as a yellow oil. This mixture was directly converted to ketone 18 in the same way as in Method A. For 18: Rf 0.49 (alumina, petroleum ether–EtOAc 1 : 1): IR (cm−1) 3422, 2932, 2853, 1715, 1460, 1442, 1367, 1340, 1288; 1H NMR (400 MHz, CDCl3) 3.43–3.38 (1H, m), 3.23–3.13 (2H, m), 2.89 (1H, dd, J = 13.6 and 6.8), 2.26–2.14 (3H, m), 1.93–1.82 (3H, m), 1.72–1.67 (1H, m), 1.61–1.48 (5H, m), 1.46–1.00 (3H, m); 13C NMR (100 MHz, CDCl3) 209.1 (s), 61.0 (d), 57.9 (d), 48.9 (d), 48.7 (t), 47.5 (t), 34.9 (t), 30.9 (t), 25.6 (t), 23.8 (t), 21.6 (t), 18.6 (t); MS (EI) m/z 193 (49, M+), 178 (22), 150 (100), 122 (80), 82 (42); HRMS calcd for C12H20NO (M + H) 194.1539, found 194.1535.(3aS*, 9aS*)-2-Methylene-dodecahydro-pyrido[2,1,6-de]quinolizine (3)
To a stirred suspension of methyltriphenylphosphonium bromide (1.0314 g, 2.89 mmol) in absolute THF (69 mL) was added 2.5M nBuLi in hexanes (1 mL, 2.5 mmol) at −78 °C and under argon atmosphere. When the addition was complete, the cold bath was removed and the yellow mixture was stirred for 75 min. A solution of ketone 18 (382 mg, 1.98 mmol) in absolute THF (10 mL) was added over 5 min. The mixture was stirred at room temperature for 1 h and then heated to 60 °C for 30 min. The mixture was allowed to cool down and water (10 mL) was added. The mixture was extracted with EtOAc (3 × 30 mL). Combined extracts were washed with brine (2 × 10 mL) and dried over Na2SO4. Evaporation gave a mixture (0.8 g) that was purified by crystallization from EtOAc and then from petroleum ether–Et2O 1 : 1 to get rid of Ph3PO. The remaining oil (0.4 g) was further purified using column chromatography on activity IV neutral alumina (71 g, hexane–Et2O 500 : 75) to afford 3 (330.45 g, 87.4%) as a yellow oil: Rf 0.59 (alumina, MeOH–EtOAc 1 : 1); IR (cm−1) 3067, 2924, 2847, 1737, 1650, 1460, 1440; 1H NMR (400 MHz, CDCl3) 4.78–4.59 (2H, m), 3.18–3.05 (2H, m), 3.00–2.86 (1H, m), 2.72–2.56 (1H, m), 2.22–1.26 (1H, m), 2.12–2.02 (1H, m), 2.00–1.85 (5H, m), 1.78–1.44 (5H, m), 1.36–1.18 (1H, m), 1.12–0.92 (2H, m); 13C NMR (100 MHz, CDCl3) 144.3 (s), 108.3 (t), 59.6 (d), 58.2 (d), 49.4 (d), 42.7 (t), 40.5 (t), 34.5 (t), 31.1 (t), 25.7 (t), 22.6 (t), 21.9 (t), 19.4 (t); MS (EI) m/z 191 (61, M+), 176 (52), 162 (38), 148 (100), 136 (82), 120 (52), 94 (57), 79 (57); HRMS calcd for C13H22N (M + H) 192.1747, found 192.1749.(±)-Hippodamine (1)
Mesitylenesulfonic acid dihydrate (245.3 mg, 1.04 mmol) was dissolved in THF (25 mL). To this solution olefin 3 (99.3 mg, 0.52 mmol) in THF (25 mL) was added in one portion. The mixture was allowed to stir at room temperature for 1 h. Then 10% palladium on carbon (100 mg) was added and hydrogen (normal pressure) was applied. The formation of 1 (Rf 0.46, alumina, MeOH–EtOAc 1 : 1) was monitored by TLC after a mini work-up. After 2 days the solids were filtered off and THF evaporated. The residue was taken into benzene and K2CO3 (1 g) was added. The mixture was stirred overnight. Solids were filtered off and the filtrate was evaporated. The remaining oil (95 mg) was further purified using column chromatography on activity IV neutral alumina (2.3 g, hexane–Et2O 5 : 1) to afford a 3.5 : 1 mixture (by 1H NMR) of 1 and 2 (93.3 mg, 93.0%). The mixture was further subjected to a multiple column chromatography (4 times) on activity IV neutral alumina (12 g, hexane–Et2O 10 : 1, sample applied in a small amount of chlorobenzene) to finally afford isomerically pure hippodamine (1) (37.1 mg, 37.0%): Rf 0.46 (alumina, MeOH–EtOAc 1 : 1); IR (cm−1) 2920, 2859, 1455, 1443, 1294, 1132, 1102; 1H NMR (400 MHz, CDCl3) 3.00–2.95 (2H, m), 2.83 (1H, dddd, J = 11.2, 11.2, 2.8, 2.8), 1.93–1.77 (4H, m), 1.70–1.63 (1H, m), 1.59–1.43 (8H, m), 1.21–1.12 (1H, m), 1.04–0.94 (2H, m), 0.87–0.78 (1H, m), 0.85 (3H, d, J = 6.4); 13C NMR (100 MHz, CDCl3) 58.8 (d), 58.1 (d), 47.8 (d), 43.4 (t), 40.0 (t), 34.6 (t), 31.4 (t), 26.1 (t), 25.7 (d), 23.1 (t), 22.5 (q), 22.3 (t), 19.7 (t); MS (EI) m/z 193 (22, M+), 192 (42), 178 (18), 164 (26), 151 (35), 150 (45), 137 (28), 136 (42), 105 (25), 77 (35), 67 (31), 55 (54), 43 (43), 41 (100); HRMS calcd for C13H24N (M + H) 194.1903, found 194.1902. Synthetic hippodamine proved identical to the natural material by IR, 1H NMR, 13C NMR, and MS spectral comparison.26,27(2R*, 3aS*, 6aS*, 9aS*)-2-Hydroxymethyl-dodecahydro-pyrido[2,1,6-de]quinolizine (22)
Olefin 3 (53.4 mg, 0.279 mmol) was dissolved in absolute THF (20 mL) and the solution was stirred at −78 °C under argon atmosphere. A 0.5 M THF solution of 9-borabicyclo[3.3.1]nonane·THF (9-BBN·THF) (136.1 mg, 1.116 mmol) was added dropwise via a septum. Stirring was continued for 1 h at −78 °C. Then the mixture was allowed to reach room temperature and stirring was continued overnight. The mixture was then refluxed for 2 h. The mixture was allowed to reach room temperature and water (1 mL) was added followed by MeOH (9.4 mL) and 3 N NaOH (0.93 mL, 1.116 mmol). The mixture was cooled to 0 °C and 11.6 M (or 30%) H2O2 (241 μL, 2.79 mmol) was added dropwise over 10 min. The mixture was stirred for 2 h. A white precipitate was filtered off. The filtrate was evaporated under reduced pressure and the residue was taken into Et2O (10 mL). This solution was washed with water (5 mL), brine (5 mL) and finally dried over Na2SO4. Evaporation gave a crude product (97 mg) that was purified using column chromatography on activity IV neutral alumina (5 g, stepwise gradient hexane–Et2O 5 : 1, pure Et2O to pure MeOH) to afford 22 (54 mg) as a white solid. The solid was further crystallized from Et2O to give 22 (49.3 mg, 84.4%) as crystals (clear, colourless, tapered rectangular prisms): Rf 0.32 (alumina, MeOH–EtOAc 1 : 1); mp 145.5–146.0 °C (Et2O): IR (cm−1) 3652, 3354, 2930, 2848, 1731, 1644, 1582, 1460, 1440; 1H NMR (400 MHz, CDCl3) 3.79 (1H, bt, J = 9.6), 3.63 (1H, bdd, J = 9.6 and 6.0), 2.96–2.84 (3H, m), 2.14–2.07 (1H, m), 1.95–1.77 (4H, m), 1.67–1.38 (9H, m), 1.28–1.13 (2H, m), 1.06–1.00 (1H, m); 13C NMR (100 MHz, CDCl3) 66.5 (t), 58.2 (d), 57.8 (d), 43.6 (d), 35.1 (t), 34.8 (d), 34.6 (t), 32.0 (t), 31.4 (t), 26.7 (t), 26.4 (t), 23.2 (t), 19.7 (t); MS (EI) m/z 209 (38, M+), 208 (100), 178 (69), 167 (47), 150 (55), 137 (40); HRMS calcd for C13H24NO (M + H) 210.1852, found 210.1855. Anal. Calcd. for C13H23NO: C, 74.59; H, 11.07; N, 6.69. Found: C, 74.65; H, 11.13; N, 6.38%.(2S*, 3aS*, 6aR*, 9aS*)-2-Hydroxymethyl-dodecahydro-pyrido[2,1,6-de]quinolizine (21) and (2R*, 3aS*, 6aS*, 9aS*)-2-hydroxymethyl-dodecahydro-pyrido[2,1,6-de]quinolizine (22)
Olefin 3 (10.0 mg, 0.052 mmol) was dissolved in absolute THF (10 mL) and the solution was stirred at −78 °C under argon atmosphere. A 1 M THF solution of borane·THF (210 μL, 0.210 mmol) was added dropwise via a septum. Stirring was continued for 1 h at −78 °C. Then the mixture was allowed to reach room temperature and stirring was continued overnight. The mixture was then refluxed for 4 h. The mixture was allowed to reach room temperature and water (11.2 μL, 0.624 mmol) was added followed by MeOH (109 μL) and 3 N NaOH (208 μL, 0.624 mmol). The mixture was cooled to 0 °C and 11.6 M (or 30%) H2O2 (44.9 μL, 0.520 mmol) was added dropwise over 10 min. The mixture was stirred for 2 h. A white precipitate was filtered off. The filtrate was evaporated under reduced pressure and the residue was taken into Et2O (10 mL). This solution was washed with water (5 mL), brine (5 mL) and finally dried over Na2SO4. Evaporation gave a crude product (20 mg) that was purified using column chromatography on activity IV neutral alumina (2.5 g, stepwise gradient hexane–Et2O 5 : 1, pure Et2O to pure MeOH) to afford a white solid. According to 1H NMR the solid was a mixture of 21 and 22 in a ratio 1 : 4.25 (9.81 mg, 90.0%).(2R*, 3aS*, 6aS*, 9aS*)-2-(p-Toluenesulfonyloxymethyl)-dodecahydro-pyrido[2,1,6-de]quinolizine (23)
A solution of alcohol 22 (10 mg, 0.05 mmol) and triphenylmethane (2 mg) in absolute THF (10 mL) was cooled to 0 °C under stirring and argon atmosphere. A 2.5 M solution of nBuLi in hexanes (21 μL, 0.053 mmol) was added from a syringe via a septum until the solution became slightly pink. The precipitated lithium alkoxide was stirred for 10 min at 0 °C. Then p-toluenesulfonyl chloride (9.1 mg, 0.05 mmol) in absolute THF (5 mL) was added dropwise. The resulting solution was stirred for 2.5 h at 0 °C. The formation of 23 was monitored by TLC. The mixture was diluted with Et2O (10 mL), washed with water (2 mL), brine (2 mL) and dried over Na2SO4. The volatiles were evaporated to give a crude product (30 mg) that was purified using column chromatography on activity IV neutral alumina (15 g, stepwise gradient of hexane–Et2O 5 : 1, 1 : 1 to 1 : 5) to afford 23 (16.5 mg, 94.8%) as a yellow oil: IR (cm−1) 2925, 2853, 1598, 1460, 1440, 1363, 1255, 1178, 1196; 1H NMR (400 MHz, CDCl3) 7.82–7.80 (2H, m), 7.38–7.36 (2H, m), 4.20 (1H, t, J = 9.6), 3.99 (1H, dd, J = 9.6 and 6.0), 2.93–2.86 (2H, m), 2.63–2.57 (1H, m), 2.46 (3H, s), 2.17–2.02 (2H, m), 1.82–1.66 (3H, m), 1.51–1.20 (9H, m), 1.18–0.78 (3H, m); 13C NMR (100 MHz, CDCl3) 144.8 (s), 133.0 (s), 129.8 (2C, d), 128.0 (2C, d), 73.3 (t), 58.1 (d), 57.3 (d), 43.2 (d), 34.5 (t), 34.4 (t), 31.5 (t), 31.2 (t), 31.0 (d), 26.6 (t), 26.3 (t), 23.0 (t), 21.7 (q), 19.5 (t); MS (EI) m/z 363 (6, M+), 208 (100), 188 (50), 176 (51), 148 (50), 91 (99); HRMS calcd for C20H30NO3S (M + H) 364.1941, found 364.1942.epi-(±)-Hippodamine (2)
To a solution of tosylate 23 (26.75 mg, 0.0736 mmol) in 2-butanone (20 mL) was added sodium iodide (110.21 mg, 0.735 mmol). The mixture was refluxed for 24 h under argon atmosphere and exclusion of light. The mixture was evaporated to dryness. Water (5 mL) was added and the resulting solution was extracted with EtOAc (3 × 10 mL). Combined extracts were washed with a saturated solution of sodium thiosulfate (5 mL), brine (2 mL) and dried over Na2SO4. The volatiles were evaporated to give crude 24 (23.4 mg) as a yellow oil (Rf 0.45, alumina, MeOH–EtOAc 1 : 1) that was used without further purification. A 5 mL round-bottom flask was charged with activated zinc25 (0.2 g, 3.06 mmol) and glacial acetic acid (3 mL) was added. The mixture was exposed to sonication for 5 min. Then crude iodide 24 (∼23.4 mg, 73.3 μmol) was added in glacial acetic acid (0.5 mL). The mixture was refluxed for 3 h. The mixture was allowed to reach room temperature and then filtered. The solids were washed with MeOH (1 mL) and the filtrate was evaporated. The residue was diluted with benzene (10 mL) and K2CO3 (1 g) was added. The mixture was stirred for 4 h. Solids were filtered off and the filtrate was evaporated to dryness to give a crude product (22 mg) that was purified using column chromatography on activity IV neutral alumina (1 g, hexane–Et2O 4 : 1) to afford 29 (11.8 mg, 83.1% over two steps) as a colourless oil: Rf 0.40 (alumina, MeOH–EtOAc 1 : 1); IR (cm−1) 2925, 2853, 1455, 1440, 1378, 1265, 1122, 1075, 1024; 1H NMR (400 MHz, CDCl3) 2.97–2.88 (3H, m), 2.13–2.06 (1H, m), 1.98–1.92 (1H, m), 1.89–1.79 (4H, m), 1.52–1.15 (10H, m), 1.10 (3H, d, J = 7.2), 1.03–0.9 (1H, m); 13C NMR (100 MHz, CDCl3) 58.4 (d), 58.3 (d), 43.1 (d), 40.5 (t), 36.6 (t), 34.7 (t), 31.5 (t), 27.0 (t), 26.7 (t), 25.7 (d), 23.4 (t), 22.1 (q), 19.8 (t); MS (EI) m/z 193 (54, M+), 192 (100), 178 (57), 165 (54), 151 (60), 150 (71), 111 (97); HRMS calcd for C13H24N (M + H) 194.1903, found 194.1905.(2R*, 3aS*, 6aS*, 9aS*)-2-Hydroxy-2-methyl-dodecahydro-pyrido[2,1,6-de]quinolizine (25)
To a stirred solution of ketone 18 (87.2 mg, 0.451 mmol) in toluene (10 mL) cooled to 0 °C under argon atmosphere was added 1.6 M MeLi in Et2O (846.0 μL, 1.353 mmol) in three portions over 3 h. Stirring was continued for another 5 h. A saturated solution of NH4Cl (1 mL) was added. The organic layer was separated and the aqueous one was extracted with benzene (3 × 5 mL). Combined extracts were dried over Na2SO4. The volatiles were evaporated to give a crude product (67 mg) that was purified using column chromatography on activity IV neutral alumina (10.6 g, stepwise gradient of hexane–Et2O 5 : 1 to 1 : 1 and finally pure Et2O). The product was allowed to crystallize from Et2O to afford 25 (57.8 mg, 61.2%) as crystals (clear, colourless plates): Rf 0.38 (alumina, MeOH–EtOAc 1 : 1); mp 146.8–147.1 °C (Et2O): IR (cm−1) 3365, 2919, 2854, 1459, 1442; 1H NMR (400 MHz, CDCl3) 3.14–3.06 (1H, m), 3.02–2.94 (2H, m), 2.36–2.24 (1H, m), 1.96–1.76 (4H, m), 1.64–1.40 (8H, m), 1.36–1.24 (1H, m), 1.20 (3H, s), 1.17–0.99 (2H, m); 13C NMR (100 MHz, CDCl3) 69.2 (s), 58.2 (d), 58.1 (d), 47.4 (t), 43.7 (d), 42.8 (t), 34.3 (t), 32.7 (q), 31.5 (t), 26.6 (t), 25.1 (t), 23.0 (t), 19.5 (t); MS (EI) m/z 209 (64, M+), 208 (100), 190 (42), 176 (35), 167 (61), 150 (66), 55 (54), 43 (90), 41 (99); HRMS calcd for C13H24NO (M + H) 210.1852, found 210.1851.Deoxygenation of 25
To a solution of tertiary alcohol 25 (23.5 mg, 0.112 mmol) in absolute THF (10 mL) 2.5 M nBuLi in hexanes (49.3 μL, 0.123 mmol) was added dropwise under stirring, argon atmosphere and at −78 °C. After 1 h carbon disulfide (26.9 μL, 0.448 mmol) was added and the mixture was allowed to reach room temperature. Stirring was continued for 3 h. MeI (27.9 μL, 0.448 mmol) was added and the mixture was stirred overnight. The mixture was filtered. The filtrate was evaporated and filtered again through a short column of neutral alumina activity IV. The crude xanthate 26 (14.0 mg) was used without any further purification. To a refluxing solution of tri-n-butyltin hydride (27.4 mg, 0.094 mmol) and AIBN (0.8 mg, 4.7 μmol) in toluene (10 mL) was added a solution of xanthate 26 (14.0 mg, 0.047 mmol) in toluene (7 mL) dropwise over 1 h. The mixture was refluxed for a further 4 h and left to stand overnight. Neutral alumina activity IV (0.5 g) was added and the mixture was evaporated to dryness under reduced pressure. The solid was purified using column chromatography on activity IV neutral alumina (10.0 g, hexane–Et2O 10 : 1) to give a mixture of 1 and 2 (combined yield 4.1 mg, 19% over 2 steps) in a ratio 2.3 : 1 (by 1H NMR).Acknowledgements
The authors would like to thank the Leverhulme Trust for funding this research. They would also like to thank Astra Zeneca for generous unrestricted funding of their research programme and the EPSRC Mass Spectrometry Service at the University of Wales, Swansea for carrying out high resolution mass spectra. The authors also acknowledge the use of the EPSRC's Chemical Database Service at Daresbury.28References
- A. G. King and J. Meinwald, Chem. Rev., 1996, 96, 1105 CrossRef CAS.
- S. Al Abassi, M. A. Birkett, J. Pettersson, J. A. Pickett, L. J. Wadhams and C. M. Woodcock, J. Chem. Ecol., 2001, 27, 33 CrossRef CAS.
- M. Langlois, J. L. Soulier, D. Yang, B. Bremont, C. Florac, V. Rampillon and A. Giudice, Eur J. Med. Chem., 1993, 28, 869 CAS.
- B. Tursch, D. Daloze, A. Pasteels, A. Cravador, J. C. Braekman, C. Hootele and D. Zimmermann, Bull. Soc. Chim. Belg., 1972, 81, 649 CAS.
- B. Tursch, D. Daloze, J. C. Braekman, C. Hootele, A. Cravador, D. Losman and R. Karlsson, Tetrahedron Lett., 1974, 5, 409 CrossRef.
- R. H. Mueller and M. E. Thompson, Tetrahedron Lett., 1980, 21, 1093 CrossRef CAS.
- R. H. Mueller, M. E. Thompson and R. M. DiPardo, J. Org. Chem., 1984, 49, 2217 CrossRef CAS.
- W. A. Ayer, R. Dawe, R. A. Eisner and K. Furuichi, Can. J. Chem., 1976, 54, 473 CAS.
- D. R. Adams, W. Carruthers and P. J. Crowley, J. Chem. Soc., Chem. Commun., 1991, 1261 RSC.
- M. Rejzek and R. A. Stockman, Tetrahedron Lett., 2002, 43, 6505 CrossRef CAS.
- W. Kitching, J. A. Lewis, M. V. Perkins, R. Drew, C. J. Moore, V. Schurig, W. A. König and W. Francke, J. Org. Chem., 1989, 54, 3893 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- R. Pappo, D. S. Jr. Allen, R. U. Lemieux and W. S. Johnson, J. Org. Chem., 1956, 21, 478 CrossRef CAS.
- D. Yang and C. Zhang, J. Org. Chem., 2001, 66, 4814 CrossRef CAS.
- B. Tse and Y. Kishi, J. Org. Chem., 1994, 59, 7807 CrossRef CAS.
- W. S. Wadsworth, J. Org. React., 1977, 25, 73 Search PubMed.
- Handbook of Reagents for Organic Synthesis: Oxidizing and Reducing Agents, ed. S. D. Burke and R. L. Danheiser, John Wiley & Sons, New York, 2000, 550 pp Search PubMed.
- J.-C. Braekman, A. Charlier, D. Daloze, S. Heilporn, J. Pasteels, V. Plasman and S. Wang, Eur. J. Org. Chem., 1999, 1749 CrossRef CAS.
- O. J. Osby, M. G. Martin and B. Ganem, Tetrahedron Lett., 1984, 25, 2093 CrossRef CAS.
- F. W. Wehrli, A. P. Marchand and S. Wehrli, Interpretation of Carbon-13 NMR Spectra, 2nd edn., John Wiley & Sons, Chichester, New York, Brisbane, Toronto, Singapore, 1983, 484 pp Search PubMed.
- W. Dieckmann, Ber. Dtsch. Chem. Ges., 1900, 33, 2670 CAS.
- Compound 25. Crystal data: C13H23NO, M = 209.3. Monoclinic, space group P21/c (no. 14), a = 9.766(3), b = 9.337(2), c = 13.767(2) Å, b = 102.38(2)°, V = 1226.1(5) Å3, Z = 4, Dc = 1.134 g cm−3, F(000) = 464, T = 293(2) K, μ(Mo-Kα) = 0.7 cm−1, λ(Mo-Kα) = 0.71069 Å. A clear, colourless plate crystal, ca. 0.62 × 0.21 × 0.05 mm, was mounted on a Nonius CAD4 diffractometer; of the 2511 reflections measured to θmax = 25°, 2146 were unique (Rint = 0.012) and 1263 ‘observed’ with I > 2σI. Refinement gave wR2 = 0.136 and R1 = 0.08129 for all 2146 reflections weighted w = [σ2(Fo2) + (0.0645P)2]−1; for the ‘observed’ data only, R1 = 0.047. CCDC reference number 248862. See http://www.rsc.org/suppdata/ob/b4/b413052a/ for crystallographic data in .cif or other electronic format.
- Compound 22. Crystal data: C13H23NO, M = 209.3. Monoclinic, space group P21/n (equiv. to no. 14), a = 7.660(2), b = 14.610(3), c = 10.367(2) Å, b = 91.13(3)°, V = 1160.0(4) Å3, Z = 4, Dc = 1.199 g cm−3, F(000) = 464, T = 140(1) K, μ(Mo-Kα) = 0.7 cm−1, λ(Mo-Kα) = 0.71069 Å. A clear, colourless tapered, rectangular prismatic crystal, ca. 0.50 × 0.25 × 0.20 mm, was mounted on a Rigaku R-Axis IIc image-plate diffractometer. Total no. of reflections recorded to θmax = 25.4°, was 6174 of which 2043 were unique (Rint = 0.052); 1851 were ‘observed’ with I > 2σI. Refinement gave wR2 = 0.103 and R1 = 0.04229 for all 2043 reflections weighted w = [σ2(Fo2) + (0.0384P)2 + 0.38P]−1 with P = (Fo2 + 2Fc2)/3; for the ‘observed’ data only, R1 = 0.039. CCDC reference number 248861. See http://www.rsc.org/suppdata/ob/b4/b413052a/ for crystallographic data in .cif or other electronic format.
- Tosylation of alcohol 22 using tosyl chloride, triethyl amine and trimethylammonium hydrochloride in dichloromethane gave a low yield of 23 (Method A in Scheme 7). For this reason 22 was first deprotonated using n-BuLi and then tosyl chloride was added giving the desired tosylate 23 in 95% yield (Method B in Scheme 7). B. L. Murr, B. J. Parkhill and A. Nickon, Tetrahedron, 1992, 48, 4845 Search PubMed.
- L. F. Fieser and M. Fieser, Reagents for Organic Synthesis, John Wiley & Sons, Inc., New York, London, Sydney, 1967, p. 1276 Search PubMed.
- B. Lebrun, J. C. Braekman and D. Daloze, Magn. Reson. Chem., 1999, 37, 60 CrossRef CAS.
- W. A. Ayer, M. J. Bennett, L. M. Browne and J. T. Purdham, Can. J. Chem., 1976, 54, 1807 CAS.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746 CrossRef.
- Both structures were determined by direct methods procedures in SHELXS and refined by full-matrix least-squares methods, on F2's, in SHELXL. In both structures, the hydroxyl hydrogen atom was located in a difference Fourier map and refined freely. In the final difference maps, the highest peaks were ca. 0.19 e Å−3 in 22 and ca. 0.15 e Å−3 in 25. G. M. Sheldrick, SHELX-97 - Programs for crystal structure determination (SHELXS) and refinement (SHELXL), University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Part 5. For Part 4 in the series see: L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371. |
| ‡ Electronic supplementary information (ESI) available: 13C NMR spectra for compounds 1, 2, 3, 7, 10, 13, 14, 18, 22, 23, and 25. X-Ray data for compounds 22 and 25. See http://www.rsc.org/suppdata/ob/b4/b413052a/ |
| This journal is © The Royal Society of Chemistry 2005 |