Application of tandem Ugi multi-component reaction/ring closing metathesis to the synthesis of a conformationally restricted cyclic pentapeptide
Silvia
Anthoine Dietrich
a,
Luca
Banfi
*a,
Andrea
Basso
a,
Gianluca
Damonte
b,
Giuseppe
Guanti
a and
Renata
Riva
a
aDepartment of Chemistry and Industrial Chemistry, via Dodecaneso 31, 16146, Genova, Italy. E-mail: banfi@chimica.unige.it; Fax: +390103536118; Tel: +390103536119
bDepartment of Experimental Medicine, Biochemistry Section, c/o Center of Excellence for Biomedical Research, University of Genova, v. le Benedetto XV, 16132, Genova, Italy
First published on 23rd November 2004
Abstract
A conformationally restricted cyclic pentapeptide, containing an unsaturated 9-membered lactam as a semi-rigid scaffold, was prepared in a very convergent manner, through tandem Ugi reaction/ring closing metathesis.
Introduction
The interaction of peptides with receptors1 or with other proteins2 has a strong influence on several physiological processes. Therefore, small molecules have been used as antagonists of protein function in the pharmaceutical industry since its very beginning. The effectiveness of protein–receptor or protein–protein interaction is strongly dependent on the secondary and tertiary structure of the peptides. Thus, small oligopeptides characterized by the same recognition sequence, but lacking the conformational restrictions typical of the natural macromolecules, may be completely inactive. Among the most important secondary motifs, the reverse-turns (e.g. the β-turns, the hairpins and so on) play a particularly important role. For this reason various research groups have tried to design and synthesize reverse-turn mimetics.This has been successfully accomplished by using cyclic penta- or hexapeptides,3,4 obtained by assembling proteinogenic amino acids. However, turn mimics composed of only amino acids are not ideal, because compounds of this kind may have unfavourable bioavailability properties.2
Another strategy involves the attachment of small peptide chains to an unnatural rigid or semi-rigid template (scaffold). In some cases this template represents the reverse-turn itself (and has been defined as “internal reverse-turn mimetic”).5 In other cases, the rigid scaffold serves to force an attached cyclic peptide chain to adopt the desired reverse-turn conformation (and has been defined as an “external reverse-turn mimetic”).5 This latter strategy seems particularly promising. The fact that the scaffold is “unnatural” should grant it a higher metabolic stability, compared to the cyclic penta- or hexapeptides. On the other hand, the fact that the recognition sequence is not placed on the template itself (as for “internal reverse-turn scaffolds”)2,6,7 allows for a higher degree of freedom in testing several different oligopeptides or peptidomimetics as recognition motifs.
In the past, various conformationally restricted reverse-turn scaffolds have been designed and synthesized. Most of them were fused bicyclic8–10 lactams, but also bridged bicyclic,11,12 spirocyclic,13 or monocyclic (with ring sizes from 3 to 7)14–18 lactams have been explored. However, only a few reports dealt with mesocyclic compounds.19–21
A drawback connected to most of the reverse turn scaffolds reported to date is that the synthetic approaches to them are poorly flexible. Even small variations of the substituents decorating the basic scaffold, which could be useful in order to finely tune the biological activity, require de novo syntheses. In the course of a project on the exploitation of isocyanide-based multi-component reactions followed by secondary transformations22–26 in diversity-oriented synthesis,27 we decided to design and synthesize a new family of “external” reverse-turn scaffolds characterized by an unsaturated 9-membered lactam (general formula 1, Scheme 1). According to our plan, various diverse members of this family could be prepared convergently in two steps by coupling a multi component reaction (the Ugi 4CR) with a subsequent ring closing metathesis (RCM). In this way, not only could the scaffolds be synthesized in just 2 steps, but several different substituents could be placed at will, as R1, R2, and R3. Although coupling of Ugi MCR with RCM has also been recently described by other groups, 17,28–31 the synthesis of 9-membered rings by this strategy was unprecedented. Also a related tandem Passerini/RCM was reported.30 In a preliminary communication32 we have described the successful synthesis of a series of compounds of general formula 1 through this approach. Now we report full details on the synthesis of a particular member of this family of scaffolds (namely 2) by two alternative synthetic routes, and its transformation into the first cyclic pentapeptides 3. The conformations of both 2 and 3 will be also thoroughly discussed, on the basis of NMR data.
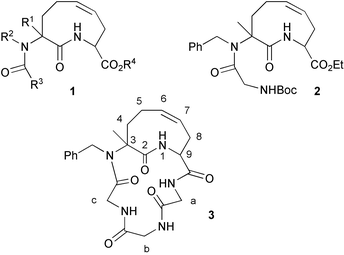 | ||
| Scheme 1 | ||
Results and discussion
Synthesis
For the preparation of the required scaffold 2, we first utilized the 2-step strategy already reported in our preliminary communication. It involved Ugi condensation of Boc-glycine 11 with isocyanoacetate 7 and the preformed imine derived from 5-hexen-2-one and benzylamine, followed by RCM of the resulting diene 12. The required isocyanide 7 was prepared, as a racemic mixture, in 4 high-yielding steps, starting from commercially available diethyl formamidomalonate, as described in Scheme 2. The Ugi condensation (Scheme 3) proceeded in high yield, but with no stereoselectivity at all, giving an inseparable mixture of the two diastereoisomers in a 1 : 1 ratio.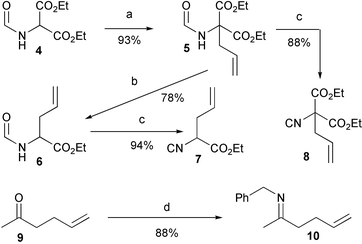 | ||
| Scheme 2 Reagents and conditions: a) NaH, allyl-Br, r.t. b) 1. NaOH, EtOH; 2. Dioxane, reflux. c) POCl3, Et3N, −30 °C. d) BnNH2, benzene, Dean–Stark. | ||
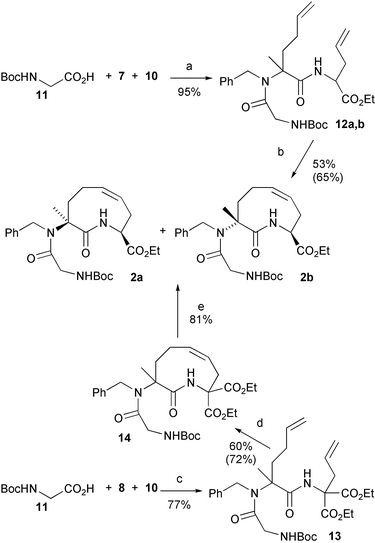 | ||
Scheme 3
Reagents and conditions: a) EtOH, r.t. b) 4 mM in CH2Cl2, PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ru(PCy3)2Cl2, reflux. c) EtOH, 65 °C. d) 6.5 mM in CH2Cl2, PhCHRu(PCy3)2Cl2, reflux. e) 1. NaOH, EtOH; 2. Dioxane, reflux. Yields in parentheses are based upon recovered starting material. Ru(PCy3)2Cl2, reflux. c) EtOH, 65 °C. d) 6.5 mM in CH2Cl2, PhCHRu(PCy3)2Cl2, reflux. e) 1. NaOH, EtOH; 2. Dioxane, reflux. Yields in parentheses are based upon recovered starting material. | ||
More critical was the ensuing ring closing metathesis. There were no examples in the literature of RCM that gives 9-membered secondary lactams. It is well known that the analogous cyclization of secondary amides to afford 8-membered lactams by RCM is unfeasible.28,33 This behaviour has been explained by the insufficient equilibrium concentration of the syn rotamer at the amide bond in the acyclic diene (only this rotamer is able to cyclize). It was suggested that, for the same reason, also secondary 9-membered lactams could not be obtained by RCM.28 On the other hand, both 8-membered28,33,34 and 9-membered lactams28 can be obtained starting from tertiary amides; in this case the relative abundance of the acyclic rotamers is expected to be closer to 1 : 1. However, while both secondary and tertiary unsaturated 8-membered lactams exist as the syn conformers, the previous report on the synthesis of tertiary unsaturated 9-membered lactams28 does not give information on the actual conformational equilibrium between syn and anti rotamers in the cyclic compounds.
On the basis of preliminary computer aided analysis,32 we expected the anti rotamer at the amide to be more stable than the syn one in secondary 9-membered unsaturated lactams 1.35 Therefore, the anti rotamer of the acyclic diene should, in this case, undergo cyclization even faster than the syn one. This prediction was later confirmed by NMR conformational analysis on the metathesis products (vide infra).
The yield of RCM was relatively good when carried out with Grubbs' 1st generation catalyst in refluxing CH2Cl2, even when at a concentration not dramatically low (4 mM). It is worth noting that formation of a 10-membered lactam by RCM was reported to afford satisfactory yields only at much lower concentrations (0.8 mM).19 It was difficult to drive the reaction to completion, mainly because the catalyst tended to become deactivated after 24–48 hours (the colour of the solution turned from pink-orange to black). This may be caused, as previously suggested,29,36 by interaction of the catalyst with one of the polar amidic groups. The overall 53% yield was raised to 65% if based upon recovered starting material. We tried to optimize the reaction by changing the solvent in order to increase the reaction temperature. However, in refluxing benzene, dichloroethane, or THF, the catalyst was rapidly deactivated. At lower temperatures these solvents were less satisfactory than CH2Cl2 anyway. We also tested Grubbs' 2nd generation catalyst. The reaction was indeed faster, and it was possible now to drive it to near-completion. However, the relative percentage of intermolecular products was increased. An analogous outcome was recently reported by Creighton and Reitz.34
The reaction was completely stereoselective with regard to the double bond, giving only Z compounds. The two diastereoisomers 2a,b were easily separated from each other, but were contaminated by several acyclic dimeric by-products derived from intermolecular processes, easily identified as such in the 13C NMR-DEPT spectra by the presence of terminal double bonds. It is worth noting that, even if only the less encumbered double bond (the one derived from the ketone), is involved in dimerization, and only E double bonds are formed, 6 different diastereoisomeric acyclic dimers may be obtained! Thus, in order to obtain 2a and 2b in pure form, 2–3 chromatographies with different eluents were necessary.
For this reason we decided to explore an alternative strategy, using this time, as the isocyanide component, the achiral malonate 8. In this approach the second stereogenic centre was planned to be generated only after the RCM reaction, thus reducing the number of diastereoisomeric products and by-products, and therefore simplifying the purification process. The Ugi condensation with 8 was slower, as expected, and required a higher temperature. However, the yield was still high enough. On the other hand, the yield and conversion of RCM were remarkably better in this case. Interestingly, the catalyst seemed to be much more stable and the solution did not change colour even after 3–4 days. The percentage of intermolecular products was also lower, and this allowed us, for preparative purposes, to increase the concentration to 6.5 mM. Purification of the product was much easier as well, since we had now only one (racemic) product, and only 2 by-products (reasonably well separated from 14) were detected by TLC. In this case, if only the less encumbered double bond is involved in dimerization, and E double bonds are formed, only 2 diastereoisomeric dimers are expected. Compound 14 was converted into a 6 : 4 mixture of 2a,b by saponification followed by decarboxylation.
The advantages of this alternative route are: a) The purification of RCM product 14 is much easier than that of RCM products 2a,b. b) A higher concentration may be used in the RCM step. c) The decarboxylation is moderately stereoselective favouring the cis isomer 2a, while from RCM of 12 a slightly higher percentage of the less wanted trans isomer 2b is obtained. e) the overall yield of the pure cis compound 2a from 5 is 24% instead of 20%; as discussed below, the cis scaffold is the most useful one.
With a good methodology for the rapid obtainment of both 2a and 2b in hand, we went on to transform these scaffolds into cyclic peptides. Since 2a and 2b were obtained in racemic forms, we chose, as the first example, to attach an achiral dipetide formed by two additional glycine units. Although the cyclic peptide 3 is not very attractive from the point of view of its biological properties, it has allowed us to check the ease of macrocycle formation for the two different diastereoisomers and to do a first preliminary conformational analysis in the absence of additional biases derived from side-chains of more complex amino acids.
Both 2a and 2b were elaborated to the acyclic triglycine derivatives 15a,b. Removal of the terminal protecting groups followed by cyclization in DMF–CH2Cl2 at 6 mM with HATU/collidine afforded the two diastereoisomeric cyclopeptides 3a and 3b (Scheme 4). In the case of the cis compound, the yield of 3a was quite good (55%) and the compound could be obtained in high purity by silica gel chromatography, as evidenced by NMR and HPLC/MS analysis. On the other hand, formation of the trans adduct 3b was much more troublesome. A crude product giving nearly a single spot in tlc was isolated in only 13% yield. Moreover, HPLC-MS analysis showed that, although the expected monomer 3b was the main component, the 2 diastereoisomers of the cyclodimer were also present (38%), as well as a small amount of 3a (8%). The actual yield of 3b was therefore only 7–8%. This result is not unexpected, since conformational analysis of 2b (vide infra) has indicated that the virtual dihedral angle between the nitrogen attached at C-3 and the carboxyl attached at C-9 as well as the distance between these two atoms is not favourable for cyclopeptide cyclization. Therefore, formation of 3b is probably disfavoured both enthalpically and entropically. The presence of 3a is probably due to traces of cis isomer not completely separated at the level of 2b. Because of the difference in cyclization yield, about 1% of 2a contaminating 2b may justify the presence of 8% 3a in crude 3b.
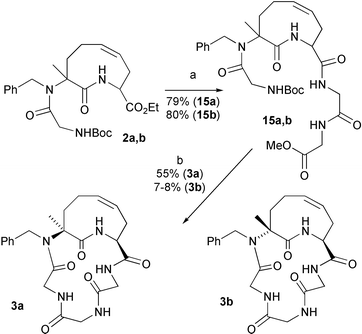 | ||
| Scheme 4 Reagents and conditions: a) 1. NaOH, EtOH, r.t.; 2. H-GlyGlyOMe, BOP, Et3N, CH2Cl2. b) 1. LiOH, THF–H2O; 2. TFA; 3. HATU, sym-collidine, DMF–CH2Cl2 (6 mM). | ||
Conformational analysis
The double bond configuration in metathesis adducts 2 and 14 as well as in their derivatives was demonstrated to be Z by the CHCH vicinal coupling constants (which were around 9–10 Hz). In compounds 2a and 2b only one set of signals was detected by NMR at r.t. This means that only one of the two possible rotamers around the ring amide bond is present. This rotamer is in both cases the anti one, as demonstrated by the presence of a strong NOE between the NH and the ortho hydrogens of the benzyl group in 2a and of a very strong NOE between the NH and the CH3 bonded at C-3 in 2b. These NOEs are obviously possible only for the anti rotamer and also allow us to determine without doubt the relative configuration of the two diastereoisomers.
From the coupling constants and NOEDIFF experiments it is also possible to get a fairly accurate idea of the preferred conformation of the 9-membered ring. In order to get some hints on the possible conformations of these tetrahydroazoninones we carried out an MM2 minimization on the parent compound with the software Chem3D (v. 4.5) from CSC, Cambridge (USA), looking for all local minima. These compounds were found to be not completely rigid. Four conformations with a comparable energy were found and three of them are depicted in Fig. 1. In the absence of substituents on the ring, conformations 16a and 16b are enantiomeric and therefore isoenergetic. The same applies for 17a,b (only one of the two conformations is shown). The difference between 16 and 17 is the position of the double bond. In 17 it is on the same side of NH; in 16 it is on the opposite side.
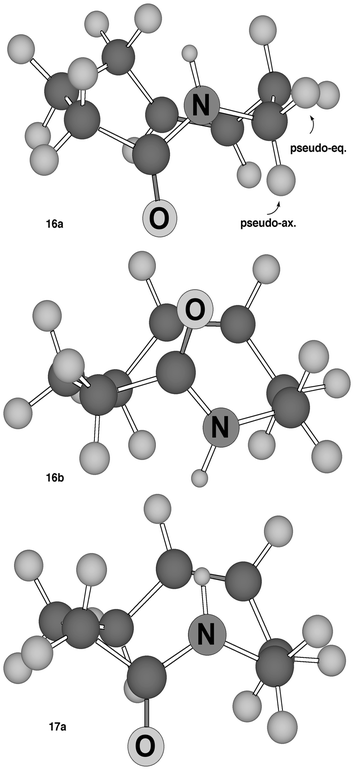 | ||
| Fig. 1 Conformations of the parent tetrahydroazoninones as minimized with Chem3D (MM2). | ||
In the following discussion, for the sake of clarity, we will consider the (9S) enantiomers of 2a and 2b, as depicted in Scheme 3. An examination of the NMR data indicates that, in both cases, a conformation analogous to 16a, placing the CO2Et in pseudo-equatorial (β) position, is actually preferred (see also Scheme 5). For example, in 2a, there is a high J1–9 of 9.3 Hz, which excludes conformation 16b, with the CO2Et in pseudo-axial position. The J8–9 (3.9 and 7.2 Hz) and J7–8 (6.7 and 9.0 Hz) are perfectly compatible with conformations 16, but not with 17. In 17 the dihedral angles related to J8–9 should be 41° and 76°, whereas those related to J7–8 should be 64° and 31° and so they are clearly not in agreement with the detected values. Moreover, NOEs between the ring NH and one of the H-8 and one of the H-5, and an NOE between H-9 and H-7 were detected. They are possible in 16, but not in 17. Also, other J values and NOEs are in agreement with the proposed conformation (see the Experimental section).
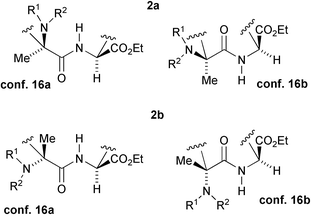 | ||
| Scheme 5 Simplified alternative conformations of 2a and 2b. | ||
For the trans isomer 2b, similar arguments demonstrate that also in this case the preferred conformation is 16a, with the CO2Et group in the pseudo-equatorial position.
The equilibrium between 16a and 16b is expected to be slow enough to allow detection of both conformations at r.t., as demonstrated in the case of malonate 14 (vide infra). The fact that only one set of signals is evident in the 1H and 13C NMR means that the preference for the conformations having the CO2Et in the pseudo-equatorial position should be quite high in both diastereoisomers.
It would be interesting to know which of the two diastereoisomers 2a and 2b is more stable. At first sight, the trans compound 2b is expected to be more stable, since the bulkier nitrogen substituent is in the pseudo-equatorial position. On the other hand, the fact that the nitrogen is planar (while CH3 is tetrahedral) and that a hydrogen bond may be formed between the carbonyl bonded to it and the ring NH could favour a pseudo-axial position of the nitrogen. The 1 : 1 ratio of the two conformers in malonate 14 (vide infra) and the only moderate stereoselectivity in the decarboxylation reaction suggest that the two stereoisomers should be quite close in energy.
Having established by NMR spectroscopy the approximate preferred conformation of the ring in 2a and 2b it was possible to check if they are well suited or not to act as external reverse-turn mimetics. Using Chem3D (MM2), we applied to 18 (a simplified analogue of 2a) the ring conformation 16a and carried out minimization, noticing that addition of the substituents does not change considerably the various bond angles and bond lengths of the ring (Scheme 6). We then compared the minimized structure with various types of β-turns. β-Turns are usually defined by two φ and two ψ angles. However, our templates have been designed in order to function as “external” reverse-turn scaffolds. So the coincidence of all of these 4 angles is not mandatory. On the contrary, it is more important to compare the virtual dihedral angles between NHAc, C-3, C-9 and CO2Me, as well as the distances between NHAc and CO2Me (Scheme 6). These values turned out to be quite similar to those determined for a type II′ β-turn (obviously if we consider the enantiomer of 18 they will be similar to a type II β-turn). Kessler has shown that a II′ β-turn may be ideal as an “external” template in preparing conformationally restricted RGD peptides to be employed as integrin ligands. In one of the most effective cyclic pentapeptides, this II′ β-turn was assured by a D-Phe-L-Val unit bonded to the RGD recognition sequence.37
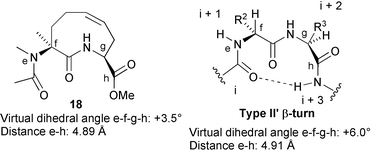 | ||
| Scheme 6 Conformational comparison of a simplified analogue of scaffold 2a and type II′ β-turn. The conformation of 18 was minimized with Chem3D and corresponds to conformation 16a (Fig. 1). Virtual dihedral angles and distances a–d for the type II′ β-turn were determined by applying standard bond length and angles and the dihedral angles typical of this β-turn. | ||
Using the same approach we calculated, for the trans (3R,9S) isomer of 18, a virtual dihedral angle and a distance of about +115° and 5.98 Å. While the angle is not far from the one typical of a type III′ β-turn, the distance is 1.4–1.5 Å higher. From these data it is clear that cis scaffold 2a is the most promising among the two isomers as an external reverse-turn mimetic. On the other hand, the low yield in the cyclization of trans15b may be explained by the excessive distance between the nitrogen at C-3 and the carbonyl at C-9 (which are the two attachment points for the tripeptide chain).
While 2a and 2b showed just one set of signals in the 1H and 13C NMR spectra, the malonate 14 gave two sets of signals, in a 1 : 1 ratio. These are most probably due to a slow conformational equilibrium between 16a and 16b. An alternative hypothesis would be a conformational equilibrium between the two rotamers at the tertiary amide, but in our opinion this is unlikely because: a) we do not see the reason why this equilibrium should be present in 14 and not in 2a,b; b) the equilibrium at the amide bond is not expected to influence in such a strong way the δ values of the ring protons and carbons; and finally, c) the NOEDIFF experiments showed, for example, that in one conformer the ring NH is close to the CH3 at C-3, while in the other one it is close to the aromatic protons. These NOEs are quite similar to those detected for 2a and 2b. All other NMR data (NOEs and J values) are in agreement with an equilibrium between the two conformations 16a and 16b. The fact that the ratio is 1 : 1 shows that the two groups at C-3 (CH3 and the nitrogen substituent) occupy the pseudo-equatorial and the pseudo-axial positions equally, suggesting also a similar energy for 2a and 2b.
Finally, the cis cyclic peptide 3a was carefully studied by NMR spectroscopy with the aid of NOEDIFF, COSY, and double resonance experiments, as well as measuring the difference in δ between d6-DMSO and CDCl3. A first important fact is that only one set of signals is present at r.t., either in d6-DMSO or in CDCl3. As already stated above, it is very unlikely that the two ring conformations related to 16a and 16b may interconvert fast enough at r.t. to give single signals. Also the two syn and anti rotamers of the tertiary amide would surely give different signals. The presence of a single set of signals means therefore that the equilibrium strongly favours a single conformer at the ring and at the tertiary amide bond.
Once again the J values and the NOEs related to the ring hydrogens gave results quite similar to those collected for the acyclic scaffolds 2a and 14. It seems therefore that the conformation of the 9-membered lactam is not strongly modified by the attachment to the tripeptide.
The signals of the NH groups of the three glycines were assigned thanks to NOEDIFF experiments: there is a NOE between the NH of glycine b and the CH2N of glycine a, and a (small) NOE between the NH of glycine c and the CH2N of glycine b (Scheme 7). The presence of a NOE between one of the CH2N groups of glycine c and the aromatic protons is consistent with an anti conformation around the tertiary amide bond.
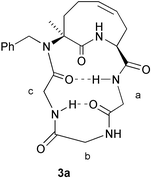 | ||
| Scheme 7 | ||
On passing from d6-DMSO to CDCl3 (0.01 M) the behaviour of the four NH protons is strikingly different: while the ring NH and the NH of glycine b displayed a lower δ in CDCl3 (−1.15 and −0.23 ppm respectively), for the NH of glycines c and a we observed a surprisingly higher δ in CDCl3 than that in DMSO (+0.91 compared to +1.00 ppm).
In order to gain further insight, we also measured the temperature dependance of these NH chemical shifts (in DMSO).38 The thermal coefficients (negative parts per billion per K) turned out to be 3.9 for ring NH, 4.9 for glycine b, 1.7 for glycine c, and −1.0 for glycine a. Moreover, the ring NH undergoes a very slow exchange with D2O (in DMSO). It takes more than 1 hour for the signal to disappear completely at r.t., while the signals of the other three NH disappear in less than 10 minutes.
The negative thermal coefficient for glycine a, and the higher δ in CDCl3 than in DMSO suggest its participation in a strong intramolecular hydrogen bond. Also for the NH of glycine c, the particularly high δ in CDCl3 and the low thermal coefficient may indicate the involvement in an intramolecular hydrogen bond. Finally, all data (including NOEs) suggest an insignificant participation of NH of glycine b in intramolecular hydrogen bonds.
As for the ring NH, both the temperature coefficient, as well as the CDCl3 − DMSO δ difference, point towards its non-involvement in hydrogen bonds. The very slow D2O exchange is somehow in contradiction with this assumption. However, this may be simply due to the fact that this NH is rather buried inside the molecule. Actually, a hydrogen bond between this NH and the carbonyl of glycine c or b would indeed prevent the observed NOEs with H-5 and H-8.
A tentative favoured conformation that seems in agreement with the collected NMR data is shown in Scheme 7 and involves a strong β-turn between glycines c (acting as amino acid i) and a (acting as aminoacid i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +3)
(in this β-turn the aminoacids i+1 and i+2 are those included in the scaffold) and a γ-turn between glycines c (i+2) and a (i).
+3)
(in this β-turn the aminoacids i+1 and i+2 are those included in the scaffold) and a γ-turn between glycines c (i+2) and a (i).
Conclusions
The cyclic peptidomimetic 3a, including a conformationally biased scaffold, has been straightforwardly prepared in only 5 steps thanks to a tandem Ugi reaction–ring closing metathesis. NMR studies have allowed us to suggest a preferred conformation for the scaffolds 2a,b and for the cyclic compound 3a. The formation of the trans cyclic peptide 3b in only low yield confirms that the corresponding scaffold is not a good external reverse-turn mimetic. On the other hand, the cis scaffold 2a mimics well a type II′ β-turn and appears to be well suited for the synthesis of conformationally biased cyclopeptides or cyclopeptidomimetics. The promising properties of 2a have been demonstrated by the good yield achieved in macrocyclization, by the fact that the macrocyclic peptide 3a seems to be conformationally defined, and by the probable presence of strong intramolecular hydrogen bonds corresponding to β- and γ-turns.Although all the compounds described in this work are racemic, and GGG is not a biologically relevant recognition sequence, these results open the way to the preparation of other cyclic peptides including enantiomerically pure scaffolds 1 and more useful tripeptide sequences (such as the RGD one) attached to it. Studies directed toward this goal are in progress in our laboratory.
Experimental
NMR spectra were taken at 200 or 300 MHz (1H), and 50 or 75 MHz (13C), using as internal standard: TMS for 1H NMR in CDCl3, the central peak of DMSO (at 2.506 ppm) for 1H NMR in d6-DMSO, the central peak of CDCl3 (at 77.02 ppm) for 13C NMR in CDCl3, the central peak of DMSO (at 39.429 ppm) for 13C NMR in d6-DMSO. Chemical shifts are reported in ppm (δ scale), coupling constants are reported in hertz. Peak assignment in 1H NMR spectra was also made with the aid of double resonance and COSY experiments. In some cases coupling constants were not directly determined from normal spectra, but with the aid of double resonance or D2O exchange experiments. In these cases they are indicated with a * symbol. In AB systems, the proton A is considered downfield and B upfield. Peak assignment in 13C spectra was made with the aid of DEPT experiments.GC-MS were carried out on an HP-5971A instrument, using an HP-1 column (12 m long, 0.2 mm wide), electron impact at 70 eV, and a mass temperature of about 170 °C. Only m/z > 33 were detected. All analyses were performed with a constant He flow of 0.9 mL min−1, and (unless otherwise stated) with initial temperature of 100 °C, initial time 2 min, rate 20 °C min−1, final temperature 260 °C, final time 4 min, injection temperature 250 °C, detector temperature 280 °C. Rt are in min.
HPLC-MS were carried out with an Agilent 1100 LC/MSD Trap SL instrument (electrospray ion trap analysis) with a C18 reverse phase Polarity column (Waters Corporation, MA, USA). In all cases, before introducing the eluent in the MS, a detection at 220 nm was performed using a diode array detector integrated in the system. The MS electrospray ion source parameters were set to maximize, from time to time, the interesting m/z ratios.
IR spectra were measured with a Perkin–Elmer 881 instrument as CHCl3 solutions. Melting points were measured on a Büchi 535 apparatus and are uncorrected. TLC analyses were carried out on silica gel plates and developed with I2 vapour. Rf values were measured after an elution of 7–9 cm. Chromatographies were carried out on 220–400 mesh silica gel using the “flash” methodology. Petroleum ether (40–60 °C) is abbreviated as PE. In extractive work-up, aqueous solutions were always reextracted thrice with the appropriate organic solvent. Organic extracts were washed with brine, dried over Na2SO4 and filtered before evaporation of the solvent under reduced pressure. Dry solvents were purchased from Fluka. All reactions employing dry solvents were carried out under a nitrogen (or argon when specified) atmosphere. The purity of all compounds was established by TLC, 1H and 13C NMR, and by GC-MS or HPLC or HPLC-MS. Diethyl formamidomalonate, 5-hexen-2-one, Boc-glycine, sodium hydride and Grubbs’ catalyst were all purchased from Fluka or Aldrich and used as such.
Diethyl (allyl)(formamido)malonate 5
Sodium hydride (60% in mineral oil) (1.652 g, 41.31 mmol) was suspended in dry dimethylformamide (DMF) (40 mL) and cooled to 0 °C. A solution of diethyl formamidomalonate (7.994 g, 39.34) in dry DMF (20 mL) was slowly added over 5 min. After 10 min, the cooling bath was removed and the mixture stirred for 30 min at r.t. Allyl bromide (3.57 mL, 4.997 g, 41.31 mmol) was then added via a syringe. After stirring for 3 h, and cooling in an ice bath, the reaction was quenched with saturated aqueous NH4Cl (100 mL). The mixture was extracted with Et2O (70 mL + 50 mL) and with AcOEt (2 × 50 mL). The united organic extracts were washed with H2O and evaporated to give an oil. It was taken up with CH2Cl2 and n-octane and evaporated again. After removing the solvent overnight at 8 × 10−2 mbar, a solid (9.92 g) was obtained. Trituration with n-pentane gave pure 5 as a white solid (8.904 g, 93%). Rf 0.57 (PE–AcOEt 1 : 1); mp 64.5–65.8 °C. Found: C, 54.45; H, 7.0; N, 5.7%. C11H17NO5 requires: C, 54.31; H, 7.04; N, 5.76%; GC-MS Rt 5.48; m/z 243 (M+, 1.8), 198 (28.2), 174 (25.7), 170 (47.1), 151 (3.4), 146 (5.9), (44.1), 124 (80.1), 123 (7.1), 118 (6.6), 114 (7.3), 112 (6.2), 97 (8.2), 96 (43.4), 74 (5.6), 69 (13.8), 68 (100.0), 67 (11.0), 56 (8.2), 47 (16.2), 43 (9.4), 42 (14.9), 41 (65.7), 39 (19.0). 1H NMR (CDCl3, 200 MHz): δ 8.17 [1 H, s, CHO]; 6.93 [1 H, s, NH]; 5.58 [1 H, ddt, CHCH2, Jd 9.4, 17.2, Jt 7.4]; 5.27–5.08 [2 H, m, CH2CH]; 4.27 [4 H, q, CH2CH3, J 7.1]; 3.10 [2 H, d, CH2CHCH2, J 7.4]; 1.27 [6 H, t, CH3, J 7.1].
Ethyl (allyl)(formamido)acetate 6
A solution of diethyl (allyl)(formamido)malonate 5 (8.33 g, 34.24 mmol) in 96% EtOH (300 mL), was treated, at r.t., with 6 M aqueous NaOH (6.85 mL, 41.09 mmol) and stirred for 140 min. Then a mixture of 32% HCl (2.69 mL, 27.39 mmol) and 96% EtOH (10 mL) was added. After evaporation of nearly all the EtOH, the residue was taken up with 0.5 M citric acid (20 mL), saturated NaCl (50 mL) and Et2O (100 mL). The phases were separated and the aqueous one re-extracted twice with Et2O and twice with AcOEt. The organic phases gave, on evaporation, a white solid (6.876 g). It was taken up in 1,4-dioxane and refluxed for 6 h. Evaporation gave an oil that was chromatographed on 90 g of silica gel (PE–AcOEt 1 : 1 to 4 : 6) to give pure 6 as a slightly yellow oil (4.629 g, 79%). Rf 0.62 (PE–AcOEt 1 : 1 + 5% AcOH). Found: C, 56.1; H, 7.75; N, 7.9%. C8H13NO3 requires: C, 56.13; H, 7.65; N, 8.18%; GC-MS Rt 3.54; m/z: 171 (M+, 1.5), 130 (59.1), 126 (45.2), 125 (16.3), 102 (16.4), 98 (100.0), 97 (12.9), 81 (9.6), 74 (86.4), 71 (6.4), 70 (44.4), 68 (8.7), 54 (6.0), 53 (6.7), 47 (5.6), 46 (21.4), 43 (23.3), 42 (12.0), 41 (19.5), 39 (15.7). 1H NMR (CDCl3, 200 MHz): δ 8.21 [1 H, s, CHO]; 6.20 [1 H, broad s, NH]; 5.69 [1 H, ddt, CHCH2, Jd 9.0, 17.6, Jt 7.1]; 5.25–5.08 [2 H, m, CH2CH]; 4.76 [1 H, dt, CHNH, Jd 8.0, Jt 6.0]; 4.32–4.12 [2 H, m, CH2CH3]; 2.73–2.46 [2 H, m, CH2CHCH2]; 1.30 [3 H, t, CH3, J 7.2].
Ethyl (allyl)(isocyano)acetate 7
A solution of 6 (2.983 g, 17.4 mmol) in dry CH2Cl2 (50 mL) was cooled to −30 °C and treated with Et3N (8.2 mL, 59.4 mmol) and POCl3 (1.75 mL, 19.2 mmol). After 25 min the reaction was quenched with saturated aqueous NaHCO3 (80 mL). The mixture was allowed to warm to r.t., extracted with Et2O, and evaporated to dryness. Chromatography (PE–Et2O 8 : 2) gave pure 7 as a colourless oil (2.510 g, 94%). Rf 0.54 (PE–Et2O 6 : 4). Found: C, 62.45; H, 7.3; N, 9.0%. C8H11NO2 requires: C, 62.73; H, 7.24; N, 9.14%; GC-MS Rt 1.55; m/z: 125 (M+ − 28, 16.8), 124 (3.9), 108 (28.4), 107 (20.2), 98 (28.4), 97 (28.0), 85 (8.3), 84 (20.0), 81 (27.2), 80 (74.8), 79 (27.7), 70 (22.6), 69 (19.0), 68 (10.1), 67 (8.5), 57 (7.0), 56 (12.9), 55 (21.1), 54 (88.5), 53 (100.0), 52 (25.2), 51 (17.1), 50 (7.3), 45 (6.3), 43 (13.6), 42 (30.5), 41 (38.7), 39 (71.5). 1H NMR (CDCl3, 200 MHz): δ 5.81 [1 H, ddt, CHCH2, Jd 9.8, 17.2, Jt 7.2]; 5.32–5.19 [2 H, m, CH2CH]; 4.28 [2 H, q, CH2CH3, J 7.2]; 2.76–2.50 [2 H, m, CH2CHCH2]; 1.32 [3 H, t, CH3CH2, J 7.2]. IR: νmax 2151, 1754, 1433, 1371, 1331, 1270, 1242, 1190, 1020, 925 cm−1.
Diethyl (allyl)(isocyano)malonate 8
Prepared from 5 (6.386 g, 26.25 mmol) following the same procedure employed for 7. Yield after chromatography: 5.192 g (88%) of a colourless oil that had some tendency to darken on standing, Rf 0.56 (PE–AcOEt 8 : 2). Found: C, 58.8; H, 6.9; N, 6.0%. C11H15NO4 requires: C, 58.66; H, 6.71; N, 6.22%; GC-MS Rt 4.04; m/z 226 (M+ + 1, 0.1), 199 (M+ − 26, 1.1), 198 (1.0), 197 (2.7), 169 (64.5), 153 (15.8), 152 (21.9), 151 (100.0), 134 (9.1), 133 (73.9), 129 (8.1), 125 (27.5), 124 (61.6), 123 (10.7), 111 (35.9), 110 (5.0), 108 (16.4), 107 (17.1), 106 (28.9), 105 (16.7), 101 (8.0), 99 (5.7), 98 (9.8), 97 (19.2), 96 (32.2), 81 (24.3), 89 (68.5), 79 (45.5), 78 (48.7), 69 (45.0), 68 (75.7), 67 (22.7), 57 (67.0), 54 (43.6), 53 (71.5), 52 (60.3), 41 (60.4), 39 (44.3). 1H NMR (CDCl3, 200 MHz): δ 5.79 [1 H, ddt, CHCH2, Jd 9.8, 17.2, Jt 7.2]; 5.35–5.20 [2 H, m, CH2CH]; 4.32 [4 H, q, CH2CH3, J 7.1]; 2.90 [2 H, d, CH2CHCH2, J 7.0]; 1.32 [3 H, t, CH3CH2, J 7.1]. IR: νmax 2142, 1752, 1368, 1298, 1192, 1147, 1095, 1036, 924 cm−1.
N-(Hex-5-en-2-ylidene)(phenyl)methanamine 10
A solution of 5-hexenone (10.16 mL, 87.73 mmol) and benzylamine (8.547 g, 79.76 mmol) in benzene was refluxed in a Dean–Stark apparatus for 4 h. Evaporation of benzene at 16 mbar, followed by distillation at 0.55 mbar, afforded 10 (pure by GC-MS) as a colourless liquid (bp 85–90 °C), which tended to darken if left in the light (13.20 g, 88%). NMR spectroscopy indicated a trans : cis ratio of 3.9 : 1. GC-MS (injection temperature: 200 °C, initial temperature: 45 °C, initial time: 2 min, rate: 20 °C min−1): Rt 7.97; m/z 187 (M+, 5.1), 186 (15.0), 172 (4.5), 104 (19.6), 91 (100.0), 65 (13.6), 42 (5.9), 41 (4.9), 39 (5.5). 1H NMR (200 MHz): δ 7.33 (cis), 7.31 (trans) [5 H, s, arom.]; 5.98–5.70 [1 H, m CHCH2]; 5.14–4.93 [2 H, m, CHCH2]; 4.52 (cis), 4.49 (trans)
[2 H, s, CH2Ph]; 2.50–2.27 [4 H, m, CH2CH2CH]; 2.09 (cis), 1.91 (trans)
[3 H, s, CH3].
Ugi adducts 12a,b
A solution of isocyanide 7 (1.784 g, 11.66 mmol) in absolute EtOH (12 mL), was treated at room temperature with imine 10 (2.609 g, 13.93 mmol) and with Boc-glycine 11 (2.442 g, 13.94 mmol). After stirring under N2 for 48 h, the solvent was evaporated, and the crude product chromatographed (PE–AcOEt 1 : 1 to 4 : 6) to give a pure 1 : 1 mixture of 12a,b as a foam (5.714 g, 95%). Found: C, 64.9; H, 8.1; N, 7.95%. C28H41N3O6 requires: C, 65.22; H, 8.01; N, 8.15%. 1H NMR (CDCl3, 200 MHz): δ 7.53–7.24 [5 H, m, arom.]; 6.35 [0.5 H, d, NH, J 7.3]; 6.32 [0.5 H, d, NH, J 7.3]; 5.84–5.68 [2 H, m, CHCH2]; 5.43 [1 H, broad s, NHBoc]; 5.20–4.90 [4 H, m, CHCH2]; 4.73–4.54 [3 H, m, CHN and CH2Ph]; 4.30–4.12 [2 H, m, CH2CH3]; 4.03–3.91 [1.5 H, CH2NHBoc]; 3.84 [0.5 H, dd, CHHNHBoc, J 4.0, 17.2]; 2.70–2.48 [2 H, m, allylic CH2]; 2.30–1.70 [4 H, m, allylic CH2 and CH2CH2CH]; 1.45 and 1.44 [3 H, 2 s, CH3]; 1.40 [9 H, s, (CH3)3C]; 1.284 [1.5 H, t, CH3CH2, J 7.2]; 1.280 [1.5 H, t, CH3CH2, J 7.2]. 13C NMR (50 MHz, CDCl3)(the number of diastereoisomers is given in brackets): δ 173.23 (1d), 173.01 (1d), 171.84 (2d), 169.94 (2d)
[CO]; 155.65 (2d)
[urethane CO]; 137.51 (2d)
[quat. arom.]; 137.38 (2d), 132.51 (2d)
[CHCH2]; 129.19 (2d), 127.61 (2d), 125.96 (2d)
[arom. CH]; 119.04 (2d), 115.21 (2d)
[CHCH2]; 79.54 (2d)
[C(CH3)3]; 65.66 (1d), 65.59 (1d)
[quat. C–N]; 61.48 (2d)
[CH2CH3]; 51.74 (1d), 51.70 (1d)
[CHN]; 47.58 (1d), 47.35 (1d)
[CH2Ph]; 43.36 (2d)
[CH2N]; 36.50 (1d), 36.43 (1d), 35.32 (1d), 34.93 (1d)
[allylic CH2]; 28.32 (2d)
[C(CH3)3
+
CH2CH2CH]; 21.45 (1d), 20.99 (1d)
[CH3]; 14.20 (2d)
[CH3CH2].
Ugi adduct 13
A solution of isocyanide 8 (3.869 g, 17.18 mmol) in absolute EtOH (17 mL), was treated at room temperature with imine 10 (2.0875 g, 11.15 mmol) and with Boc-glycine 11 (2.000 g, 11.41 mmol). After stirring under N2 at 65 °C for 11 h, and at r.t. for 48 h, the solvent was evaporated, and the crude product chromatographed (PE–AcOEt 6 : 4 to 1 : 1) to give pure 13 as a foam (5.130 g, 77%). Found: C, 63.05; H, 7.8; N, 7.05%. C31H45N3O8 requires: C, 63.35; H, 7.72; N, 7.15%. 1H NMR (CDCl3, 200 MHz): 7.52–7.22 [5 H, m, arom.]; 7.05 [1 H, s, NH]; 5.92–5.62 [2 H, m, CHCH2]; 5.41 [1 H, broad s, NHBoc]; 5.24–4.91 [4 H, m, CHCH2]; 4.60 and 4.56 [2 H, AB system, CH2Ph, J 19.4]; 4.30–4.15 [4 H, m, CH2CH3]; 4.02 and 3.80 [2 H, AB part of ABX system, CH2NHBoc, JAB 17.2, JAX 4.4, JBX 4.0]; 3.10 [2 H, d, C–CH2CH, J 7.4]; 2.37–1.70 [4 H, m, CH2CH2CH]; 1.45 [3 H, s, CH3]; 1.40 [9 H, s, C(CH3)3]; 1.25 and 1.23 [6 H, 2 t, CH3CH2, J 7.2]. 13C NMR (CDCl3, 50 MHz): δ 172.11, 169.78, 167.82, 167.71 [CO]; 155.61 [urethane CO]; 137.53 [quat. arom.]; 137.40, 132.05 [CHCH2]; 129.19, 127.50, 125.92 [arom. CH]; 119.53, 115.28 [CHCH2]; 79.56 [C(CH3)3]; 66.30, 65.39 [quat. CN]; 62.48, 62.38 [CH2CH3]; 47.64 [CH2Ph]; 43.31 [CH2NHBoc]; 36.97, 34.99 [allylic CH2]; 28.34 [C(CH3)3]; 28.20 [CH2CH2CH]; 21.36, 14.02 [CH3].
(Z)rac-3-(((N-Benzyl-t-butoxycarbonyl)amino)acetamido)-9,9-bis(ethoxycarbonyl)-3-methyl-4,5,8,9-tetrahydro-1H-azonin-2(3H)-one 14
A solution of diene 13 (2.250 g, 3.83 mmol) in dry CH2Cl2 (600 mL) was placed under argon, and treated with benzylidene bis(tricyclohexylphosphine)ruthenium dichloride (393 mg, 0.478 mmol). In order to ensure removal of all oxygen present, a series of vacuum/argon cycles was carried out. The orange solution was refluxed for 48 h. Et3N (1 mL) was added, and most of the solvent was evaporated, until about 100 mL were left. This solution was introduced in a column of silica gel packed with CH2Cl2. Elution with CH2Cl2–AcOEt–EtOH–Et3N 74 : 20 : 5 : 1 to 69 : 25 : 5 : 1 gave a black crude product containing both substrate and products (2.362 g). It was chromatographed with AcOEt–PE–EtOH 34.5 : 64.5 : 0.5 to 64.5 : 34.5 : 0.5 to give pure 14 as a grey foam (1.278 g, 60%). Also 382 mg of starting material (17%) were recovered. Yield based on recovered starting material was 72%. Apart from 14 and the substrate 13, the crude product showed the presence of 2 impurities (faster eluting than 14 in PE–AcOEt). They were both intermolecular dimers, characterized by the presence of terminal double bonds (clearly detected with 13C/DEPT NMR spectroscopy). Rf 0.18 (AcOEt–PE 6 : 4), 0.74 (AcOEt–PE 9 : 1). Found: C, 62.2; H, 7.4; N, 7.45%. C29H41N3O8 requires: C, 62.24; H, 7.38; N, 7.51%. 1H NMR (CDCl3, 300 MHz) (the spectrum shows 2 distinct conformers in a 1 : 1 ratio): δ 7.52–7.22 [5 H, m, arom.]; 6.89, 6.74 [1 H, 2 s, NH of 2 conf.]; 5.74–5.55 [1 H, m, H-6 of 2 conf.]; 5.52 [1 H, broad s, NHBoc]; 5.55–5.42 [0.5 H, m, H-7 of 1 conf.]; 5.14 [0.5 H, dt, H-7 of 1 conf., Jd 7.2, Jt 10.3]; 4.62 and 4.55 [1 H, slightly broad AB system, CH2Ph of 1 conf., JAB 17.1]; 4.50 [1 H, s, CH2Ph of 1 conf.]; 4.46–4.22 [4 H, m, CH2CH3]; 4.15–4.01 [1 H, m, CHHNHBoc of 2 conf.]; 3.85 [0.5 H, broad d, CHHNHBoc of 1 conf., J 15.9]; 3.50 [0.5 H, slightly broad dd, CHHNHBoc of 1 conf., J 1.9, 17.0]; 3.39 [0.5 H, t, pseudo-eq. H-8 of 1 conf., J 13.8]; 3.35 [0.5 H, t, pseudo-eq. H-8 of 1 conf., J 13.8]; 2.59 [0.5 H, dd, pseudo-ax. H-8 of 1 conf., J 6.9, 13.8]; 2.50 [0.5 H, dd, pseudo-ax. H-8 of 1 conf., J 6.5, 13.8]; 2.45–2.34 [0.5 H, m, H-5 of 1 conf.]; 2.10–1.80 [3.5 H, m, H-5, H-4]; 1.67 [1.5 H, s, CH3 of 1 conf.]; 1.41 [6 H, s, CH3 of 1 conf. + C(CH3)3 of 1 conf.]; 1.37 [4.5 H, s, C(CH3)3 of 1 conf.]; 1.36–1.23 [6 H, m, CH3CH2]. From COSY we could establish that the signal at δ 5.14 is coupled with those at δ 3.39 and 2.59. Irradiating the NH at δ 6.74 there was an NOE of 3.2% on the methyl at δ 1.67, and an NOE of 0.5% on H-8 at δ 2.50. On irradiating the NH at δ 6.89 there was a NOE of 7% on the ortho aromatic H (doublet at δ 7.28 with J 7.0) and a NOE of 1% on H-7 at δ 5.14 ppm. Therefore the signals at δ 6.89, 5.14, 3.39, 2.59, 1.41 are all due to the conformer with the CH3 on the opposite side of the ring NH; the signals at δ 6.74, 5.55–5.42, 3.35, 2.50, 1.67 are due to the conformer with the CH3 group on the same side as the ring NH. 13C NMR (CDCl3, 50 MHz): 171.68, 171.25, 170.26, 169.51, 168.60, 168.00, 167.25, 166.96 [CO]; 155.64 [urethane CO, 2 conf.]; 137.53 [quat. arom., 2 conf.]; 137.31, 135.71 [CHCH]; 129.48, 129.10, 127.74, 127.37, 125.71, 125.39 [arom. CH]; 124.04, 121.75 [CHCH]; 79.50, 79.27 [C(CH3)3]; 65.48, 64.80, 64.19, 64.08 [quat. CN]; 63.00, 62.76, 62.17 (2 conf.)
[CH2CH3]; 46.99, 46.07 [CH2Ph]; 43.51, 42.93 [CH2NHBoc]; 38.62, 37.78 [CH2CH]; 31.92, 29.19 [CH2CH]; 28.30 [C(CH3)3]; 23.84, 19.88 [CH2CH2CH]; 22.40, 20.50 [CH3]; 14.03, 13.98 [CH3CH2].
(Z,3R*,9R*) and (Z,3R*,9S*)-3-(((N-Benzyl-t-butoxycarbonyl)amino)acetamido)-9-(ethoxycarbonyl)-3-methyl-4,5,8,9-tetrahydro-1H-azonin-2(3H)-ones 2a and 2b
2a
(cis): Rf 0.26 (PE–AcOEt 3 : 7). Found: C, 63.9; H, 7.7; N, 8.4%. C26H37N3O6 requires: C, 64.05; H, 7.65; N, 8.62%. 1H NMR (300 MHz, CDCl3): δ 7.42 [2 H, t, meta-H, J 7.5]; 7.32 [1 H, t, para-H, J 7.5]; 7.25 [2 H, d, ortho-H, J 7.7]; 6.30 [1 H, d, NH, J 9.3]; 5.54 [1 H, broad s, NHBoc]; 5.54–5.40 [1 H, m, H-6]; 5.33 [1 H, slightly broad q, H-7, J
≈ 9.0]; 4.74–4.52 [2 H, m, CH2Ph]; 4.58 [1 H, ddd, CHN, J
≈ 3.9, 7.2, 9.3]; 4.22 [2 H, q, CH2CH3, J 7.2]; 4.10 and 4.02 [2 H, AB part of ABX system, CH2NHBoc, JAB 16.9, JAX 4.7, JBX 3.9]; 2.79 [1 H, broad ddd, H-8 α, J7–8
≈ 9.0*]; 2.17 [1 H, dt, H-8 β, Jd 13.5, Jt 6.7]; 2.10–1.88 [3 H, m, H-5 (2) and H-4 (1)]; 1.88–1.74 [1 H, m, H-4 (1)]; 1.44 [3 H, s, CH3C]; 1.41 [9 H, s, (CH3)3C]; 1.30 [3 H, t, CH3CH2, J 7.1]. NOEDIFF: on irradiating the NH at δ 6.30: 5.0% overall NOE on benzyl ortho-H; 2.4% NOE on H-8 β; 4.6% NOE on one of the H-5; no NOE on H-8 β or on CH3C. On irradiating the CH3 at δ 1.44: 1.2% NOE on benzyl ortho-H; 0.9% NOE on H-6; 1.2% on H-4 downfield; no NOE on NH. On irradiating H-7: 1.2% NOE on H-9. 13C NMR (75 MHz, CDCl3): δ 171.68, 171.56, 170.39 [CO]; 155.65 [urethane CO]; 136.92 [quat. arom.]; 135.28 [CHCH]; 129.36, 127.92, 125.71 [arom. CH]; 122.51 [CHCH]; 79.57 [C(CH3)3]; 65.68 [CNBn]; 61.47 [CH2CH3]; 51.12 [CHN]; 47.45, 43.65 [CH2N]; 38.83 [CH2CH]; 29.86 [CH2CH]; 28.26 [C(CH3)3]; 21.91 [CH2CH2CH]; 21.56 [CH3]; 14.11 [CH3CH2]. IR: νmax 3427, 2971, 1728, 1680, 1489, 1448, 1402, 1369, 1163, 1054 cm−1.
2b
(trans): Rf 0.18 (PE–AcOEt 3 : 7). Found: C, 63.85; H, 7.6; N, 8.35%. C26H37N3O6 requires: C, 64.05; H, 7.65; N, 8.62%. 1H NMR (300 MHz, CDCl3): δ 7.54 [2 H, d, ortho-H, J 7.6]; 7.38 [2 H, t, meta-H, J 7.3]; 7.26 [1 H, t, para-H, J 7.4]; 6.18 [1 H, d, NH, J 9.6]; 5.65–5.30 [3 H, m, CH2CH2CHCH and NHBoc]; 4.71 [1 H, dt, CHN, Jd 3.0, Jt 9.8]; 4.56 [2 H, s, CH2Ph]; 4.32–4.06 [3 H, m, CH2CH3 and CHHNHBoc]; 3.59 [1 H, dd, CHHNHBoc, J 3.0, 17.2] 2.62–2.42 [1 H, m H-8 α, small J with H-9 (from COSY)]; 2.40–2.10 [2 H, m, H-8 β
+
H-5]; 2.10–1.80 [3 H, m, H-5 (1) and H-4 (2)]; 1.67 [3 H, s, CH3C]; 1.37 [9 H, s, (CH3)3C]; 1.31 [3 H, t, CH3CH2, J 7.1]. NOEDIFF: on irradiating the NH at δ 6.18: 3.4% NOE on C–CH3; 5.0% NOE on H-8 β. On irradiating the CH3 at δ 1.65: 1.1% NOE on benzyl ortho-H; 17.5% NOE on NH; 6.0% NOE on one of the H-5. 13C NMR (50 MHz, CDCl3): δ 172.26, 171.62, 169.73 [CO]; 155.65 [urethane CO]; 137.57 [quat. arom.]; 134.40 [CHCH]; 129.05, 127.36, 125.79 [arom. CH]; 124.06 [CHCH]; 79.33 [C(CH3)3]; 64.22 [CNBn]; 61.65 [CH2CH3]; 51.66 [CHN]; 45.87, 42.99 [CH2N]; 38.14 [CH2CH]; 33.00 [CH2CH]; 28.26 [C(CH3)3]; 23.02 [CH2CH2CH]; 20.90 [CH3]; 14.13 [CH3CH2].
(Z,3R*,9R*)-3-(((N-Benzyl-t-butoxycarbonyl)amino)acetamido)-9-(((((methoxycarbonylmethyl)amino)carbonylmethyl)amino)carbonyl)-3-methyl-4,5,8,9-tetrahydro-1H-azonin-2(3H)-one 15a
N-Boc-glycylglycine methyl ester (179.4 mg, 0.728 mmol) was dissolved in CH2Cl2 (1.2 mL), and treated, at r.t., with CF3CO2H (400 µL). After 1 h 15 min, the solvent was evaporated to dryness. The residue was taken up in CH2Cl2 and evaporated again three times to give crude glycylglycine methyl ester TFA salt. Meanwhile, a solution of ester 2a (213.2 mg, 0.437 mmol) in EtOH (5.5 mL), was treated, at r.t., with 3 N aq. NaOH (190 µL, 0.57 mmol). After stirring for 30 min, the solution was poured into 5% aq. (NH4)H2PO4 (30 mL) + 1 N HCl (1.3 mL). After saturation with NaCl, the mixture was extracted 4 times with AcOEt. Evaporation furnished the crude acid. It was taken up in CH2Cl2 (3 mL) and added to the flask containing the crude glycylglycine methyl ester. Et3N (273 µL, 1.96 mmol) and BOP (benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate) (298.2 mg, 0.674 mmol) were added in this order. The solution was stirred at r.t. overnight and then poured into 5% aq. (NH4)H2PO4 saturated with NaCl (30 mL), and extracted with AcOEt (3 times). The organic layer was washed with saturated aq. NaHCO3 and evaporated to dryness. Chromatography with CH2Cl2–AcOEt–EtOH 46.5 : 46.5 : 7 to 45 : 45 : 10 followed with another chromatography with AcOEt–MeOH 98 : 2 gave pure 15a as a white solid (203 mg, 79%). Rf 0.52 (AcOEt–CH2Cl2–MeOH 69 : 30 : 1). Found: C, 59.1; H, 7.1; N, 11.8%. C29H41N5O8 requires: C, 59.27; H, 7.03; N, 11.92%. 1H NMR (DMSO, 300 MHz): δ 8.58 [1 H, t, NH, J 5.7]; 7.75 [1 H, d, NH, J 10.2]; 7.50–7.25 [5 H, m, arom.]; 7.09 [1 H, broad t, NH]; 6.87 [1 H, t, NH, J 5.7]; 5.43 [1 H, slightly broad q, CHCH, J 9.0]; 5.25 [1 H, dt, CHCH, Jd 3.5, Jt 11.1]; 4.98 [1 H, d, CHHPh, J 18.3]; 4.26 [1 H, d, CHHPh, J 18.3]; 4.16–3.78 [7 H, m, CHN and CH2N]; 3.64 [3 H, s, OCH3]; 2.60–2.40 [2 H, m, CH2]; 2.35–1.95 [4 H, m, CH2]; 1.74–1.57 [1 H, m, CH2]; 1.38 [9 H, s, C(CH3)3]; 1.15 [3 H, s, CH3]. 13C NMR (DMSO, 50 MHz): δ 172.86, 170.88, 169.93, 169.06, 168.80 [CO]; 156.17 [urethane CO]; 137.94 [quat. arom.]; 133.39 [CHCH]; 128.56, 127.29, 126.60 [arom. CH]; 124.56 [CHCH]; 77.74 [C(CH3)3]; 64.40 [quat. CN]; 52.37 [CHN]; 51.68 [CH3O]; 47.82 [CH2Ph]; 43.25, 41.98, 40.37 [CH2N]; 38.28, 29.67 [CH2CH]; 28.09 [(CH3)3C]; 23.01 [CH2CH2CH]; 22.80 [CH3]. IR: νmax 3360 (broad), 2953, 2864, 2816, 1740, 1650, 1543, 1487, 1448, 1368, 1326, 1279, 1145, 1079, 1037, 983, 949 cm−1.
(Z,3R*,9R*)-3-(((N-Benzyl-t-butoxycarbonyl)amino)acetamido)-9-(((((methoxycarbonylmethyl)amino)carbonylmethyl)amino)carbonyl)-3-methyl-4,5,8,9-tetrahydro-1H-azonin-2(3H)-one 15b
Prepared in 80% yield by the same procedure described above for 15a. Rf 0.45 (AcOEt–CH2Cl2–MeOH 69 : 30 : 1). Found: C, 58.9; H, 7.2; N, 11.6%. C29H41N5O8 requires: C, 59.27; H, 7.03; N, 11.92%. 1H NMR (CDCl3, 200 MHz): δ 8.41 [1 H, t, NH, J 5.9]; 8.27 [1 H, broad s, NH]; 7.62 [2 H, d, ortho-H, J 7.4]; 7.44–7.19 [3 H, m, meta- and para-H]; 6.75 [1 H, d, ring NH, J 10.0]; 6.57 [1 H, broad t, NHBoc]; 5.43 [1 H, slightly broad q, CHCH, J 9.1]; 5.21–5.02 [1 H, broad m, centre 5.11, CHCH]; 4.62 [2 H, s, CH2Ph]; 4.41 [1 H, t, CHN, J 9.9]; 3.95–3.77 [4 H, m, CH2N]; 3.64 [3 H, s, OCH3]; 3.70–3.60 [1 H, m, CHHNHBoc]; 3.29 [1 H, dd, CHHNHBoc, J 5.4, 16.7]; 2.50–2.16 [3 H, m, CH2]; 2.00–1.85 [2 H, m, CH2]; 1.74–1.57 [1 H, m, CH2]; 1.59 [3 H, s, CH3]; 1.33 [9 H, s, C(CH3)3]. 13C NMR (DMSO, 50 MHz): δ 172.31, 170.89, 170.15, 169.06, 168.80 [CO]; 155.35 [urethane CO]; 139.50 [quat. arom.]; 133.53 [CHCH]; 128.35, 126.55, 125.87 [arom. CH]; 124.83 [CHCH]; 77.72 [C(CH3)3]; 63.74 [quat. CN]; 52.20 [CHN]; 51.62 [CH3O]; 44.80 [CH2Ph]; 42.25, 41.63, 40.45 [CH2N]; 37.77, 32.24 [CH2CH]; 28.06 [(CH3)3C]; 21.94 [CH2CH2CH]; 19.97 [CH3].
Cyclic peptide 3a
A solution of compound 15a (110.1 mg, 0.187 mmol) in tetrahydrofuran (5 mL), was treated at r.t. with 0.5 M aq. LiOH (1.13 mL, 0.565 mmol) and stirred for 1 h. The solution was poured into 5% aq. (NH4)H2PO4 (15 mL) + 1 N HCl (0.5 mL). After saturation with NaCl, the mixture was extracted 3 times with AcOEt and evaporated to dryness. It was taken up in CH2Cl2 (1 mL) and treated with CF3CO2H (250 µL). After stirring for 90 min at r.t., the solvents were evaporated. In order to remove completely the CF3CO2H, the residue was taken up twice with n-heptane and evaporated again. Then it was finally taken up in dry DMF (13 mL) and dry CH2Cl2 (17 mL). Sym-collidine (104 µL, 0.786 mmol) and HATU [(O-7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate] (150.6 mg, 0.396 mmol) were added in sequence and the mixture stirred for 2 days at r.t. The solvents were evaporated. Then the residue was taken up in CHCl3, and washed with 5% aq. (NH4)H2PO4 saturated with NaCl, and with 10% K2CO3 (15 mL) + 0.1 M NaOH (5 mL). Evaporation and chromatography (CH2Cl2–EtOH 9 : 1) gave pure 3a as a white solid (47.2 mg, 55%). Rf 0.42 (CH2Cl2–MeOH 9 : 1). Found: C, 60.4; H, 6.4; N, 15.25%. C23H29N5O8 requires: C, 60.65; H, 6.42; N, 15.37%.1H NMR (DMSO, 300 MHz): δ 8.70 [1 H, t, NH-b, J 5.7]; 7.64 [1 H, d, ring NH, J 13.2]; 7.46–7.35 [3 H, m, meta-H and NH-c]; 7.34–7.26 [3 H, m, ortho- and para-H]; 6.92 [1 H, d, NH-a, J 7.3]; 5.43 [1 H, slightly broad q, H-7, J 9.0]; 5.27 [1 H, dt, H-6, Jd 5.2, Jt 10.8]; 5.00 [1 H, d, CHHPh, J 18.3]; 4.38 [1 H, dd CHHN of glycine c, J 5.1, 15.3*] 4.35 [1 H, d, CHHPh, J 18.3]; 4.15–4.03 [2 H, m, H-9 and CHHN of glycine a]; 3.81 [1 H, dd, CHHN of glycine b, J 6.0, 15.6]; 3.60* [1 H, dd, CHHN of glycine c, JAB 15.6*]; 3.57* [1 H, dd, CHHN of glycine b, JAB 15.6*]; 3.35* [1 H, dd, CHHN of glycine a, JAB 14.1*]; 2.47 [1 H, ddd, H-8 α, J ≈ 2.0, 7.8, 12.6]; 2.55–2.10 [3 H, m, H-5 β, H-8 β, H-4 (1)]; 2.10–1.95 [1 H, m, H-5 α]; 1.33 [1 H, dd, H-4 (1), J 9.9, 15.6]; 1.18 [3 H, s, CH3]. NOEDIFF: on irradiating the signal at δ 3.65 (H of glycines c and b): 3.3% NOE on ortho-H (doublet at δ 7.27); on irradiating the signal at δ 3.81 (H of glycine b): 1% NOE of NH of glycine c; on irradiating the ring NH at δ 7.64: 4.44% NOE on H-5 β (at δ 2.35) and 3.33% NOE on H-8 β (at δ 2.16); on irradiating the signal at δ 4.10 (CHN and CH of glycine a): 3% NOE on H-7, 1.4% NOE on NH of glycine b; on irradiating NH of glycine b (at δ 8.70): 1.9% NOE on CHHN of glycine a at δ ≈ 4.06 ppm and 1.5% NOE on CHHN of glycine a at δ 3.35 ppm. On irradiating the signal of the methyl at δ 1.18 ppm: 3.7% NOE on ortho-H at δ 7.27 and 1% on meta-H at δ 7.36. 1H NMR (anhydrous CDCl3, 300 MHz, 0.01 M)(attribution of ring NH is certain (COSY). Attribution of glycine a-c NH is only guessed): δ 8.47 [1 H, broad t, NH b, J ≈ 5.0]; 8.31 [1 H, broad s, NH c]; 7.92 [1 H, d, NH a, J 8.1]; 7.42 [2 H, t, meta-H, J 7.2]; 7.36–7.24 [3 H, m, ortho- and para-H]; 6.49 [1 H, d, ring NH, J 9.9]; 5.55 [1 H, slightly broad q, H-7, J 9.1]; 5.34 [1 H, dt, H-6, Jd 4.5, Jt 10.8]; 5.29 [1 H, d, CHHPh, J 18.0]; 4.61 [1 H, d, CHHPh, J 18.0]; 4.66–4.36 [4 H, m, H-9 + 3 CHH glycines]; 3.52 [1 H, dd, CHH glycine b, J 4.5, 15.3]; 3.42 [1 H, slightly broad d, CHH glycine a or c, J 14.0]; 3.41 [1 H, slightly broad d, CHH glycine a or c, J 14.1]; 2.59 [1 H, slightly broad q, H-8 β, J 11.4]; 2.42–2.22 [3 H, m, (from left to right): H-5, H-4 β, H-8 α]; 2.06–1.90 [1 H, m, H-5]; 1.58 [1 H, dd, H-4 α, J 10.5, 15.9]; 1.47 [3 H, s, CH3]. Δδ (CDCl3 − DMSO): ring NH: −1.15; NH-a: +0.91; NH-b: −0.23; NH-c: +1.00.
13C NMR (DMSO, 75 MHz): δ 172.01, 170.84, 170.21, 169.57, 168.96 [amidic CO]; 137.91 [quat. arom.]; 135.51 [CHCH]; 128.60, 127.31, 126.76 [arom. CH]; 124.35 [CHCH]; 64.95 [quat. CN]; 51.80 [CHN]; 48.37 [CH2Ph]; 43.44, 43.32, 41.77 [CH2N]; 38.15, 28.89 [allylic CH2]; 22.92 [CH2]; 22.42 [CH3]. 13C NMR (CDCl3, 75 MHz): δ 172.53, 172.44, 172.28, 171.90, 171.23 [amidic CO]; 137.27 [quat. arom.]; 133.26 [CHCH]; 129.25, 127.96, 126.07 [arom. CH]; 124.47 [CHCH]; 65.74 [quat. CN]; 51.91 [CHN]; 49.28 [CH2Ph]; 44.50, 42.73, 42.48 [CH2N]; 39.02, 30.03 [allylic CH2]; 24.08 [CH2]; 23.04 [CH3].
HPLC-MS was performed with the following eluent: 5 min isocratic 100% A (0.1% TFA in H2O); then linear gradient from 0% to 70% B (0.1% TFA in CH3CN) over 20 min. Temperature: 15 °C for 15 min; then gradient to 5 °C in 1 min; then 5 °C for the rest of analysis; flow: 0.35 mL min−1 for 16 min; then 0.1 mL min−1. Detection by TIC and DAD (220, 254 nm) showed only 1 peak, with Rt = 31.4 min: m/z (positive mode): 456.24 (M + H+) (base peak) (calculated mass: 456.22). MS/MS of this peak: 428.0 (62%), 411.0 (44), 399.9 (100), 382.8 (9), 370.9 (8), 353.7 (13), 320.8 (54), 296.7 (5), 292.8 (12), 284.8 (12), 263.7 (14), 228.8 (13), 211.8 (16), 206.9 (11), 178.8 (5), etc. In addition, other smaller peaks (<10%) were detected: m/z = 478 (M + Na+), 494 (M + K+), 911 (dimeric artifact), 933 and 949 (Na+ and K+ of dimeric artifact). The fact that the 911 peak was an instrument artifact is demonstrated by the following evidence: a) This dimeric artifact was present also in the trans cyclic peptide 3b, where the real dimer was detected at different Rt (see below). b) The fragmentation energy used to perform MS/MS analysis on the 911 ion co-eluting with the monomers was lower (0.6 V) and therefore consistent with a non-covalent interaction, than that needed to fragment the 911 ion of the real trans dimer (9 V). c) The obtained MS/MS mass spectrum of the dimeric artifact revealed a poor fragmentation: 456.0 (100%), 428.0 (31), 411.0 (4), 399.9 (8); on the contrary, the MS/MS spectrum of the real trans dimers gave a rich fragmentation (see below). d) The dimeric artifact gaved a poorly resolved isotopic cluster, probably due to the presence of multicharged (e.g. tetramers) instrumental overlapping signals.
HRMS was carried out on an ibrid q-TOF geometry tandem mass spectrometer (Q-STAR XL MS/MS system – Applied Biosystems MSD Sciex, Toronto, Canada) equipped with a MALDI ion source. 2,5-Dihydroxybenzoic acid at a final concentration of 5 mg mL−1in 70 : 30 0.1% TFA in H2O–0.05% TFA in CH3CN was used as matrix. All the measures were carried out mixing 500 fmol of an in-house obtained hexapeptide (ALELFR, MW = 747.4272) with 500 fmol of sample. Internal instrument calibration was performed using the main mono-charged matrix fragment at m/z = 137.0239 (from DHB) and mono-charged ion at m/z = 748.4352 of the above mentioned hexapeptide. The main peak obtained was that of the mono-sodium adduct of the monomer at m/z = 478.2034. Calculated for C23H29N5O5Na: 478.2066 (accuracy: 6 ppm)
Cyclic peptide 3b
Prepared from 15b following the same procedure above described for 3a. After careful chromatographic purification, a product apparently giving a single spot in tlc (Rf 0.38 (CH2Cl2–MeOH 9 : 1) was obtained in 13% yield. Both 1H and 13C NMR showed the presence of 3b as the major product, but it was contaminated by other three similar substances. HPLC-MS (see 3a above for the conditions) showed a main sharp peak corresponding to the expected monomer 3b (Rt 32.2 min), a peak corresponding to the cis isomer 3a (Rt 31.4 min), and two broader peaks (Rt 37.5 and 39.5 min), corresponding to the 2 diastereoisomeric cyclodimers. Attempts to isolate pure 3b by silica gel chromatography with various eluents failed. The relative ratio of 3b, 3a and cyclodimers in this mixture, determined by HPLC/UV, is 55 : 8 : 37. Therefore the yield of 3b is 7%.HPLC-MS [for the conditions see 3a]: the peak at Rt 32.2 gave m/z (positive mode): 456.22 (M + H+) (base peak). Also other smaller peaks (<10%) were detected: 478 (M + Na+), 494 (M + K+), 911 (dimeric artifact), 933 and 949 (Na+ and K+ of dimeric artifact). MS/MS of the peak at 456: 428.0 (42%), 411.0 (100), 399.9 (69), 382.8 (18), 370.9 (17), 353.7 (15), 320.8 (26), 296.7 (10), 292.8 (11), 284.8 (26), 263.7 (26), 251.7 (10), 246.6 (10), 228.8 (12), 211.8 (10), 206.9 (19), 178.8 (18), etc. MS/MS of the peak at 911 of the cyclodimer with Rt 37.5: 740.6 (100), 712.6 (10), 696.6 (40), 563.3 (20), 456.0 (38), 428.0 (11), 411.0 (5), 399.9 (13), 284.8 (14). MS/MS of the peak at 911 of the cyclodimer with Rt 39.5: 740.6 (100), 712.6 (10), 696.6 (44), 563.3 (24), 456.0 (52), 428.0 (13), 411.0 (4), 399.9 (13), 284.8 (12). HRMS: (for conditions see 3a) the main peak obtained was that of the mono-sodium adduct of the monomer at m/z = 478.2118. Calculated for C23H29N5O5Na: 478.2066 (accuracy: 10 ppm).
We tried to separate a mixture of 3b and 3a (8 : 2) from the cyclodimers by reverse-phase preparative HPLC (conditions were similar to those employed for analytical HPLC). The separation was successful but, surprisingly, on evaporation to dryness (at 30–40 °C), extensive decomposition of 3b was observed giving hydrolysed adducts (as demonstrated by the presence in the MS of peaks with m/z = 474). After a further silica gel chromatography, we isolated a small amount of an 83 : 17 mixture of 3a and 3b. Thus it seems that 3b is much more sensitive to hydrolysis than 3a.
From careful analysis of the NMR spectra of the above mentioned mixtures we could extract the signals of 3b: 1H NMR (DMSO, 300 MHz): δ 8.44 [1 H, t, NH, J 6.1]; 7.74 [1 H, d, NH, J 6.3]; 7.48–7.22 [6 H, m, arom. and NH]; 7.11 [1 H, d, NH, J 6.9]; 5.66–5.36 [2 H, m, CHCH]; 4.71 and 4.59 [2 H, AB system, CH2Ph, J 17.7]; 4.45–3.60 [6 H, m, CHN and CH2N]; 3.43–3.30 [1 H, m, CHHN]; 2.50–1.60 [5 H, m, allylic CH2 and other CHH]; 1.40–1.20 [1 H, m, CHH]; 1.01 [3 H, s, CH3]. 13C NMR (DMSO, 75 MHz): δ 171.68, 171.35, 170.94, 170.17, 169.21 [amidic CO]; 138.11 [quat. arom.]; 134.57 [CHCH]; 128.68, 127.28, 126.81 [arom. CH]; 125.87 [CHCH]; 64.39 [quat. CN]; 53.02 [CHN]; 46.04 [CH2Ph]; 44.72, 43.90, 42.15 [CH2N]; 36.62, 23.11 [allylic CH2]; 19.63 [CH3]; 19.23 [CH2].
Acknowledgements
We gratefully thank Federico De Pellegrini and Carlo Scapolla for their very valuable collaboration, and MIUR (PRIN 00 and 02) for financial support.References
- T. R. Gadek and J. B. Nicholas, Biochem. Pharm., 2003, 65, 1 CrossRef CAS.
- K. Burgess, Acc. Chem. Res., 2001, 34, 826 CrossRef CAS.
- R. Haubner, D. Finsinger and H. Kessler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1375 CrossRef CAS.
- M. A. Dechantsreiter, E. Planker, B. Mathä, E. Lohof, G. Hölzemann, A. Jonczyk, S. L. Goodman and H. Kessler, J. Med. Chem., 1999, 42, 3033 CrossRef CAS.
- A. Golebiowski, J. Jozwik, S. R. Klopfenstein, A. O. Colson, A. L. Grieb, A. F. Russell, V. L. Rastogi, C. F. Diven, D. E. Portlock and J. J. Chen, J. Comb. Chem., 2002, 4, 584 CrossRef CAS.
- W. Qiu, X. Gu, V. A. Soloshonok, M. D. Carducci and V. J. Hruby, Tetrahedron Lett., 2001, 42, 145 CrossRef CAS.
- M. Eguchi, M. S. Lee, M. Stasiak and M. Kahn, Tetrahedron Lett., 2001, 42, 1237 CrossRef CAS.
- S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Tetrahedron, 1997, 53, 12789 CrossRef CAS.
- L. Halab, F. Gosselin and W. D. Lubell, Biopolymers, 2000, 55, 101 CrossRef CAS.
- L. Belvisi, A. Caporale, M. Colombo, L. Manzoni, D. Potenza, C. Scolastico, M. Castorina, M. Cati, G. Giannini and C. Pisano, Helv. Chim. Acta, 2002, 85, 4353 CrossRef CAS.
- A. Trabocchi, E. G. Occhiato, D. Potenza and A. Guarna, J. Org. Chem., 2002, 67, 7483 CrossRef CAS.
- T. K. Chakraborty, A. Ghosh, A. Ravi Sankar and A. C. Kunwar, Tetrahedron Lett., 2002, 43, 5551 CrossRef CAS.
- M. F. Braña, M. Garranzo, B. de Pascual-Teresa, J. Pérez-Castells and M. Rosario Torres, Tetrahedron, 2002, 58, 4825 CrossRef CAS.
- S. N. Filigheddu and M. Taddei, Tetrahedron Lett., 1998, 39, 3857 CrossRef CAS.
- T. K. Chakraborty, A. Ghosh, R. Nagaraj, A. Ravi Sankar and A. C. Kunwar, Tetrahedron, 2001, 57, 9169 CrossRef CAS.
- K. Brickmann, P. Somfai and J. Kihlberg, Tetrahedron Lett., 1997, 38, 3651 CrossRef CAS.
- A. D. Piscopio, J. F. Miller and K. Koch, Tetrahedron, 1999, 55, 8189 CrossRef CAS.
- A. Nouvet, M. Binard, F. Lamaty, J. Martinez and R. Lazaro, Tetrahedron, 1999, 55, 4685 CrossRef CAS.
- B. E. Fink, P. R. Kym and J. A. Katzenellenbogen, J. Am. Chem. Soc., 1998, 120, 4334 CrossRef CAS.
- A. J. Souers and J. A. Ellman, Tetrahedron, 2001, 57, 7431 CrossRef CAS.
- J. Bondebjerg, Z. Xiang, R. M. Bauzo, C. Haskell-Luevano and M. Meldal, J. Am. Chem. Soc., 2002, 124, 11046 CrossRef CAS.
- L. Banfi, G. Guanti and R. Riva, Chem. Commun., 2000, 985 RSC.
- L. Banfi, G. Guanti, R. Riva, A. Basso and E. Calcagno, Tetrahedron Lett., 2002, 43, 4067 CrossRef CAS.
- A. Basso, L. Banfi, R. Riva, P. Piaggio and G. Guanti, Tetrahedron Lett., 2003, 44, 2367 CrossRef CAS.
- L. Banfi, A. Basso, G. Guanti and R. Riva, Mol. Diversity, 2003, 6, 227 Search PubMed.
- L. Banfi, A. Basso, G. Guanti and R. Riva, Tetrahedron Lett., 2004, 45, 6637 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef.
- G. Vo-Thanh, V. Boucard, H. Sauriat-Dorizon and F. Guibé, Synlett, 2001, 37 CAS.
- C. Hebach and U. Kazmaier, Chem. Commun., 2003, 596 RSC.
- B. Beck, G. Larbig, B. Mejat, M. Magnin Lachaux, A. Picard, E. Herdtweck and A. Dömling, Org. Lett., 2003, 5, 1047 CrossRef CAS.
- R. Krelaus and B. Westermann, Tetrahedron Lett., 2004, 45, 5987 CrossRef CAS.
- L. Banfi, A. Basso, G. Guanti and R. Riva, Tetrahedron Lett., 2003, 44, 7655 CrossRef CAS.
- C. J. Creighton and A. B. Reitz, Org. Lett., 2001, 3, 893 CrossRef CAS.
- C. J. Creighton, G. C. Leo, Y. Du and A. B. Reitz, Bioorg. Med. Chem., 2004, 12, 4375 CrossRef CAS.
- S. R. Wilson and R. A. Sawicki, J. Org. Chem., 1979, 44, 330 CrossRef CAS.
- A. Fürstner and K. Langemann, Synthesis, 1997, 792 CrossRef CAS.
- R. Haubner, R. Gratias, B. Diefenbach, S. L. Goodman, A. Jonczyk and H. Kessler, J. Am. Chem. Soc., 1996, 118, 7461 CrossRef CAS.
- N. J. Baxter and M. P. Williamson, J. Biomol. NMR, 1997, 9, 359 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |