Amination of ethers using chloramine-T hydrate and a copper(I) catalyst
David P.
Albone
a,
Stephen
Challenger
b,
Andrew M.
Derrick
b,
Shaun M.
Fillery
c,
Jacob L.
Irwin
a,
Christopher M.
Parsons
a,
Hiroya
Takada
a,
Paul C.
Taylor
*a and
D. James
Wilson
a
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: p.c.taylor@warwick.ac.uk; Fax: 44 24 76524112; Tel: 44 24 76524375
bPfizer Central Research, Sandwich, Kent, UK CT13 9NJ
cAstraZeneca Pharmaceuticals, Mereside, Alderley Park, UK SK10 4TG
First published on 23rd November 2004
Abstract
Amination of C–H bonds activated by ether oxygen atoms is facile with chloramine-T as nitrene source and copper(I) chloride in acetonitrile as catalyst. For cyclic ethers the hemiaminal products are generally stable and can be isolated pure. For acyclic ethers, the hemiaminal products, as expected, fragment with elimination of alcohol to yield imines. When activation of benzylic positions is remote through a conjugated system, stable benzylamine derivatives are isolated. Mechanistic studies are consistent with concerted insertion of an electrophilic nitrenoid into the C–H bond in the rate-determining step, though in an asynchronous manner with a more activated substrate.
Introduction
In 1967 Kwart and Kahn reported the copper-catalysed decomposition of benzenesulfonyl azide in the presence of cyclohexene to yield products consistent with a nitrene-transfer mechanism (Scheme 1).1 Both the aziridine 1 and the allylic amination 2 products are synthetically very interesting, and throughout the 1980s the groups of Breslow and of Mansuy sought catalysts which led to higher yields and selectivities for each of these two classes of product.2 It was not until 1991 that an efficient and general nitrene-transfer aziridination procedure was developed by the group of Evans.3 Simple copper(I) and copper(II) salts were found to give high yields of aziridines from a wide range of alkenes and TsN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) IPh (Scheme 2), with only isolated examples of allylic insertions. A nitrene-transfer mechanism was formally proposed by Jacobsen in 1995.4
IPh (Scheme 2), with only isolated examples of allylic insertions. A nitrene-transfer mechanism was formally proposed by Jacobsen in 1995.4
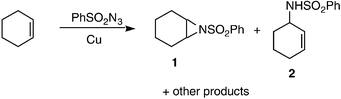 | ||
| Scheme 1 | ||
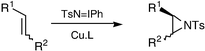 | ||
| Scheme 2 | ||
More recently, the need to find catalysts for amination, as opposed to aziridination, has attracted a good deal of attention.5 For example, the Müller group described the amination of hydrocarbons with NsNIPh as the nitrene source and rhodium(II) catalysts. High yields of amination products were observed with cycloalkenes (Scheme 3) and with benzylic substrates (Scheme 4), but very large excesses of hydrocarbon were required.6 The Che group has introduced manganese and ruthenium porphyrins as catalysts for amination using TsNIPh.7 Enantioselective intermolecular amination procedures6b have been reported by the groups of Müller,6 Che7c and Katsuki8 with ees of up to 89%.
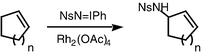 | ||
| Scheme 3 | ||
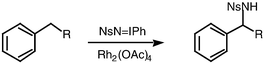 | ||
| Scheme 4 | ||
The main drawback with many of these procedures is the troublesome two-step synthesis of the nitrene-transfer agent. Che's group have addressed this to some extent with a modified procedure that exploits the in situ generation of the iodinane from TsNH2 and PhI(OAc)2.9 The Du Bois group has shown that enantioselective intramolecular amination with “in situ” iodinanes is a highly effective strategy for elaboration of relatively unfunctionalised alcohols.10
The use of chloramine-T trihydrate as a nitrene source has been a focus of activity both in our group11 and in others.12 In contrast to iodinanes such as NsIPh and TsNIPh, which require two-step syntheses,13 chloramine-T is an inexpensive, commercially available reagent. We have been largely unsuccessful in our search for catalysts for benzylic and allylic amination using chloramine-T, this being consistent with the report from the Bedekar group that bromamine-T, which is not commerically available, was superior to chloramine-T in such reactions.12b On the other hand, the amination of methylene groups activated by oxygen is facile using chloramine-T as nitrene source with copper(I) catalysts. Our results in this area are described herein. We note that an accidental amination of dioxane, when chloramine-T was heated with a sulfoxide in the presence of copper metal in dioxane as solvent, was reported long ago.12a However, we believe this to be the first systematic study of amination with chloramine-T.
Results and discussion
Scope and limitations
The previous report of amination of dioxane with chloramine-T12a led us to start our investigation with cyclic ethers. It quickly became apparent that a very inexpensive catalyst, copper(I) chloride with acetonitrile as ligand and solvent, was effective in these reactions. Indeed, unless stated otherwise, all the reactions described herein were carried out with a small excess of chloramine-T and 10 mol% of copper(I) chloride in acetonitrile.Amination of isochromane, tetrahydrofuran and dioxane proceeded smoothly to yield the hemiaminal products (Schemes 5 and 6), with tosyl amide TsNH2 being the only significant by-product. With the exception of 5, the products were isolated and fully characterised, although it was difficult to avoid decomposition on chromatography, in particular with 5, hence our inability to purify this compound. We were delighted to find that our standard conditions were also effective for the amination of acyclic ethers (Scheme 7). In these cases, the hemiaminal products could not be isolated pure, since elimination of alcohols leading to imines was observed. The fact that hemiaminals are imine equivalents has been reported.14 Although it is clear that the same elimination reaction will occur from the cyclic hemiaminal products, in that case there is no entropic driving force to the elimination.
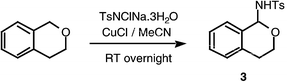 | ||
| Scheme 5 | ||
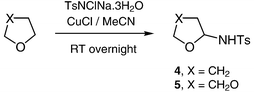 | ||
| Scheme 6 | ||
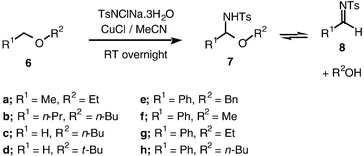 | ||
| Scheme 7 | ||
Insertion into the α-methylene group of simple dialkyl ethers 6a–d was observed. Interestingly, the reaction appears to be much more sensitive to steric rather than electronic factors. Hence, while the hemiaminal 7a resulting from amination of diethyl ether could be isolated, though impure, in around 50% yield, amination of di-n-butyl ether 6b proceeded with less than 30% conversion using the standard procedure. Similarly, as judged by the crude 1H NMR spectrum of the crude reaction mixtures at the end of the standard procedure, amination of n-butyl methyl ether 6c proceeded with around 50% conversion, whereas amination of tert-butyl methyl ether 6d gave only traces of the expected products of degradation of hemiaminal 7d.
With benzyl ethers 6e–h amination was, as expected, particularly facile, proceeding quantitatively. A trace of the benzylic hemiaminal product 7e was observed in the 1H NMR spectrum of the reaction mixture after ca. one hour, but decomposition to the tosyl imine 8, which was then hydrolysed to benzaldehyde under the reaction conditions, was rapid. As mentioned above, the hemiaminal 7a derived from diethyl ether is sufficiently stable to be isolated as part of a mixture and was tentatively characterised by its crude 1H NMR spectrum. We did not observe any of the other proposed hemiaminal intermediates 7b, 7c and 7f–h; their presence is inferred from the formation of the corresponding imine 8, the presence of which was clear from the characteristic imine protons at ca. 9 ppm in the crude 1H NMR spectrum.
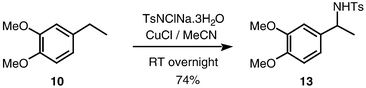
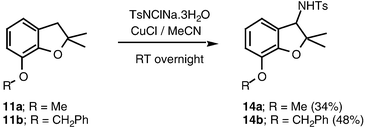
Assuming that the activating effect of the oxygen atom is from a lone pair, it is to be expected that benzyl positions will be activated towards amination not just by ether oxygen atoms in the α-position, but by ether substituents in the ortho or para positions of the aryl substituent, from where the effect of the lone pair can be transmitted by conjugation. Indeed, substrates 9 to 11, including mono- and di-alkoxy substituted benzylic substrates, were transformed to benzyl amines in acceptable yields (Schemes 8, 9 and 10). It should be noted that amination of 11b, which has two benzylic sites, occurred solely at the remotely activated heterocyclic position (Scheme 10). Unlike the hemiaminals described above, the sulfonamide products 12–14 are of course configurationally stable. Hence, there is an opportunity for inducing enantioselectivity in these reactions, this being the subject of current research in our laboratories.
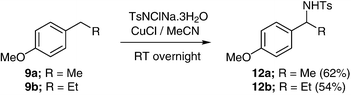 | ||
| Scheme 8 | ||
 | ||
| Scheme 9 | ||
 | ||
| Scheme 10 | ||
Finally, attempted amination of nitrogen analogues of the substrates studied here yielded complex reaction mixtures and we have not pursued these further.
Mechanistic studies
The mechanism of the amination procedure described herein was also of interest. Müller showed that amination using NsNIPh and dirhodium tetraacteate most probably proceeded via concerted insertion of an intermediate nitrenoid into the C–H bond.6a On the other hand, the Che group have demonstrated that amination using TsNIPh and ruthenium porphyrin catalysts is a radical process, with the radical recombination step occurring with retention of stereochemistry.7d
The amination of substrates 15 lends itself to a Hammett-type analysis. Competition experiments were carried out under our standard conditions, but in addition to the usual one equivalent of benzyl methyl ether, a further one equivalent of a substituted benzyl methyl ether was added. The relative rates of reaction of the two ethers were determined from the integrals of the benzylic protons of the residual starting materials in the 1H NMR spectrum. To permit comparison with the work of the Müller group, Hammett plots were constructed for both CuCl and Rh2(OAc)4 as catalysts. It should be noted that, while the rhodium(II) catalyst was effective for amination of a range of the substrates described above, the rates of the reactions were consistently slower.
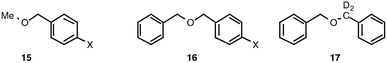
The Hammett plot for the CuCl reactions is shown in Fig. 1 and that for Rh2(OAc)4 in Fig. 2. In both cases, an approximately linear correlation with σ was observed and ρ was found to be both small in magnitude and negative. These data are clearly inconsistent with the presence of highly charged or radical benzylic intermediates in the rate-determining step. Instead, following Müller6a and Du Bois,15 we propose that the data are more consistent with a relatively concerted insertion of an electrophilic nitrenoid into the C–H bond (Fig. 3). It should be noted that intermolecular competition experiments cannot give an accurate ρ value, since the relative concentrations of the substrates vary during the reaction. We had hoped that intramolecular competition with substrates 16 would overcome this limitation, but disappearance of starting material cannot be used to monitor the relative rates in this case and quantitative determination of the plethora of products from the decomposition of the intermediate hemiaminals and susbsequent hydrolysis of the imine products 8 was not possible in our hands. Nevertheless, the approximate ρ values with substrates 16 were similar to those from the intermolecular experiments.
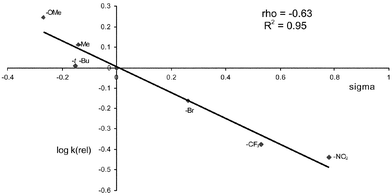 | ||
| Fig. 1 k(rel) vs.σ for amination of 15 with copper(I) catalysis. | ||
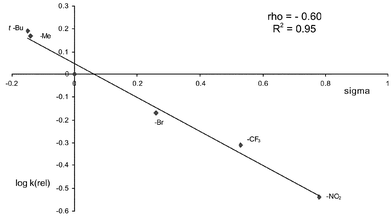 | ||
| Fig. 2 k(rel) vs. σ for amination of 15 with rhodium(II) catalysis. | ||
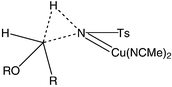 | ||
| Fig. 3 Proposed concerted transition state for C–H insertion. | ||
If insertion into the activated C–H bond is indeed the rate determining step, further information can be gleaned from kinetic isotope effects. We first carried out an intermolecular competition experiment using THF and perdeutero THF. It should be noted that in this experiment there is a significant excess of the substrate relative to chloramine-T. The relative rates of consumption of the H and D starting materials was followed by gated 13C NMR spectroscopy with a very long (2 min) delay time. The kH/kD value of 1.5 is low, but this is consistent with a highly concerted transition state in which the C–H–N bond angle is much less than the 180° required for a maximum kH/kD value. Indeed, a similar value was reported by Du Bois.15 It should be noted that the existence of an isotope effect excludes the mechanism proposed to operate for reaction of pentafluorophenylnitrene with THF, where the first, rate-determining step is thought to be formation of an O–N ylide.16
Interestingly, an intramolecular competition experiment using 17 as substrate yielded a kH/kD value of around 4, This is consistent with an asynchronous concerted mechanism as described in the literature,17 falling part way between a truly concerted three-centre transition state in which the C–H–N bond angle is much less than 180° (as in Fig. 3) and a two-step mechanism, such as reported by Che,7d in which the C–H–N bond angle is close to 180°. In this analysis kH/kD should increase, as in our case, with more highly activated substrates.
Conclusion
In summary, amination of C–H bonds activated by ether oxygen atoms is facile with chloramine-T and a simple copper(I) salt in acetonitrile. For cyclic ethers the hemiaminal products are generally stable and can be isolated pure. For acyclic ethers, the hemiaminal products, as expected, fragment with elimination of alcohol to yield imines, with the exception of remotely activated benzylic sites, when stable benzylamine derivatives are isolated. Mechanistic studies are consistent with concerted insertion of an electrophilic nitrenoid into the C–H bond in the rate-determining step, though in an asynchronous manner with a more activated substrate.Experimental
Copper(I) chloride (99+%) was purchased from Aldrich. Ethers 15 were prepared by the standard Williamson synthesis.18 Their NMR data were compared with literature reports.19 Ethers 16 and 17 were prepared by the literature methods.20 Chemical shifts are relative to the residual solvent signal. Coupling constants are in Hz.General procedure for amination of ethers
Substrate (1 mmol, 1 equiv), Cu(I)Cl (0.1 equiv) and chloramine-T trihydrate (1.3 equiv) were dissolved in degassed MeCN (6 cm3) under nitrogen. After stirring overnight, the green suspension was filtered through a short plug of silica and the solvent was removed under reduced pressure. The product was purified by flash chromatography on silica gel (10% EtOAc in DCM).Intermolecular kinetic isotope experiment
A Schlenk flask was charged with chloramine-T hydrate (211 mg, 750 µmol), 1,3,5-tris(tert-butyl)benzene (197 mg, 800 µmol)—as an internal standard—and d3-acetonitrile (2.0 cm3) and the mixture was thoroughly degassed before addition of THF (122 µL, 1.50 mmol) and perdeutero THF (122 µL, 1.50 mmol). An aliquot of ca. 1 cm3 was removed and passed through a small plug of silica gel into an NMR tube which was then sealed under N2 and stored at −18 °C prior to analysis. The reaction was initiated by the addition of copper(I) chloride (10 mg, 100 µmol) and the reaction stirred at 25 °C. After 3 h another aliquot of ca. 1 cm3 was removed and filtered through a small plug of silica into an NMR tube, which was sealed under N2. Quantitative analysis was performed using a gated 13C NMR experiment with a relaxation delay of 120 s.Intramolecular kinetic isotope experiment
In a procedure analogous to that for the intermolecular experiment, d2-dibenzyl ether (200 mg, 1.00 mmol) was reacted in d3-acetonitrile (2.0 cm3) with chloramine-T (281 mg, 1.00 mmol) and copper(I) chloride (10 mg, 100 µmol). Again, 1,3,5-tris(tert-butyl)benzene (81 mg, 330 µmol) was employed as the internal standard.Acknowledgements
We thank Astra Zeneca, Pfizer and the EPSRC for financial support, Dr Christopher Samuel for his mechanistic insight and Dr Adam Clarke for help with NMR analysis.References
- H. Kwart and A. A. Kahn, J. Am. Chem. Soc., 1967, 89, 1951 CrossRef CAS.
- R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400 RSC; J-P. Mahy, G. Bedi, P. Battioni and D. Mansuy, Tetrahedron Lett., 1988, 29, 1927 CrossRef CAS.
- D. A. Evans, M. M. Faul and M. T. Bilodeau, J. Org. Chem., 1991, 56, 6744 CrossRef CAS.
- Z. Li, R. W. Quan and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5889 CrossRef CAS.
- M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2003, 125, 12078 CrossRef CAS.
- (a) I. Nägeli, C. Baud, G. Bernadinelli, Y. Jacquier, M. Moran and P. Müller, Helv. Chim. Acta, 1997, 80, 1087 CrossRef CAS; (b) for a useful recent review see P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905 Search PubMed.
- (a) J-L. Liang, J-S. Huang, X-Q. Yu, N-Y. Zhu and C-M. Che, Chem. Eur. J., 2002, 8, 1563 CrossRef CAS; (b) X-Q. Yu, J-S. Huang, X-G. Zhou and C-M. Che, Org. Lett., 2000, 2, 2233 CrossRef CAS; (c) X-G. Zhou, X-Q. Yu, J-S. Huang and C-M. Che, Chem. Commun., 1999, 2377 RSC; (d) S-M. Au, J-S. Huang, W-Y. Yu, W-H. Fung and C-M. Che, J. Am. Chem. Soc., 1999, 121, 9120 CrossRef CAS.
- Y. Kohmura and T. Katsuki, Tetrahedron Lett., 2001, 42, 3339 CrossRef CAS.
- S-M. Au, J-S. Huang, C-M. Che and W-Y. Yu, J. Org. Chem., 2000, 65, 7858 CrossRef CAS.
- C. G. Espino and J. Du Bois, Angew. Chem., Int. Ed., 2001, 40, 598 CrossRef CAS; C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935 CrossRef CAS; J. J. Fleming, K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2003, 125, 2028 CrossRef CAS . See also C. Fruit and P. Müller, Tetrahedron: Asymmetry, 2004, 15, 1019 Search PubMed.
- D. P. Albone, P. S. Aujla, P. C. Taylor, S. Challenger and A. M. Derrick, J. Org. Chem., 1998, 63, 9569 CrossRef CAS ; it should be noted that the catalyst reported does not function with the currently available CuOTf benzene complex.
- (a) D. Carr, T. P. Seden and R. W. Turner, Tetrahedron Lett., 1969, 477 CrossRef CAS; (b) B. M. Chanda, R. Vyas and A. V. Bedekar, J. Org. Chem., 2001, 66, 30 CrossRef CAS; (c) L. Simkhovich and Z. Gross, Tetrahedron Lett., 2001, 42, 8089 CrossRef CAS; (d) J. Gullick, S. Taylor, P. McMorn, D. Bethell, P. C. B. Page, F. E. Hancock, F. King and G. Hutchings, J. Mol. Catal., A, 2002, 180, 85 CrossRef CAS . see also J. U. Jeong, B. Tao, I. Sagasser, H. Henniges and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 6844 Search PubMed.
- Y. Yamada, T. Yamamoto and M. Okawara, Chem. Lett., 1975, 361 CAS; G. Besenyei, S. Nemeth and L. I. Simandi, Tetrahedron Lett., 1993, 34, 6105 CrossRef CAS.
- S. D. Derrick, R. Boehme, K. M. Wong, F. Nemeth, K. Tanaka, B. Rumberg, R. A. Beekman and P. W. Dibble, Tetrahedron, 1996, 52, 7679 CrossRef CAS.
- J. Du Bois, presented at 225th ACS National Meeting, New Orleans, LA, March 23–27, 2003.
- R. Poe, K. Schnapp, M. J. T. Young, J. Grayzar and M. S. Platz, J. Am. Chem. Soc., 1992, 114, 5054 CrossRef CAS.
- H. M. L. Davies, Q-H. Jin, P-D. Ren and A. Yu. Kovalevsky, J. Org. Chem., 2002, 67, 4165 CrossRef CAS; for an excellent review see C. G. Espino, http://www.organicdivision/essays_2002/espino.pdf.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, 5th edn., Wiley, New York, 2001 Search PubMed.
- X = MeO: J. I. Bhat, W. Clegg, H. Maskill, M. R. J. Elsegood, I. D. Menneer and P. C. Miatt, J. Chem. Soc., Perkin Trans. 2, 2000, 1435 Search PubMed ; X = tert-butyl: R. L. Huang, T-W. Lee and H. Ong, J. Chem. Soc. C, 1969, 2522 RSC ; X = Br: K. Rengan and R. Engel, J. Chem. Soc., Perkin Trans. 1, 1991, 987 Search PubMed ; X = CF3: J. W. Hilborn, E. MacKnight, J. A. Pincock and P. J. Wedge, J. Am. Chem. Soc., 1994, 116, 3337 RSC.
- J. W. Boyd, P. W. Schmalzel and L. L. Miller, J. Am. Chem. Soc., 1980, 102, 3856 CrossRef CAS.
- T. Soeta, K. Nagai, H. Fujihara, M. Kuriyama and K. Tomioka, J. Org. Chem., 2003, 68, 9723 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |