Biotransformation of the sesquiterpene (+)-valencene by cytochrome P450cam and P450BM-3
Rebecca J.
Sowden
,
Samina
Yasmin
,
Nicholas H.
Rees
,
Stephen G.
Bell
and
Luet-Lok
Wong
*
Department of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QR. E-mail: luet.wong@chem.ox.ac.uk
First published on 18th November 2004
Abstract
The sesquiterpenoids are a large class of naturally occurring compounds with biological functions and desirable properties. Oxidation of the sesquiterpene (+)-valencene by wild type and mutants of P450cam from Pseudomonas putida, and of P450BM-3 from Bacillus megaterium, have been investigated as a potential route to (+)-nootkatone, a fine fragrance. Wild type P450cam did not oxidise (+)-valencene but the mutants showed activities up to 9.8 nmol (nmol P450)−1 min−1, with (+)-trans-nootkatol and (+)-nootkatone constituting >85% of the products. Wild type P450BM-3 and mutants had higher activities (up to 43 min−1) than P450cam but were much less selective. Of the many products, cis- and trans-(+)-nootkatol, (+)-nootkatone, cis-(+)-valencene-1,10-epoxide, trans-(+)-nootkaton-9-ol, and (+)-nootkatone-13S,14-epoxide were isolated from whole-cell reactions and characterised. The selectivity patterns suggest that (+)-valencene has one binding orientation in P450cam but multiple orientations in P450BM-3.
Introduction
Terpenoid hydrocarbons (C5H8)n are biosynthesised by the coupling of isoprene units in the form of isopentenyl pyrophosphate (IPP) to give polyenyl pyrophosphates which are then transformed to the terpenes by terpene synthases. These synthase enzymes catalyze the dissociation of pyrophosphate to form polyenyl carbocations which can undergo complex rearrangements that are controlled by the enzyme active site, resulting in the diverse structures of the terpenes (linear, cyclic, polycyclic). Coupling of two IPP molecules gives geranyl pyrophosphate (GPP, Fig. 1), which is the precursor to all monoterpenes (C10H16). Addition of another isoprene unit to GPP gives farnesyl pyrophosphate (FPP), from which all sesquiterpenes (C15H24) are derived. Diterpenoid compounds include the gibberellins, which are plant hormones, while the triterpene squalene is the precursor to all sterols.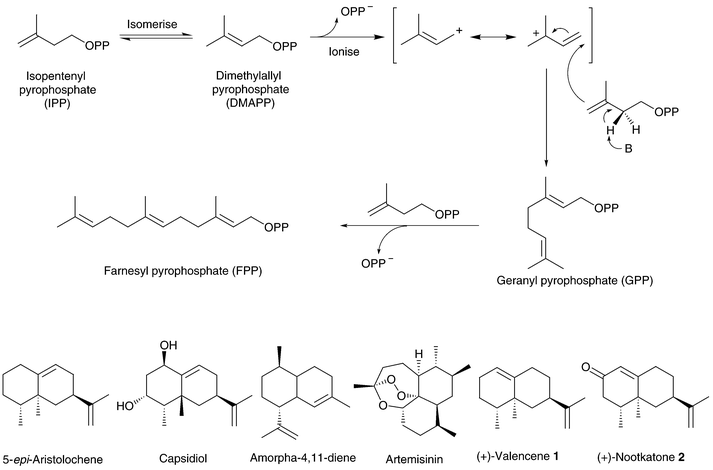 | ||
| Fig. 1 The biosynthesis pathway of geranyl (GPP) and farnesyl pyrophosphate (FPP) via the coupling of isoprene units. The structures of (+)-valencene 1, (+)-nootkatone 2, and related examples of sesquiterpenoid compounds are shown. | ||
The sesquiterpenes are the largest class of terpenoid compounds and common constituents of plant essential oils. Many sesquiterpenes and their alcohol, aldehyde and ketone derivatives, are biologically active or precursors to metabolites with biological functions, while others have desirable fragrance, flavouring and medicinal properties.1 For example the phytoalexin capsidiol is derived from the sesquiterpene 5-epi-aristolochene,2 and amorpha-4,11-diene is the precursor to the potent antimalarial compound artemisinin (Fig. 1).3,4 (+)-Valencene (1) is found in orange oil (Fig. 1) while its ketone derivative (+)-nootkatone (2), is a sought after fragrance compound found in grapefruit juice. Recently the valencene synthase gene from orange has been cloned and functionally expressed in Escherichia coli.5,6
The oxidation of (+)-valencene is an attractive synthetic route to the high added-value compound (+)-nootkatone, and chemical oxidation of (+)-valencene has been studied.7–9 Cu(I)-mediated oxidation by alkyl hydroperoxides is one route,10 and supported metal catalysts have been reported.11 Biological oxidation of (+)-valencene is another possibility, but microbial oxidation to (+)-nootkatone commonly suffers from inhibition by nootkatone which is a known inhibitor of P450 enzymes.12 However, given the diverse range of compounds oxidised by the superfamily of P450 enzymes, it is unlikely that (+)-nootkatone will inhibit all P450 enzymes. Cytochrome P450cam from Pseudomonas putida, and P450BM-3 from Bacillus megaterium, are the most well characterised P450 enzymes.13–18 The natural substrate of P450cam is D-(+)-camphor while P450BM-3 catalyses the oxidation of medium to long chain fatty acids. Both enzymes are stable, highly active, and have high substrate specificity. However, they will also oxidise compounds that are structurally different from the natural substrate, although at much lower rates. Hence P450cam and P450BM-3 are attractive targets for potential biotransformation applications, and site-directed mutagenesis as well as forced evolution methods have been applied to engineer both enzymes for the oxidation of non-natural substrates.19–32 The oxidation of (+)-valencene by P450 enzymes has not been described. We report here a preliminary study into the oxidation of this sesquiterpene by engineered mutants of P450cam and P450BM-3, potentially for the direct synthesis of (+)-nootkatone.
Results and discussion
Oxidation of (+)-valencene by cytochrome P450cam
Wild type P450cam showed no valencene oxidation activity, probably because the enzyme active site could not accommodate this larger and more hydrophobic molecule. We have shown that hydrophobic substitutions at Y96 (Fig. 2), a prominent polar residue in the P450cam active site, enhanced the activity of the enzyme for the oxidation of hydrophobic organic compounds.33–37 The oxidation of large substrates such as polycyclic aromatic hydrocarbons is further promoted by substitution of active site residues with smaller amino acids, in particular the F87A and Y96A mutations.28,38 Mutations at V247 also affect the binding of non-natural substrates. The V247L mutation can close the top of the substrate pocket and thus constrain substrates to bind closer to the heme.28,31,39 The L244A mutation alters substrate binding by creating space close to the heme.28,30,31,40,41 Since valencene is significantly larger than camphor (Fig. 2), the binding and oxidation of valencene by a number of P450cam mutants with combinations of mutations at these residues were investigated. The results are summarised in Table 1.| P450cam enzyme | % High spin heme (±5%) | K d/µM | NADH oxidation rate | Product formation rate | Overall yield |
|
|
|---|---|---|---|---|---|---|---|
| Wild type | <5% | — | — | — | — | — | — |
| F87A/Y96F | 80% | 3.09 ± 0.26 | 36 ± 5.3 | 7.2 ± 2.0 | 20.0% | 70% | 12% |
| F87L/Y96F | 85% | 4.23 ± 0.31 | 22 ± 2.4 | 2.8 ± 1.0 | 12.7% | 65% | 20% |
| F87A/Y96F/V247L | 25% | 1.88 ± 0.30 | 30 ± 1.8 | 5.7 ± 0.9 | 19.0% | 78% | 6.7% |
| F87A/Y96F/L244A | 80% | 2.70 ± 0.16 | 63 ± 13 | 9.8 ± 1.6 | 15.6% | 60% | 12% |
| F87V/Y96F/L244A | 65% | 3.40 ± 0.45 | 48 ± 5.4 | 9.7 ± 1.7 | 20.2% | 38% | 47% |
| F87A/Y96F/L244A/V247L | 100% | 2.76 ± 0.35 | 27 ± 6.7 | 6.9 ± 0.9 | 25.6% | 86% | 4.2% |
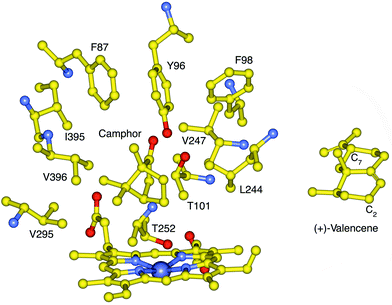 | ||
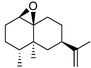
| Fig. 2 The active site structure of P450cam with bound camphor showing the side chains that contact the natural substrate, and the location of T252 which is required for oxygen binding and O–O bond cleavage. The F87, Y96 and V247 side chains are located high in the active site while L244 is closer to the heme. The structure of (+)-valencene 1 is shown in an orientation whereby the C7 isopropenyl group would be in the vicinity of the 87 side-chain if C2 is placed close to the heme, to account for the high regioselectivity of P450cam mutants for C2 oxidation. | ||
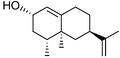
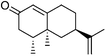
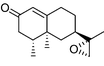
The Y96F mutation did not confer any valencene oxidation activity to P450cam, nor did the Y96A (data not shown) even though this latter mutation should generate a larger active site. On the other hand the F87A/Y96F mutant showed valencene oxidation activity, with a catalytic NADH turnover rate of 36 nmol (nmol P450)−1 min−1. The heme spin state shifted from low spin in the absence of substrate to 80% high spin when valencene was bound. However, valencene binding (Kd = 3.09 µM) was substantially weaker than camphor binding by the wild type enzyme (1.15 µM). Analysis of the organics by gas chromatography (GC) showed that valencene was oxidised by the F87A/Y96F mutant to one major and two minor products. There were other small peaks (<2% each) in the chromatogram. The major product (70%) was characterised as (+)-trans-nootkatol, 3 (Fig. 3), by co-elution with an authentic sample prepared by hydride reduction of nootkatone. One minor product (12%) was readily identified as (+)-nootkatone by co-elution with an authentic sample. The other minor product (4%) had a much longer GC retention time, and it was shown by control incubations to arise from the slow oxidation of nootkatone by the enzyme. This compound was isolated from a preparative scale, whole-cell oxidation of nootkatone by a P450BM-3 mutant (see below) and characterised as (+)-trans-nootkaton-9-ol, 4 (Fig. 3). Although nootkatone is a known inhibitor of P450 enzymes, activity assays with equal concentrations of both compounds showed that nootkatone did not inhibit the oxidation of valencene (data not shown).
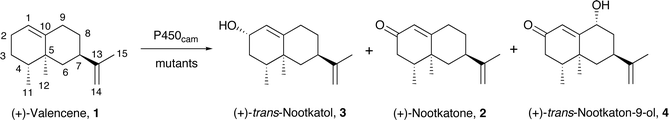 | ||
| Fig. 3 The main products from the oxidation of (+)-valencene by P450cam mutants. | ||
The availability of the three main products allowed the product yield based on NADH consumed (the coupling efficiency, see Experimental) to be determined. The F87A/Y96F mutant was found to catalyse valencene oxidation at the rate of 7.2 min−1 with 20% yield. This activity is a large increase over the wild type enzyme although it is low compared to the 1000 min−1 rate and >95% yield of camphor oxidation by wild type P450cam under identical conditions.
The role of the residue at the 87 position was investigated with the F87L mutation. The F87L/Y96F mutant showed that a larger side-chain at this position weakened valencene binding (Kd 4.23 µM) and lowered both the NADH oxidation activity and product yield. However, the product distribution was slightly shifted towards nootkatone (65% trans-nootkatol and 20% nootkatone). Adding the V247L mutation to the F87A/Y96F mutant tightened valencene binding but did not increase the rate of valencene oxidation (Table 1), although the extent of further oxidation of nootkatol to nootkatone was reduced. Introducing the L244A mutation had little effect on valencene binding but nearly doubled the NADH turnover activity and also increased the valencene oxidation rate to 9.7 min−1. There was more oxidation of nootkatol to nootkatone and then to nootkaton-9-ol (10% of products). Therefore the V247L mutation retarded further oxidation while the L244A mutation promoted further oxidation of both nootkatol and nootkatone. The L244A mutation was also added to the Y96A and Y96F mutants, but the Y96A/L244A and Y96F/L244A mutants did not show significant activity (data not shown). The data showed that a small side chain at the 87, but not the 96, position is highly beneficial to valencene oxidation.
The L244A and V247L mutations were combined in the F87A/Y96F/L244A/V247L mutant. The activity of this mutant was similar to the F87A/Y96F, but the selectivity was shifted to 86% (+)-trans-nootkatol, with only 4% nootkatone. Hence the effect of the V247L mutation on selectivity dominated over that of L244A. Since the F87L mutation promoted the oxidation of nootkatol to nootkatone, compared to the F87A mutation, the effect of the F87V mutation was examined. The F87V/Y96F/L244A mutant showed one of the highest activities of the P450cam mutants studied (9.7 min−1), and the product distribution was also significantly different, with 38% (+)-trans-nootkatol, 47% nootkatone, and 5% (+)-trans-nootkaton-9-ol.
Valencene can form 17 different monooxygenation products. The observation that the large majority (>85%) of the first oxidation step occurred at C2 suggests that the valencene binding orientation places this allylic carbon close to the heme iron. Since a small side chain at the 87-position appears to be important for valencene binding and oxidation, it seems reasonable that the C7 isopropenyl group is accommodated in the space created by the F87 mutations (Fig. 2). Further oxidation of both nootkatol and nootkatone was observed to various extents for all P450cam mutants even in the presence of an excess of valencene, suggesting that the mutations altered the binding of valencene and its oxidation products. The effect of the 87 side-chain is subtle because the F87V mutation promoted the oxidation of nootkatol but not nootkatone, while both smaller (F87A) and larger (F87L) side chains retarded nootkatol oxidation.
The valencene oxidation activities of the P450cam mutants are low compared to camphor oxidation by the wild type. However, it is encouraging that these mutants show high regioselectivity and the F87V/Y96F/L244A mutant gives a high proportion of nootkatone. The high selectivity of the F87A/Y96F/L244A/V247L mutant for (+)-trans-nootkatol may also be useful because a dehydrogenase enzyme can be expressed in the same host to oxidise nootkatol to nootkatone and form NADH from NAD+. This combined-enzyme route has the potential advantage of regenerating some of the NADH consumed in the P450cam mediated oxidation step such that there is less metabolic stress on the host organism during the biotransformation of valencene to nootkatone.
(+)-Valencene oxidation by P450BM-3
Wild type P450BM-3 did not show a significant heme spin state shift when valencene was bound, but the enzyme–substrate combination oxidised NADPH at a rate of 40 min−1 (Table 2). This activity is substantially below the ca. 5000 min−1 activity observed with the fatty acid natural substrates. GC analysis of the reactions showed that P450BM-3 oxidised valencene to numerous products (Table 3). There was no dominant product. With wild type P450BM-3, nootkatone was not observed although trans-nootkatol was present (12.9%). The product with a retention time (9.1 min) comparable to those of the nootkatols (cis 9.7 min, trans 9.9 min) was probably another alcohol derivative of valencene. Only very small peaks were observed at longer GC retention times, indicating that the initial monooxygenation products were not further oxidised by the wild type enzyme.| P450BM-3 enzyme | NADPH rate | Oxidation rate | Overall yield |
|---|---|---|---|
| WT | 40 ± 5.2 | 12 ± 2.5 | 30.4% |
| (516 ± 36) | (112 ± 14.2) | (21.4%) | |
| R47L/Y51F | 45 ± 2.3 | 30 ± 2.8 | 65.9% |
| (296 ± 23) | (79 ± 4.1) | (26.8%) | |
| R47L/Y51F/F87A | 82 ± 10 | 34 ± 2.1 | 41.8% |
| (203 ± 12.8) | (70 ± 16) | (34.5%) | |
| R47L/Y51F/I263A | 30 ± 1.5 | 10 ± 2.6 | 33.9% |
| (234 ± 26) | (25 ± 3.1) | (10.8%) |
| P450BM-3 enzyme | R.T. 9.1 min |
|
|
|
R.T. 12.3 min |
|
R.T. 12.8 min |
|
|---|---|---|---|---|---|---|---|---|
| Wild type | 11.4% | — | 12.9% | — | 7.0% | 16.3% | 23.8% | — |
| R47L/Y51F | 10.4% | 8.1% | — | — | — | 14.4% | 33.6% | — |
| R47L/Y51F/F87A | — | 8.1% | — | 7.7% | 22% | 27.9% | — | 14.3% |
| R47L/Y51F/I263A | 13.8% | 6.5% | 20.5% | 7.3% | — | 10.2% | 17.8% | — |
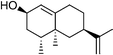
Whole-cell biotransformation reactions were carried out with wild type P450BM-3 and mutants,42 for product isolation and characterisation. Since terpene hydrocarbons are known to be toxic to microorganisms,43,44n-hexadecane was added to the E. coli minimal medium used for the whole-cell reactions at 2% v/v in order to partition excess organics into a separate phase and minimise the aqueous concentration of the terpenoid compounds. Hexadecane is benign to most host organisms commonly used for biotransformations, including E. coli. Control experiments showed that hexadecane was not oxidised at any appreciable rate by any of the P450BM-3 enzymes studied. We found that the presence of the second phase significantly enhanced both substrate conversion and product yield so that complete oxidation of 2 mM valencene was possible even under the un-optimised conditions employed. In addition, GC analysis showed that the extent of further oxidation in vivo was also lower than for the reactions in vitro.
(+)-trans-Nootkatol was isolated by silica chromatography from the organic extract of the in vivo oxidation of valencene by wild type P450BM-3. The product at retention time 12.6 min was also isolated in a sufficiently pure form to be characterised as (+)-cis-valencene-1,10-epoxide 5. The stereochemistry was determined from the NOESY spectrum by a strong cross peak between the C1 hydrogen and the C12 methyl group. Repeated attempts to isolate the product with the retention time of 12.8 min were unsuccessful; fractions containing this compound were always contaminated with 5, small amounts of 4, and other impurities. Quantitative GC analysis gave a valencene oxidation rate of 12.2 min−1 for wild type P450BM-3in vitro, with an overall yield of 30.4%. Both the rate and product yield of wild type P450BM-3 were higher than those observed for the P450cam mutants.
We had shown previously that the R47L/Y51F combination of mutations enhanced the activity of P450BM-3 for the oxidation of hydrophobic organic compounds.42 The R47 and Y51 side chains are located at the entrance to the substrate access channel (Fig. 4). In addition, the F87A and I263A mutations, which create space close to the heme, promote the oxidation of large molecules such as polyaromatic hydrocarbons.42 As shown in Table 2, the R47L/Y51F mutations increased the valencene oxidation activity. The F87A mutation increased the NADPH oxidation rate but reduced the yield, while the I263A mutation decreased the activity.
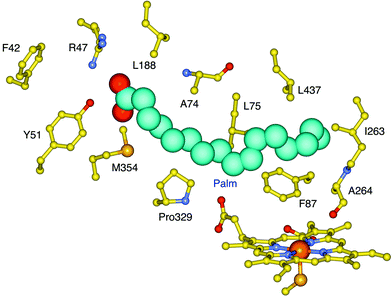 | ||
| Fig. 4 The active site structure of P450BM-3 with palmitoleic acid (Palm) bound. The residues that line the substrate access channel are also shown. The residues R47, Y51, F87 and I263 were targeted for mutagenesis. | ||
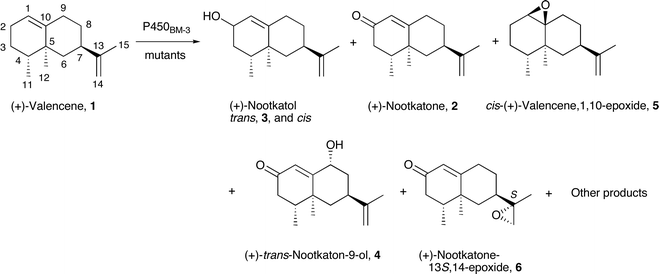 | ||
| Fig. 5 The characterised products from the oxidation of (+)-valencene by wild type and mutants of P450BM-3. Numerous other products were also formed. | ||
The mutations significantly altered the product distribution (Table 3). Although the R47 and Y51 residues are far from the heme, the nootkatol product from the R47L/Y51F mutant was the cis rather than the trans isomer found for the wild type. There was much more of the 12.8 min product (33.6%), but again no nootkatone was observed (Fig. 5). These selectivity changes agreed with previous observations that the R47L/Y51F combination of mutations appear to have structural effects that are transmitted to the vicinity of the heme to alter the orientation of substrate binding and hence the product distribution.42 The F87A mutation greatly reduced the proportions of the cis-1,10-epoxide 5 and the product at 9.1 min (Table 3), and nootkatone was present. There was also a new product with a longer retention time of 14.4 min. This compound was isolated from a whole-cell reaction with the R47L/Y51F/F87A mutant and characterised as nootkatone-13S,14-epoxide 6. Repeated attempts to isolate the 12.3 min product from this reaction were unsuccessful. The I263A mutation had very different effects on the selectivity. Like the wild type, the nootkatol formed was the trans-isomer (20.5%). Nootkatone was present, but there was much less of the nootkatone epoxide 6. Hence the I263A mutation reversed the effect of the R47L/Y51F mutations on the diastereoselectivity of nootkatol formation and also slowed down the oxidation of both nootkatol and nootkatone. Overall, the formation of numerous products from valencene oxidation by P450BM-3 indicated that this substrate has multiple binding orientations.
The observation of the nootkatone epoxide 6 led us to investigate nootkatone oxidation by P450BM-3. The wild type enzyme showed much higher NADPH turnover and product formation rates with nootkatone than with valencene (Table 2), and gave 83% 6 (data not shown). The R47L/Y51F mutations reduced the activity, which was consistent with the hydrophobic substitutions disfavouring the binding and oxidation of the more polar nootkatone compared to valencene, and the epoxide 6 was again the major product. Nootkatone was oxidised by the R47L/Y51F/F87A mutant at a rate of 70 min−1, twice that for valencene (Table 2). There was 70% of the epoxide 6 (data not shown) and 10% of trans-nootkaton-9-ol 4, which was isolated from a whole-cell oxidation reaction and characterised. Small amounts of this compound were detected in the products of valencene oxidation by the P450BM-3 mutants, but it was more prominent in the reaction catalysed by the P450cam mutants because the P450cam system gave much more nootkatone. Hence, far from being an inhibitor, nootkatone was actually a good substrate of P450BM-3. The low activity of the R47L/Y51F/I263A mutant suggested that the region around the 263 side-chain might be important for nootkatone binding.
Conclusions
Cytochrome P450cam and P450BM-3 have been engineered for the oxidation of (+)-valencene. These two enzymes show very different product selectivity patterns. The P450cam mutants oxidise valencene with high regioselectivity for C2 oxidation (>85%). The relative proportions of nootkatol and nootkatone vary from 86% (+)-trans-nootkatol and 4% nootkatone for the F87A/Y96F/L244A/V247L mutant, to 38% nootkatol and 47% nootkatone for the F87V/Y96F/L244A. P450BM-3 shows higher valencene oxidation activity than P450cam but it is much less selective. The selectivity trends suggest a specific valencene binding orientation in P450cam but multiple orientations in P450BM-3. Nootkatone did not inhibit these two bacterial P450 enzymes; in fact nootkatone was a better substrate than valencene for wild type P450BM-3. Both enzymes require optimisation for applications in nootkatone synthesis, but the good yields and total substrate conversions in whole-cell reactions utilising the two-phase method are promising. The protein engineering challenges are different for the two systems: P450cam has the desired selectivity but the activity has to be increased while both the substrate specificity and selectivity of P450BM-3 require improvement.Experimental
General and materials
General molecular biology manipulations were carried out according to literature methods.45 Restriction enzymes and T4 DNA ligase were from New England Biolabs and used with buffers supplied by the manufacturer. General chemicals, solvents and buffer components were from Sigma-Aldrich and were of biochemical grade or the highest purity available. (+)-Valencene, (+)-nootkatone and (+)-carvone were from Sigma-Aldrich and Fluka. UV/Vis spectra, enzyme activity assays and substrate binding titrations were performed at 30 ± 0.5 °C on a CARY 1E spectrophotometer. 1H and 13C NMR spectra were acquired on a Varian UnityPlus 500 MHz spectrometer. Mass spectra were obtained on a Micromass GCTof mass spectrometer with a solids probe inlet system and operating in electron impact (EI) mode. Haptacosafluorotributylamine (FC43) was used to calibrate the instrument. Gas chromatography (GC) analyses were carried out on a ThermoFinnigan Trace instrument equipped with a flame-ionisation detector (FID) using DB-1 fused silica capillary columns with helium as the carrier gas. Both the injector and FID were maintained at 250 °C.Molecular biology
P450BM-3 and the three proteins in the P450cam system (putidaredoxin reductase, putidaredoxin and P450cam) were expressed and purified as described previously.42,46–48 All proteins were stored in buffer containing 50% v/v glycerol. Glycerol and salts were removed immediately before experiments by eluting the protein down a 5 cm3 PD-10 column (Amersham Pharmacia) with 50 mM Tris buffer, pH 7.4 for P450cam, and 100 mM potassium phosphate buffer, pH 8.0 for P450BM-3. Putidaredoxin reductase was used directly from 25 µM stocks. All the mutants used in this study had been reported previously,38,42,49 except for the F87V/Y96F/L244A mutant of P450cam. Site-directed mutagenesis of the Y96F/L244A gene was carried out with the Stratagene Quik-Change kit. The oligonucleotide 5′-TCCAGCGAGTGCCCGḠ![[T with combining macron]](https://www.rsc.org/images/entities/char_0054_0304.gif)
![[C with combining macron]](https://www.rsc.org/images/entities/char_0043_0304.gif) ATCCCTCGTGAAGCC-3′ and its reverse complement (MWG Biotech) were used to generate the F87V mutation (the V87 codon is underlined). Potential mutants were selected on agar plates using the chloramphenicol resistance marker of the pCHC expression plasmid.46 Colonies were grown in Luria-Bertani broth. Positive mutants were identified by DNA sequencing, and then fully sequenced by the DNA Sequencing Facility in the Department of Biochemistry, University of Oxford.
ATCCCTCGTGAAGCC-3′ and its reverse complement (MWG Biotech) were used to generate the F87V mutation (the V87 codon is underlined). Potential mutants were selected on agar plates using the chloramphenicol resistance marker of the pCHC expression plasmid.46 Colonies were grown in Luria-Bertani broth. Positive mutants were identified by DNA sequencing, and then fully sequenced by the DNA Sequencing Facility in the Department of Biochemistry, University of Oxford.
Preparation of (+)-nootkatol
(+)-Nootkatone (1.0 g, 4.6 mmol) was dissolved in diethyl ether (10 cm3) and added dropwise, with stirring, to a suspension of 1 g (26.3 mmol) of LiAlH4 in 100 cm3 diethyl ether cooled at −78 °C. The mixture was allowed to warm to ambient temperature, refluxed for 1 h, and then cooled on ice. Ethanol (10 cm3) then water (100 cm3), followed by 0.1 M sulfuric acid (10 cm3), were added to work up the product and destroy the remaining LiAlH4. The mixture was extracted with diethyl ether (3 × 150 cm3) and the combined extracts dried over anhydrous MgSO4. Solvents were removed at 40 °C, 800 mbar and then 240 mbar. The residue was dissolved in CH2Cl2 and applied to a silica column (0.06–0.2 mm, 70–230 mesh). The (+)-nootkatols were eluted with hexane–EtOAc mixtures containing increasing concentrations of EtOAc, and fractions were analysed by GC. (+)-trans-Nootkatol (retention time, R.T., 9.9 min) was isolated as the pure compound but fractions of the cis isomer (R.T. 9.7 min) always contained small amounts of the trans isomer. Solvents were removed from the fractions at 40 °C and 240 mbar. The compounds were then maintained at 40 °C and 100 mbar until no change in mass occurred. (+)-trans-Nootkatol 3 (250 mg, 25%) was obtained as a pale yellow oil; δH(500 MHz; C6D6; Me4Si; for numbering see Fig. 3) 0.45 (1 H, br s, 2-OHeq), 0.71 (3 H, d, J 6.5, 4-Me), 0.82 (3 H, s, 5-Me), 0.91 (1 H, dd, J 12.8, 12.8, 6-Hax), 1.15 (1 H, dddd, J 4.3, 12.5, 12.5, 13.9, 8-Hax), 1.27 (1 H, ddd, J 9.5, 11.7, 12.8, 3-Hax), 1.32 (1 H, ddq, J 1.6, 6.5, 12.8, 4-H), 1.58 (1 H, ddd, J 1.6, 7.6, 11.7, 3-Heq), 1.62 (3 H, dd, J 0.8, 1.0, 13-Me), 1.68 (1 H, dddd, J 2.4, 4.6, 5.7, 12.5, 8-Heq), 1.81 (1 H, ddd, J 2.7, 2.7, 12.8, 6-Heq), 1.96 (1 H, ddd, J 2.4, 4.1, 13.9, 9-Heq), 2.09–2.16 (1 H, m, 9-Hax), 2.12–2.19 (1 H, m, 7-H), 4.10 (1 H, br s, 2-Hax), 4.77–4.78 (2 H, m, 2 × 14-H), 5.31 (1 H, ddd, J 1.3, 1.6, 1.9, 1-H); δC(125.7 MHz; C6D6; Me4Si) 14.93 (q, 4-Me), 17.74 (q, 5-Me), 20.40 (q, 13-Me), 32.13 (t, 9), 32.76 (t, 8), 37.29 (t, 3), 38.14 (q, 5), 39.17 (d, 4), 40.57 (d, 7), 44.33 (t, 6), 67.47 (d, 2), 108.61 (t, 14), 125.34 (d, 1), 144.66 (s, 10), and 150.05 (s, 13). m/z (EI) 220.1784 (M+, C15H24O requires 220.1827).(+)-Valencene binding by P450cam mutants
The total assay volume was 1.5 cm3. Substrate-free P450cam was prepared by gel filtration using 5 cm3 PD-10 columns pre-equilibrated with 50 mM Tris, pH 7.4. The protein was diluted to 3 µM, and KCl was added to 200 mM. The spectrum was recorded between 300 and 450 nm. Both the sample and reference cuvettes contained the substrate free enzyme at the start of the experiment. Then 1 µL, 2 µL, 3 µL, 4 µL and 7 µL aliquots of a 1 mM solution of (+)-valencene in ethanol were added to the sample cuvette only, and the spectra recorded. These difference spectra showed a peak at 420 nm and a trough at 387 nm. The peak to trough difference in absorbance was fitted to a hyperbolic function against the substrate concentration by the Origin 7.0 software (Origin Labs) to give the binding constant, Kd.NADH turnover rate determination
Gas chromatography analysis of (+)-valencene oxidation products
Quantitative gas chromatography analysis
The concentration of valencene oxidation products in an incubation mixture was determined by calibrating the FID response to monooxygenated derivatives using (+)-nootkatone as a representative compound. The reasonable assumption was that these isomeric compounds would have near identical responses on the FID. Mixtures containing all components of a standard valencene oxidation incubation except NAD(P)H and the substrate, but additionally 5, 10, 50, 100, and 250 µM of (+)-nootkatone, were extracted and analysed as for a normal incubation. The ratio of the area of the (+)-nootkatone peak to that of the internal standard was plotted against the (+)-nootkatone concentration to give a straight-line calibration plot that passed through the origin. The use of an internal standard significantly reduced the spread of the data. The calibration plot based on (+)-nootkatone did not apply for the further oxidation products such as nootkaton-9-ol and nootkatone-13,14-epoxide; these doubly oxygenated compounds had much higher FID responses than the monooxygenated valencenes. (+)-trans-Nootkaton-9-ol, which was isolated and purified from a preparative scale whole-cell reaction, was used as a representative example of these further oxidation products and a calibration was repeated. The two calibration plots were used to calculate the concentration of the products formed in a reaction. For P450BM-3 there were products that were not characterised; those that had similar retention times to the nootkatols were assumed to be monooxygenated valencenes while the ones with longer retention times than the valencene-1,10-epoxide (>13 min) were assumed to be doubly oxygenated compounds, and the appropriate calibration plots were used to estimate their concentrations. Since the enzyme concentration is 1 µM, if the total product concentration is a µM and the NAD(P)H was consumed in t min, the substrate oxidation rate is defined as a/t nmol (nmol P450)−1 min−1. If the initial NAD(P)H concentration is b µM, the yield is a/b and expressed as a percentage. This calculation ignores the fact that more than one molecule of NAD(P)H may be required for the formation of one molecule of a product. For example, if nootkatone was the only product and every molecule of NAD(P)H consumed was utilised for nootkatone formation, the yield would be 50%.Whole-cell substrate oxidation
A number of (+)-valencene and (+)-nootkatone oxidation products were prepared by whole-cell oxidation by E. coli expressing the P450BM-3 holoenzyme.42 In a typical reaction, a 500 cm3 culture of the E. coli DH5α harbouring the expression plasmid was grown at 30 °C until the turbidity at 600 nm reached ∼1.0 and then induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The cultures were then grown for another 16 h. Cells were harvested by centrifugation (1000g, 4 °C, 5 min) and resuspended in 500 cm3E. coli minimal media (per litre: 7 g K2HPO4, 3 g KH2PO4, 0.5 g sodium citrate, 0.1 g MgSO4, 1 g (NH4)2SO4, 1 cm3 glycerol, 50 cm3 1 M Tris buffer, pH 7.4, 0.1% w/v glucose, and 10 mg thiamine). Hexadecane was added to 2% v/v to partition the valencene oxidation products and organics that might be harmful to the bacterial host, into the organic phase. The (+)-valencene or (+)-nootkatone substrate was added in 50 mg aliquots in 1 cm3 ethanol and the cultures returned to the orbital shaker (120 rpm, 30 °C). After 24 h, more substrate and glucose were added. Cells were pelleted after 48 h and the supernatant extracted with EtOAc (3 × 200 cm3). The combined extracts were dried and the solvent was evaporated off at 40 °C and 240 mbar. The oily residue was dissolved in CH2Cl2 and applied to a silica column (0.06–0.2 mm, 70–230 mesh). Organics were eluted with hexane–EtOAc mixtures containing increasing concentrations of EtOAc and fractions were analysed by GC to locate the products. The appropriate fractions containing the target compounds were combined and the solvents removed at 40 °C and 240 mbar. The compounds were then maintained at 40 °C and 100 mbar until no change in mass occurred.(+)-trans-Nootkaton-9-ol 4 (15 mg, 13.5% based on the total amount of nootkatone added to the whole-cell reaction) was obtained as a pale yellow oil; δH(500 MHz; C6D6; Me4Si) 0.53 (3 H, d, J 6.7, 4-Me), 0.82 (1 H, dd, J 12.5, 12.6, 6-Hax), 1.10 (3 H, s, 5-Me), 1.21 (1 H, ddd, J 3.2, 12.7, 13.5, 8-Hax), 1.54 (1 H, ddq, J 5.5, 6.7, 12.0, 4-H), 1.62 (3 H, dd, J 0.9, 1.3 13-Me), 1.74 (1 H, ddd, J 2.7, 2.8, 12.9, 6-Heq), 1.98 (1 H, dddd, J 2.7, 2.8, 2.8, 13.5, 8-Heq), 2.09–2.12 (2 H, m, 2 × 3-H), 2.22 (1 H, br s, 9-OHeq), 2.83 (1 H, dddd, J 2.8, 2.8, 12.5, 12.6, 7-Hax), 4.03 (1 H, dd, J 2.9, 2.9, 9-Hax), 4.77 (1 H, dq, J 0.9, 1.6, 14-H-cis-Me), 4.79 (1 H, dq, J 1.3, 1.6, 14-H-trans-Me), 5.79 (1 H, s, 1-H); δC(125.7 MHz; C6D6; Me4Si) 13.99 (q, 4-Me), 17.58 (q, 5-Me), 20.56 (q, 13-Me), 34.16 (d, 7), 37.91 (t, 8), 38.62 (s, 5), 40.89 (d, 4), 42.29 (t, 3), 43.70 (t, 6), 72.95 (d, 9), 109.23 (t, 14), 127.06 (d, 1), 149.11 (s, 13), 167.65 (s, 10) and 199.32 (s, 2). m/z (EI) 234.1613 (M+, C15H22O2 requires 234.1620), 216 (9%), 190 (3).
(+)-cis-Valencene-1,10-epoxide 5 (19.1 mg, 16% based on the total amount of valencene added to the whole-cell reaction) was obtained as a pale yellow oil; m/z (EI) 220.1768 (M+, C15H24O requires 220.1827).
(+)-Nootkatone-13S,14-epoxide 6 (52.1 mg, 47% based on the total amount of nootkatone added to the whole-cell reaction) was obtained as a pale yellow oil; δH(500 MHz; C6D6; Me4Si) 0.50 (3 H, d, J 6.7, 4-Me), 0.57 (3 H, d, J 0.4, 5-Me), 0.71 (1 H, dd, J 12.8, 12.8, 6-Hax), 0.77 (1 H, dddd, J 4.6, 12.6, 12.6, 13.6, 8-Hax), 0.94 (3 H, d, J 0.7, 13-Me), 1.21 (1 H, dddd, J 3.2, 3.3, 12.6, 12.7, 7-Hax), 1.37 (1 H, ddddd, J 2.5, 3.1, 3.3, 5.2, 12.6, 8-Heq), 1.55 (1 H, ddq, J 4.0, 6.7, 14.2, 4-H), 1.76 (1 H, ddd, J 2.7, 2.8, 12.8, 6-Heq), 1.81 (1 H, ddd, J 2.7, 4.3, 14.9, 9-Heq), 1.87 (1 H, dddd, J 2.0, 5.0, 13.6, 14.9, 9-Hax), 2.00 (1 H, dd, J 14.2, 16.7, 3-Hax), 2.14 (1 H, ddd, J 1.0, 4.0, 16.7, 3-Heq), 2.16 (1 H, d, J 5.0, 14-H-cis-Me), 2.21 (1 H, dq, J 0.7, 5.0, 14-H-trans-Me), 5.79 (1 H, d, J 0.6, 1-H); δC(125.7 MHz; C6D6; Me4Si) 14.30 (q, 4-Meax), 15.86 (q, 5-Meeq), 17.43 (q, 13-Me), 28.37 (t, 8), 31.81 (t, 9), 38.61 (s, 5), 39.16 (d, 7), 40.10 (t, 6), 40.10 (d, 4), 41.82 (t, 3), 52.30 (t, 14), 58.09 (s, 13), 125.02 (d, 1), 167.41 (s, 10) and 197.45 (s, 2). m/z (EI) 234.1626 (M+, C15H22O2 requires 234.1620), 216 (12%), 206 (14), 176 (4).
Acknowledgements
RJS thanks the EPSRC, UK for a studentship and SY thanks the Lady Noon Foundation for a scholarship.References
- B. M. Fraga, Nat. Prod. Rep., 2004, 21, 669–693 RSC.
- L. Ralston, S. T. Kwon, M. Schoenbeck, J. Ralston, D. J. Schenk, R. M. Coates and J. Chappell, Arch. Biochem. Biophys., 2001, 393, 222–235 CrossRef CAS.
- D. L. Klayman, Science, 1985, 228, 1049–1055 CrossRef CAS.
- Y. J. Chang, S. H. Song, S. H. Park and S. U. Kim, Arch. Biochem. Biophys., 2000, 383, 178–184 CrossRef CAS.
- L. Sharon-Asa, M. Shalit, A. Frydman, E. Bar, D. Holland, E. Or, U. Lavi, E. Lewinsohn and Y. Eyal, Plant J., 2003, 36, 664–674 CrossRef CAS.
- J. Chappell, Trends Plant Sci., 2004, 9, 266–269 CrossRef CAS.
- G. W. Shaffer, E. H. Eschinasi, K. L. Purzycki and A. B. Doerr, J. Org. Chem., 1975, 40, 2181–2185 CrossRef CAS.
- A. G. Davies and I. G. E. Davison, J. Chem. Soc., Perkin Trans. 2, 1989, 825–830 RSC.
- I. Bombarda, E. M. Gaydou, J. Smadja and R. Faure, J. Agric. Food Chem., 1996, 44, 217–222 CrossRef CAS.
- S. F. Arantes, A. Farooq and J. R. Hanson, J. Chem. Res. (S), 1999, 248–248 RSC.
- J. A. R. Salvador and J. H. Clark, Green Chem., 2002, 4, 352–356 RSC.
- W. Tassaneeyakul, L. Q. Guo, K. Fukuda, T. Ohta and Y. Yamazoe, Arch. Biochem. Biophys., 2000, 378, 356–363 CrossRef CAS.
- I. C. Gunsalus and G. C. Wagner, Methods Enzymol., 1978, 52, 166–188 CAS.
- Y. Miura and A. J. Fulco, Biochim. Biophys. Acta, 1975, 388, 305–317 CrossRef CAS.
- E. J. Mueller, P. J. Loida and S. G. Sligar, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, New York, 1995, p. 83–124 Search PubMed.
- A. J. Fulco, Annu. Rev. Pharmacol. Toxicol., 1991, 31, 177–203 CrossRef CAS.
- C. A. Hasemann, R. G. Kurumbail, S. S. Boddupalli, J. A. Peterson and J. Deisenhofer, Structure, 1995, 2, 41–62 CrossRef CAS.
- T. L. Poulos, J. Cupp-Vickery and H. Li, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, New York, 1995, p. 125–150 Search PubMed.
- D. Appel, S. Lutz-Wahl, P. Fischer, U. Schwaneberg and R. D. Schmid, J. Biotechnol., 2001, 88, 167–171 CrossRef CAS.
- T. W. Ost, C. S. Miles, J. Murdoch, Y. Cheung, G. A. Reid, S. K. Chapman and A. W. Munro, FEBS Lett., 2000, 486, 173–177 CrossRef CAS.
- C. S. Miles, T. W. Ost, M. A. Noble, A. W. Munro and S. K. Chapman, Biochim. Biophys. Acta, 2000, 1543, 383–407 CrossRef CAS.
- H. Joo, Z. Lin and F. H. Arnold, Nature, 1999, 399, 670–673 CrossRef CAS.
- A. Glieder, E. T. Farinas and F. H. Arnold, Nat. Biotechnol., 2002, 20, 1135–1139 CrossRef CAS.
- M. W. Peters, P. Meinhold, A. Glieder and F. H. Arnold, J. Am. Chem. Soc., 2003, 125, 13442–13450 CrossRef CAS.
- K. J. French, M. D. Strickler, D. A. Rock, G. A. Bennett, J. L. Wahlstrom, B. M. Goldstein and J. P. Jones, Biochemistry, 2001, 40, 9532–9538 CrossRef CAS.
- D. A. Grayson, Y. B. Tewari, M. P. Mayhew, V. L. Vilker and R. N. Goldberg, Arch. Biochem. Biophys., 1996, 332, 239–247 CrossRef CAS.
- L. L. Wong, A. C. G. Westlake and D. P. Nickerson, Struct. Bonding, 1997, 88, 175–207 CAS.
- X. Chen, A. Christopher, J. P. Jones, S. G. Bell, Q. Guo, F. Xu, Z. Rao and L. L. Wong, J. Biol. Chem., 2002, 277, 37519–37526 CrossRef CAS.
- M. P. Mayhew, A. E. Roitberg, Y. Tewari, M. J. Holden, D. J. Vanderah and V. L. Vilker, New J. Chem., 2002, 26, 35–42 RSC.
- K. J. French, D. A. Rock, J. I. Manchester, B. M. Goldstein and J. P. Jones, Arch. Biochem. Biophys., 2002, 398, 188–197 CrossRef CAS.
- S. G. Bell, X. Chen, R. J. Sowden, F. Xu, J. N. Williams, L. L. Wong and Z. Rao, J. Am. Chem. Soc., 2003, 125, 705–714 CrossRef CAS.
- S. G. Bell, E. Orton, H. Boyd, J.-A. Stevenson, A. Riddle, S. Campbell and L.-L. Wong, Dalton Trans., 2003, 2133–2140 RSC.
- S. M. Fowler, P. A. England, A. C. G. Westlake, D. R. Rouch, D. P. Nickerson, C. Blunt, D. Braybrook, S. West, L. L. Wong and S. L. Flitsch, Chem. Commun., 1994, 2761–2762 RSC.
- D. P. Nickerson, C. F. Harford-Cross, S. R. Fulcher and L. L. Wong, FEBS Lett., 1997, 405, 153–156 CrossRef CAS.
- J.-A. Stevenson, A. C. G. Westlake, C. Whittock and L.-L. Wong, J. Am. Chem. Soc., 1996, 118, 12846–12847 CrossRef CAS.
- P. A. England, C. F. Harford-Cross, J. A. Stevenson, D. A. Rouch and L. L. Wong, FEBS Lett., 1998, 424, 271–274 CrossRef CAS.
- J. P. Jones, E. J. O'Hare and L. L. Wong, Chem. Commun., 2000, 247–248 RSC.
- C. F. Harford-Cross, A. B. Carmichael, F. K. Allan, P. A. England, D. A. Rouch and L.-L. Wong, Protein Eng., 2000, 13, 121–128 CrossRef CAS.
- S. G. Bell, J.-A. Stevenson, H. D. Boyd, S. Campbell, A. D. Riddle, E. L. Orton and L.-L. Wong, Chem. Commun., 2002, 490–491 RSC.
- J. J. De Voss, O. Sibbesen, Z. Zhang and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 1997, 119, 5489–5498 CrossRef CAS.
- J. J. De Voss, O. Sibbesen, Z. P. Zhang and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 1997, 119, 5489–5498 CrossRef CAS.
- A. B. Carmichael and L. L. Wong, Eur. J. Biochem., 2001, 268, 3117–3125 CrossRef CAS.
- J. Sikkema, J. A. de Bont and B. Poolman, J. Biol. Chem., 1994, 269, 8022–8028 CAS.
- J. Sikkema, J. A. de Bont and B. Poolman, Microbiol. Rev., 1995, 59, 201–222 CAS.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 1989 Search PubMed.
- A. C. Westlake, C. F. Harford-Cross, J. Donovan and L. L. Wong, Eur. J. Biochem., 1999, 265, 929–935 CrossRef CAS.
- J. A. Peterson, M. C. Lorence and B. Amarneh, J. Biol. Chem., 1990, 265, 6066–6073 CAS.
- T. Yasukochi, O. Okada, T. Hara, Y. Sagara, K. Sekimizu and T. Horiuchi, Biochim. Biophys. Acta, 1994, 1204, 84–90 CrossRef CAS.
- J. P. Jones, E. J. O'Hare and L. L. Wong, Eur. J. Biochem., 2001, 268, 1460–1467 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |