Synthesis and photophysical evaluation of charge neutral thiourea or urea based fluorescent PET sensors for bis-carboxylates and pyrophosphate
Thorfinnur Gunnlaugsson*a, Anthony P. Davisab, John E. O'Briena and Mark Glynna
aDepartment of Chemistry, Trinity College Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie; Fax: 00 353 1 671 2826; Tel: 00 353 1 608 3459
bSchool of Chemistry, University of Bristol, Cantock’s Close, Bristol, UK BS8 1TS
First published on 9th November 2004
Abstract
The synthesis of the fluorescent photoinduced electron transfer (PET) chemosensors 1–3 for bis-anions such as bis-carboxylates and pyrophosphate in organic solvents is described herein. These sensors are based on the receptor–spacer–fluorophore–spacer–receptor motif where the receptors are charge neutral aromatic thiourea or urea receptors and the fluorophore is anthracene. The anion recognition was evaluated using 1H NMR as well as absorption and fluorescence spectroscopy in DMSO. For simple anions such as acetate or fluoride, the recognition was shown to be through hydrogen bonding of the corresponding anion to the receptors. This gave rise to only minor changes in the absorption spectra, but significant changes in the fluorescence emission spectra, which was substantially (70–95%) quenched. Analysis of these recognition events implied a 1 : 2 (sensor : anion) binding and ideal PET behaviour for ions such as AcO− and H2PO4−. For F−, the luminescent quenching indicated a 1 : 1 binding, but we deduced that this was due more to complete quenching of the excited state after the addition of one equivalent of the anion. For all of the anions, the quenching contributed to enhanced efficiency of PET from the receptors to the excited state of the fluorophore. In the case of the bis-anions (ambient), such as di-carboxylates, similar fluorescence quenching was observed. However, here either a 1 : 1 or a 1 : 2 binding was observed depending on the length of the spacer separating the two carboxylate moieties and the nature of the receptor. Whereas both pyrophosphate and malonate gave rise to a 1 : 1 binding, glutarate gave rise to ∼1 : 2 binding for the thiourea sensors 1 and 2. However, for the urea based sensor 3, the binding was found to be 1 : 1 for all the bis-anions. For such a 1 : 1 binding we propose that the anion most likely bridges the fluorophore moiety. This was also evident from the 1H NMR (DMSO-d6) spectrum where the anthracene resonances were significantly affected. By simply modifying the electronic structure of the receptor, the sensitivity of the recognition process could also be modified; e.g. compound 1, bearing the trifluoromethyl substituent, showed stronger binding to the bis-anions than 2, which possessed a simple phenyl moiety.
Introduction
The sensing and monitoring of ions and molecules is currently of major interest, as anions are important from industrial (such as fertilisers), biological/medicinal and environmental points of view.1 Unlike cationic recognition and sensing,2 which is now a well-established area of research, anion recognition has been much less explored. However, recently a large number of excellent examples of anion receptors and luminescent sensors for anions have been produced and several reviews have appeared over the last few years.3 Major challenges in anion sensing are; they exhibit a variety of structures and conformations, they are often highly hydrated and they may be pH dependent, making it difficult to detect them in an aqueous environment.4 Consequently, structurally complicated and synthetically demanding receptors have been developed, which may not be realistic for use in practical anion sensors applicable to, for example, in vitro or in vivo monitoring.5We have recently focused our research towards the development of structurally simple sensors for anion recognition; developing lanthanide luminescent sensors, where coordinated unsaturated Tb(III) complexes6 were used to detect carboxylates such as salicylic acid in water at pH 7; as well as using charge neutral PET (photoinduced electron transfer) anion sensors, which employ thiourea based receptors.7 For the latter, recognition occurs through hydrogen bonding between the thiourea protons and the anion. This causes the fluorescence of the fluorophore to be quenched by electron transfer from the anion–receptor complex.8 We have demonstrated this by employing anions such as AcO−, H2PO4− and F− in DMSO, whereas anions such as ClO4−, Cl−, Br− and I− were not detected, presumably due to the simple nature of the receptor. However, the success of our original idea led us to extend our approach to anion sensing to the detection of anions that have more than one binding site, such as bis-carboxylates and phosphates such as pyrophosphate, etc.9,10 Such fluorescence sensing is of great biological and medicinal relevance as many dicarboxylates take part in metabolic processes together with pyrophosphate, which itself is essential for energy formation as it is the product of the hydrolysis of ATP under cellular conditions. Herein we present the full results from these investigations.11
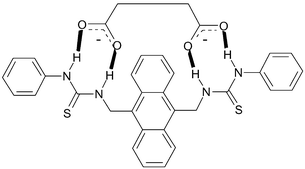
Our approach, which we call second-generation PET anion sensors, are based on the receptor–spacer–fluorophore–spacer–receptor motif shown in Fig. 1, originally developed by de Silva and coworkers for the sensing of bis-ammonium species and for mimicking logic gates.12 Here however, we have simply used two anion receptors that are connected in a linear fashion via the two spacers to an anthracene fluorophore through the 9 and 10 positions. This design gave the two thiourea based fluorescent sensors 1 and 2, and the urea analogue 3.
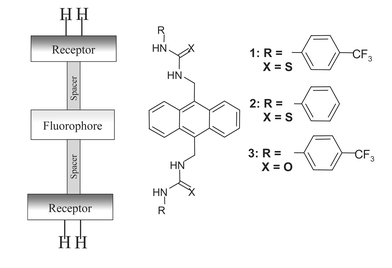 | ||
| Fig. 1 The receptor–spacer–fluorophore–spacer–receptor model used and the corresponding sensors 1–3. | ||
Even though these are structurally simple sensors, by simply altering the nature of the receptor site, e.g. by incorporating electron withdrawing substituents such as CF3, we hoped to be able to ‘tune’ the selectivity and sensitivity of anion recognition.8 This would be due to a modulation of the strength of the hydrogen bonding interactions between the thiourea/urea N–H and the anions, resulting in a receptor–anion complex which would be substantially affected and would subsequently affect the PET quenching of the fluorophore.13 To the best of our knowledge, these chemosensors are the first examples of charge neutral fluorescent PET sensors that show ideal PET behaviour for bis-anions.13,14
Results and discussion
Synthesis of 1–4
The chemosensors 1–3 were prepared from the corresponding isothiocyanates (5, 6 and 7) and 9,10-diaminomethylanthracene 4 in CH2Cl2 by stirring at room temperature (Scheme 1). The products were formed as precipitates that were isolated by filtration, washed with cold CH2Cl2 and recrystallised from CHCl3, giving 1–3 in ca. 80% yield. The isothiocyanates used in the synthesis of 1 and 2 were 4-(trifluoromethyl)-phenyl- 5 and phenyl-isothiocyanate 6 respectively, whereas for 3, 4-(trifluoromethyl)phenyl-isocyanate 7 was used. All the sensors were characterised using conventional methods, with the exception of MS-analysis due to fragmentations (see Experimental section). The 1H NMR spectra (400 MHz) of 1–3, when recorded in either DMSO-d6 or CDCl3, showed that the sensors were all highly symmetrical, as is evident from Fig. 2 (for 2), where only two resonances for the thiourea protons are observed at 9.36 and 8.06 ppm respectively. The latter is observed as a triplet due to the coupling of the methylene spacer, which is observed as a doublet at 5.75 ppm (the NMR of 1 is discussed later).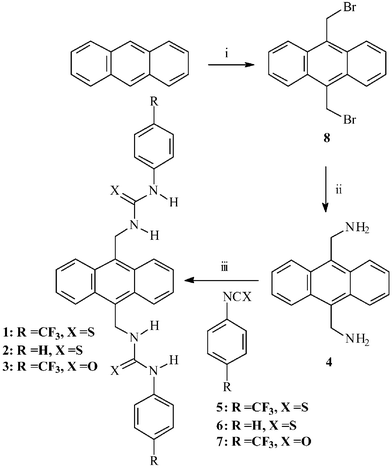 | ||
| Scheme 1 The synthesis of sensors 1–3. i) 1,3,5-Trioxane, HBr, CH3CO2H; tetradecyltrimethyl ammonium bromide; ii) hexamethylenetetramine in anhydrous CHCl3 followed by HCl, ethanol, H2O; iii) CH2Cl2 room temperature. | ||
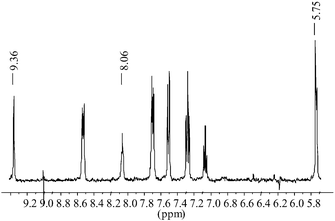 | ||
| Fig. 2 The 1H NMR (DMSO-d6, 400 MHz) of 2. | ||
The synthesis of the starting material 4 was more troublesome than expected. A preparation described previously employed the classical Gabriel synthesis.15 We, however, found that the first step in this synthesis resulted in the formation of the insoluble bis-phthalimide anthracene derivative and the conversion of this intermediate to 4 was extremely poor. More successful was an alternative method (Scheme 1) which involved the initial synthesis of 9,10-bisbromomethylanthracene 8 in one step from anthracene (75% yield) using a method derived by Altava et al.16a This involved reacting anthracene with 48% HBr and 1,3,5-trioxane, in glacial acetic acid in the presence of a catalytic amount of tetradecyltrimethyl ammonium bromide. The desired product was removed by filtration and dried over P2O5 followed by recrystallisation from toluene. From 8, 4 could be formed in ca. 85% yield as a crude product that could be used without further purification. This synthesis involved the treatment of 8 with hexamethylenetetramine in anhydrous CHCl3 under an inert atmosphere, followed by refluxing the resulting solution for five hours before the off-white precipitate formed was isolated by filtration.16b The resulting solid was then treated with a 20 : 4 : 5 mixture of EtOH, H2O and concentrated HCl and heated at 70 ºC for twelve hours, causing the HCl salt to redissolve. Upon cooling the mixture to room temperature a yellow precipitate was formed which was filtered and washed with 10% KHCO3 and then extracted into CHCl3. As stated earlier, the quality of this crude material was sufficient for use in the synthesis of 1–3.
Photophysical evaluation of 1–3 using monodentate anions
The ground and the excited state properties of 1–3 were investigated in DMSO. The absorption spectrum for 1, shown in Fig. 3, exhibits the same fine structure as is commonly observed for anthracene at 400, 379 and 359 nm respectively, with an extinction coefficient ε = 4 × 105 cm−1 M−1. For comparative purposes the absorption spectrum for 9,10-dimethyl-anthracene (DMA) is superimposed on the spectrum of 1 in Fig. 3. DMA spectra has peaks at 397, 375 and 356 nm respectively, illustrating that the two thiourea receptors in 1 have a slight but significant influence on the absorption bands, which become red-shifted. In comparison, the fluorescence emission spectra of 1 consisted of three bands at 409, 430 and 455 nm when excited at 379 nm (with quantum yield of fluorescence, ΦF = 0.047)17. These were significantly more red-shifted than those observed for DMA, which had emission bands at 395, 419 and 444 nm. However, a ΦF = 0.87 was measured under the same conditions for DMA, indicating significant quenching of the excited state of 1 which we assign to photoinduced electron transfer (PET) quenching taking place from the receptors in 1 to the excited state of the anthracene. For 2 (λabs = 400, 379 and 360 nm and λF = 409, 431 and 454 nm), the quantum yield was measured to be 0.11, which is substantially higher than that determined for 1, suggesting that the rate of PET is somewhat less in the free sensor of 2 in comparison to that observed for 1.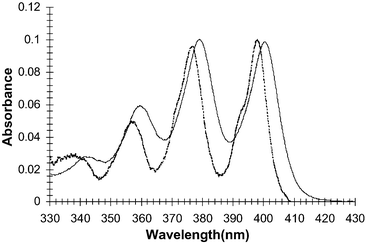 | ||
| Fig. 3 The absorption spectra of 1 (solid) and DMA (dashed) in DMSO. | ||
The effect on the absorption and the fluorescence emission spectra was recorded in the presence of several anions, using tetrabutyl ammonium (TBA) salts in DMSO. The absorption spectrum of 1 in the presence of increasing concentrations of TBAOAc (in DMSO) is shown in Fig. 4. As can be seen from this titration, there are only minor changes observed for the anthracene absorption, indicating that insignificant ground state interactions occur between the two receptors and the fluorophore, as no π–π or n–π interactions occur owing to the presence of the covalent spacers. Unfortunately, these changes were so small that we were unable to obtain a reliable binding profile from these results.
![The changes (uncorrected for dilution) in the absorption spectra of 1 in DMSO upon addition of AcO− ([AcO−]
= 0–58 mM).](/image/article/2005/OB/b409018g/b409018g-f4.gif) | ||
| Fig. 4 The changes (uncorrected for dilution) in the absorption spectra of 1 in DMSO upon addition of AcO− ([AcO−] = 0–58 mM). | ||
In contrast to these results, the changes in the fluorescence emission spectra were very different, as can be seen in Fig. 5, upon excitation of the anthracene fluorophores at 378 nm. From these changes it can be seen that emission was substantially quenched, ca. 70% at 430 nm, upon anion recognition at the receptor sites and the subsequent formation of the electron rich anion–receptor complexes. These changes are due to an enhanced photoinduced electron transfer (PET) rate from the anion bound receptors to the excited state of the anthracene. Furthermore, the addition of 10 mM of AcO− to a solution of DMA did not affect the ΦF of DMA; hence the emission was not quenched by either electron transfer or other dynamic processes, confirming that the quenching of 1 was indeed due to the recognition of the anion on the receptor site, with concomitant enhancements in PET quenching. This process was reversible, as upon addition of competitive hydrogen bonding solvents, such as ethanol or water, the emission was restored (switched on), indicating that the anion–receptor complexes were broken and hence reversible.
![The changes in the fluorescence emission spectra of 1 in DMSO upon addition of AcO− ([AcO−]
= 0–58 mM). The emission is switched off upon ion recognition.](/image/article/2005/OB/b409018g/b409018g-f5.gif) | ||
| Fig. 5 The changes in the fluorescence emission spectra of 1 in DMSO upon addition of AcO− ([AcO−] = 0–58 mM). The emission is switched off upon ion recognition. | ||
As these are monodentate anions we expected them to bind in the ratio of 1 : 2 (sensor : anions) as each receptor can recognise the acetate. This was indeed found to be the case as can be seen in Fig. 6 for the changes in fluorescence at 430 nm, where the emission is switched off over ca. four logarithmic units, from –log[Ac−] 2–6. In contrast to our earlier work, a 1 : 1 binding occurs over two concentration units. We thus conclude that this binding is 1 : 2 (see later in Fig. 14). It is also important to notice that no other significant spectral changes occur in the emission spectrum and, as a result, the presence of any red- or blue-shifts, or exiplex or eximer emission formation is not observed. Hence, only the intensity, and subsequently the quantum yield, of fluorescence is modulated upon anion sensing. Similar results were observed using H2PO4−. From these results it is clear that the sensor is behaving like an ideal PET sensor.13,14,18
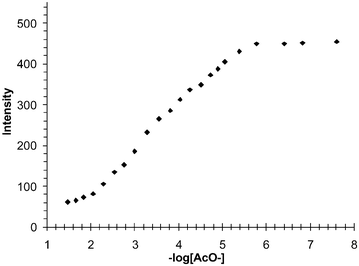 | ||
| Fig. 6 The changes in the fluorescence emission spectra of 1 at 430 nm in DMSO upon addition of AcO−. From these changes it can be deduced that the recognition of the anion occurs in a two-step process over four logarithmic concentration units. | ||
To confirm the selectivity of these sensors towards linear anions such as acetates, we also titrated the sensors with other anions. Upon addition of spherical anions such as Cl− or Br−, no significant quenching was observed excluding quenching by the heavy atom effect. Moreover, no quenching was observed upon addition of ClO4−. To investigate the selectivity of 1 for AcO− over Cl−, a known amount of AcO− was added to a solution of 1 with a 40 mM background concentration of Cl−. Whereas in the presence of Cl− the emission was switched on, it was quenched upon the addition of AcO− clearly confirming that 1 was selectively binding AcO− over Cl−. The same results were obtained for 2 when studied under identical experimental conditions.
Even though these spherical anions did not give rise to any change in either the ground or the excited states, the titration of 1 or 2 with F− was very different as the fluorescence emission was almost fully quenched (ca. 98%) after F− recognition. This was due to its small size and high charge density, which makes the hydrogen bonding between the receptors and the anion strong and, hence, the efficiency of the PET process is enhanced. However, the 1 : 2 binding was not so clear in this case as for the acetate or the phosphate titration. This could possibly be due to effective quenching of the excited state by only one of the receptor–F− complexes, prior to the binding of the second F−. Such an effect has recently been shown by Lippard et al.19 to occur in PET chemosensors for Zn(II), e.g. the sensor had two receptors but only 1 : 1 binding was confirmed by luminescence, as the emission was fully switched on after one equivalent of Zn(II). Here, a similar effect is possible, e.g. the emission is fully quenched by F− after only one equivalent. We do not believe that the changes observed in the fluorescence spectra are due to deprotonation of one or more of the thiourea protons as could possibly occur. We base this on the fact that we have recently developed naphthalimide based sensors that show dual fluorescence/colorimetric changes by F−, where the former was caused by hydrogen bonding recognition at a thiourea receptor and the colorimetric changes were due to deprotonation of an aryl amine by F−.7b Similar results were seen for 3 (λabs = 398, 377 and 358 nm and λF = 404, 427 and 453 nm) as observed for 1, upon titrations with these anions in DMSO. The changes for the titration of the latter using AcO− is shown in Fig. 7. Analysis of these changes indeed gave rise to a 1 : 2 binding as seen previously for 1 and 2, where the changes occurred from ca. −log[Ac−] of 1.8–4.8 (see later in Fig. 15 and Fig. 16). However, here the quenching is only ca. 60% as the urea–acetate complex is somewhat less effective in PET processes, most likely due to weaker binding as the urea protons are less acidic than the thiourea protons of 1.
![The changes in the fluorescence emission spectra of 3 in DMSO upon addition of AcO− ([AcO−]
= 0–40 mM). As in the case of 1, the emission of 3 is switched off upon ion recognition.](/image/article/2005/OB/b409018g/b409018g-f7.gif) | ||
| Fig. 7 The changes in the fluorescence emission spectra of 3 in DMSO upon addition of AcO− ([AcO−] = 0–40 mM). As in the case of 1, the emission of 3 is switched off upon ion recognition. | ||
Evaluating the 1 : 2 binding of 1 using 1H NMR
To confirm the 1 : 2 binding observed in the above investigations, for both acetate and phosphate, we conducted a 1H NMR titration of 1 using various anions and observed the changes in the 1H NMR of the thiourea protons in DMSO-d6. As mentioned previously, the high level of symmetry makes the 1H NMR of 1 and 2 very simple, as seen in Fig. 2 for 2. For 1, the thiourea protons appeared as two characteristic resonances at 9.68 and 8.38 ppm, respectively, Fig. 8 (top spectrum) and, as for 2 (Fig. 2), the spacer appears as a doublet. The position of the thiourea protons changed dramatically upon addition of anions such as AcO− and H2PO4− to a solution of the sensors in DMSO-d6. With either of these anions (0–5 equivalents of TBA salts), the thiourea resonances were gradually shifted downfield by 2.5–3 ppm, confirming the formation of the anion–receptor complexes through hydrogen bonding. Analysis of the change at 8.38 ppm as a function of concentration is shown in Fig. 9 for titration of H2PO4−. These results clearly show that after only two equivalents of H2PO4− the thiourea resonance stops shifting, indicating that a 1 : 2 binding is observed in this case (some line broadening was also observed). Similar results were seen for the titration of AcO−. However, the clear plateau after the addition of two equivalents of the anion was not as sharp. In addition to monitoring these changes, the position and characteristics of the resonances assigned to the aromatic protons and the methylene spacers were also monitored (see Fig. 8). In comparison to the changes in the two thiourea protons, these resonances did not change significantly, except for those assigned to the phenyl ring of the receptor as the anion recognition effects the electron density of the ring upon anion recognition. This indicates that there were only minor interactions between the anion–receptor complex and the anthracene moiety. From these results we conclude that for the simple monodentate anions the stoichiometry between the sensor and the anion is 1 : 2. We did not however determine the binding constant for this anion recognition event.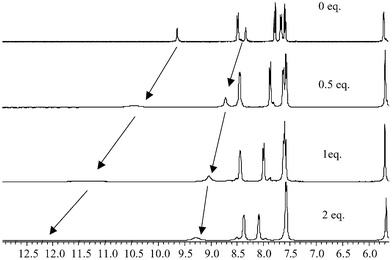 | ||
| Fig. 8 The changes in the 1H NMR (400 MHz) spectra of 1 in DMSO-d6 upon addition of H2PO4− using, 0, 0.5 and 1 equivalent of the anion. | ||
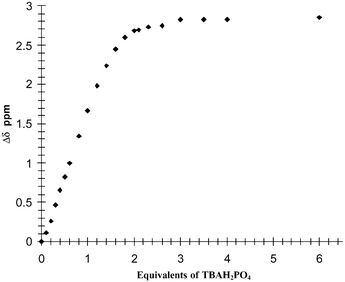 | ||
| Fig. 9 The changes in the 1H NMR resonance (Δδ) of the 8.38 ppm proton of 1 in DMSO-d6 upon addition of H2PO4− (shown in equivalents). | ||
Photophysical evaluation of 1–3 using bidentate anions
In a similar manner to that described above, the ground state and the excited state of 1–3 were investigated in the presence of biologically important bis-anions such as glutarate, malonate, and pyrophosphate [using tris(tetrabutylammonium) hydrogen pyrophosphate] in DMSO.20 The changes in the absorption spectra of 1 in the presence of pyrophosphate are shown in Fig. 10. As previously observed, there are only minor changes observed in the anthracence region upon anion recognition. In comparison to that observed in Fig. 4, the changes here are more pronounced particularly in the 358 and 378 nm transitions. Furthermore, analysis of the changes at longer wavelengths clearly showed that an isosbestic point was observed at 406 nm (Fig. 11). This indicates that the anthracence moiety experiences some perturbation in its electronic structure, most likely due to direct interactions with the anion. Similar results were observed for both 2 and 3.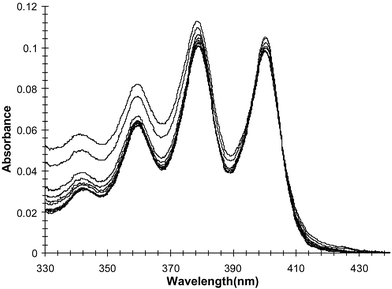 | ||
| Fig. 10 The changes (corrected for dilution) in the absorption spectrum of 1 upon titration with pyrophosphate. | ||
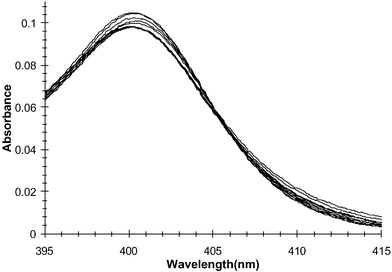 | ||
| Fig. 11 The changes in the absorption spectrum of 1 upon titration with H2PO4− between 390–410 nm only. | ||
The concomitant changes in the fluorescence emission spectra of 1 are shown in Fig. 12. Unlike that seen for the changes in the ground state, the excited state of the anthracene is substantially affected upon anion recognition, being switched off by ca. 95%. As before in the case of the mono-anions, no other significant spectral changes were observed in the emission spectra, i.e. no change in the emission wavelength or the formation of excimer emission bands. Similar changes were seen for 2, the quenching for 2 being ca. 90%. For these changes the ΦF values of 0.001 and 0.017 were determined after these titrations for 1 and 2 respectively. Similar changes were seen for 3 upon titration with pyrophosphate. As before the emission is switched off similarly to that seen for the mono-anions. This efficient fluorescence quenching is due to the enhancements in the reduction potential of the thiourea receptor moieties upon anion recognition. Hence, the rate of the electron transfer from the HOMO of the receptor to the anthracene excited state is increased as ΔGET becomes more thermodynamically favourable. As in previous cases, the emission was switched on again upon addition of EtOH.
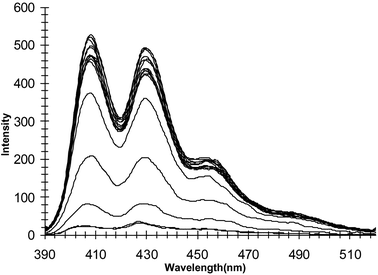 | ||
| Fig. 12 The changes in the fluorescence spectrum of 1 upon titration with pyrophosphate, which is switched off upon anion recognition. | ||
The changes in the fluorescence emission of 1 upon titration with glutarate in DMSO were similar to that observed in pyrophosphate (Fig. 12). As previously demonstrated, the emission was switched off (ΦF = 0.012) upon recognition of this anion. The fluorescence emission of 1 was also switched off when using malonate (ΦF = 0.011) ([anion] = 40 mM in all cases). No other spectral changes were observed in the emission spectra. Similar results were seen using 2, where the emission was however, less affected, with ΦF = 0.039 and 0.035 after the addition of 40 mM of glutarate and malonate respectively. This is not surprising as the receptor is less electron deficient compared to 1 and consequently the hydrogen bonding to the anion can be predicted to be weaker. The changes in 3 were similar to those observed for 1 and 2. However, the quenching was somewhat smaller than that seen for 1 because the urea protons are less acidic than the thiourea protons. This means that by simply changing the nature of the substituent and the substituent pattern can have substantial affect on both the binding sensitivity as well as the selectivity. On each occasion the reversibility of this binding was demonstrated by adding competitive solvents to the media.
To evaluate the selectivity and the sensitivity of this anion sensing, the changes at different wavelengths were monitored. The relative fluorescence changes for the anions tested for 1 are shown in Fig. 13 (Cl− and Br− are omitted as the fluorescence changes were too small), when observed at 430 nm as a function of −log[anion]. As can be seen from these changes, all the anions give rise to sigmoidal curves. Importantly, the emission is switched off over two logarithmic units for pyrophosphate and malonate, which would suggest a 1 : 1 binding and a simple equilibrium. From these changes, using the equation log[(Imax − IF)/(IF − Imin)] = log[anion] − logβ,13,22 the binding constants (logβ) of 2.34 (± 0.05) and 3.40 (± 0.05) were determined, for malonate and pyrophosphate respectively, for 1.
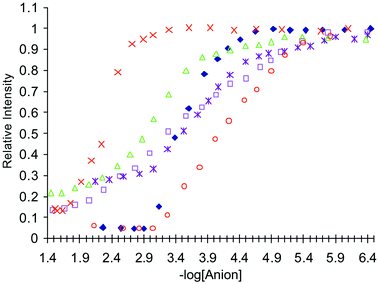 | ||
| Fig. 13 The relative intensity changes in the fluorescence spectrum of 1, when measured at 430 nm, upon addition of: ×= malonate; △ = H2PO4−; □ = AcO−; ◆ = pyrophosphate; * = glutarate; ○ = F−. | ||
As stated earlier, the changes in the fluorescence were modulated over more than two logarithmic units for the mono-anions.13 These results are also included in Fig. 13 for 1. Similar trends were found for 2 and 3, as shown in Figs. 14 and 15 respectively. For the 1 : 1 binding of the bis-anions to 2, the binding constant was measured to be 3.07 (± 0.05) and 2.02 (± 0.05) for pyrophosphate and malonate respectively. The gluatarte binding was also observed but not with a clear 1 : 1 binding. These overall changes can be seen in Figs. 13–15, and treading the data observed using a 1 : 1 fitting gave binding constants of 3.74, 3.15 and 3.77 for 1, 2 and 3 respectively.
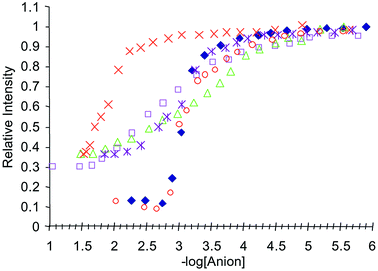 | ||
| Fig. 14 The relative intensity changes in the fluorescence spectrum of 2, when measured at 430 nm, upon addition of: ×= malonate; △ = H2PO4−; □ = AcO−; ◆ = pyrophosphate; * = glutarate; ○ = F−. | ||
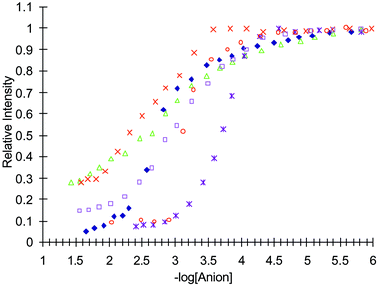 | ||
| Fig. 15 The relative intensity changes in the fluorescence spectrum of 3, when measured at 430 nm, upon addition of: ×= malonate; △ = H2PO4−; □ = AcO−; ◆ = pyrophosphate; * = glutarate; ○ = F−. | ||
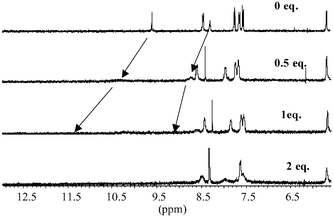 | ||
| Fig. 16 The changes in the 1H NMR (400 MHz) spectra of 1 in DMSO-d6 upon addition of pyrophosphate using 0, 0.5, 1 and equivalent of the anion. Unlike that seen in Fig. 8, the aromatic resonances are substantially affected upon binding. | ||
For 3, the binding of the anions was similar to that seen for both 1 and 2. The changes for the binding of both the mono- and the bis-anions are shown in Fig. 15. As mentioned previously, the mono-anions gave rise to a 1 : 2 binding for AcO− and H2PO4−. It is also clear that for F− the emission is ca. 90% quenched after one equivalent of the anion. For malonate and pyrophosphate the binding was also determined to be in a 1 : 1 stoichiometry, with logβ values of 2.6 (± 0.05) and 2.8 (± 0.05) respectively. However, unlike that seen for either 1 or 2, the detection of glutarate gives rise to ca. 1 : 1 binding. These binding results are summarized in Table 1.
| 1 | 2 | 3 | |
|---|---|---|---|
| a All measured in DMSO at room temperature and were repeated 2 or 3 times. Using data from different wavelengths gave, on all occasions, same binding constants with in 5–10% error.b Determined using the equation: log[(Imax − IF)/(IF − Imin)] = log[anion] − logβ.22 | |||
| F− | 4.13 | 3.03 | 3.30 |
| Pyrophosphate | 3.40 | 3.07 | 2.72 |
| Malonate | 2.34 | 3.15 | 2.66 |
| Glutarate | 3.74 | 3.27 | 3.77 |
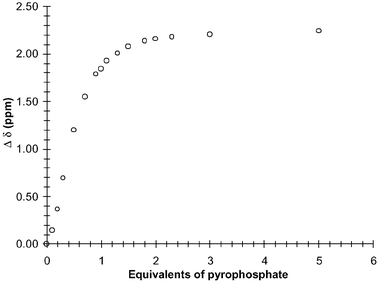 | ||
| Fig. 17 The changes in the 1H NMR resonance (Δδ) of the 8.38 ppm proton of 1 in DMSO-d6 upon addition of pyrophosphate. | ||
These results indicate that for 1 and 2 there is a clear trend, where all the anions were detected at a lower concentration for 1 in comparison to 2, with the exception of malonate. This is due to the electron withdrawing effect of the CF3 substituent on the receptor, making the N–H more acidic. However, for 3 the binding is usually weaker than for either 1 or 2. Overall, the presence of the second binding site in 1–3 gives rise to the selective 1 : 1 binding of anions that can potentially bridge the anthracene moiety. We foresee this occuring in a manner as shown in Scheme 2. This is supported by the fact that an isosbestic point was observed for these titrations, whereas in the 1 : 2 binding of the more simple anions, such as acetate and phosphate, it was absent. We also showed that this 1 : 2 binding was observed in the 1H NMR, where the resonances assigned to the anthracene fluorophore were not affected. Consequently, we evaluated the changes in the 1H NMR spectra of 1 in the presence of pyrophosphate, which had shown the best fit to 1 : 1 binding for 1–3.
 | ||
| Scheme 2 The possible binding of the bis-anions by 2 in a 1 : 1 binding stoichiometry. | ||
Evaluating the 1 : 1 binding of pyrophosphate by 1 using 1H NMR
The 1H NMR titrations were carried out in the same manner as described for the mono-anions. As before, the position of the thiourea protons changed dramatically upon addition (greater than 3 ppm) of pyrophosphate in DMSO-d6. There was also a substantial line broadening observed at the later stage of the titration. These changes are seen in Fig. 16 for 1. By plotting the changes in the thiourea proton at 8.38 as a function of the added anion, a binding profile was observed (Fig. 17) which demonstrated the formation of 1 : 1 binding between the anion and the sensor. From these changes, a logβ of 3.61(± 0.05) was determined, with a clear 1 : 1 binding, which is in good agreement with that seen using fluorescence (logβ of 3.45). For such recognition to take place the anion would have to bridge the anthracene moiety in the manner we have proposed in Scheme 2. Hence, such bridging would most likely affect the resonances of the anthracene moiety. As discussed earlier, the anthracene resonances were not affected upon addition of either AcO− or H2PO4− (Fig. 8). However, as can be seen in Fig. 16, this is not the case for pyrophosphate, where both shifts and line broadening occur upon anion recognition. Therefore, these results support the possibility that the anion bridges across the anthracene, ruling out the possibility of obtaining the 1 : 1 ratio via two sensors binding to two anions simultaneously.As we did not obtain the formation of any excimer emission in the fluorescence spectra, it also supports the notion that a 2 : 2 binding is not occurring. Recently however the interactions between pyrophosphate and pyrene fluorophores have been demonstrated between the two pyrene units and pyrophosphate.21
Conclusion
We have demonstrated the synthesis of several new PET sensors for anions. These sensors were designed to target anions flanked with two binding sites such as bis-carboxylates and pyrophosphate. The anion sensing of these compounds was demonstrated using fluorescence. For sensors 1–3 the emission was switched off upon titration with various anions. This was due to the enhancement in the PET quenching of the anthracene excited state upon anion recognition, which occurred through hydrogen bonding between the thio or the urea protons and the anions. This binding mode was confirmed by observing the changes in the thiourea protons of 1 upon titration with several anions. By monitoring the changes in the fluorescence emission spectra as a function of anion concentration, we demonstrated that for simple anions such as AcO− and H2PO4− the recognition was due to the formation of a 1 : 2 binding between the sensors and the anions. For halides such as Cl− and Br− no significant fluorescence changes occurred. However, for F− the emission was almost fully quenched after only one equivalent of the anion.The recognition of bis-anions such as pyrophosphate, malonate and glutarate was also demonstrated. For the first two of these anions, the sensing was shown to be in the 1 : 1 stoichiometry, whereas for glutarate the binding was most likely 1 : 2 for 1 and 2. The 1 : 1 binding of pyrophosphate was also demonstrated using 1H NMR.
To the best of our knowledge these are the first PET sensors for such bis-anions where the anion recognition occurs through hydrogen bonding between the anion and the charge neutral receptors, with concomitant enhancements in the quenching of the fluorophore excited state. We are currently developing other anion sensors for bis-anions with the aim of improving both selectively and the sensitivity of the sensors, as well as making this sensing process possible in aqueous environments.
Experimental
General
Reagents (obtained from Aldrich) and solvents were purified using standard techniques. Solvents were dried over the appropriate drying agent before use using standard procedures. Melting points were determined using a GallenKamp melting point apparatus. Infrared spectra were recorded on a Mattson Genesis II FTIR spectrophotometer equipped with a Gateway 2000 4DX2-66 workstation. Oils were analysed using NaCl plates and solid samples were dispersed in KBr and recorded as clear pressed discs. 1H NMR spectra were recorded at 400 MHz using a Bruker Spectrospin DPX-400 instrument. 13C NMR were recorded at 100 MHz using a Bruker Spectrospin DPX-400 instrument. Fluorescent measurements were made on a Perkin Elmer LS 50B. UV-Vis absorption spectra were recorded at room temperature using a Shimadzu UV-2401PC.Syntheses of 1–4
Acknowledgements
We thank Enterprise Ireland, Kinerton Ltd and TCD for financial support and Dr Hazel Moncrieff and Dr Juliann Tierney for their help.References
- (a) Chemical Sensors and Biosensors for Medical and Biological Applications, ed. U. S. Spichiger-Keller, Wiley-VCH,Weinheim, Germany, 1998 Search PubMed; (b) C. F. Mason, Biology of Freshwater Pollution, Longman, New York, 2nd edn., 1991 Search PubMed.
- (a) A. P. de Silva, B. McCaughan, B. O. F. McKinney and M. Querol, Dalton Trans., 2003, 1902 RSC; (b) K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116 RSC; (c) K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 216 CrossRef CAS.
- (a) Special issue on anion recognition, Coord. Chem. Rev., 2003, 240 Search PubMed; (b) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef CAS; (c) C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192 RSC; (d) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef; (e) P. A. Gale, Coord. Chem. Rev., 2001, 213, 79 CrossRef CAS; (f) P. A. Gale, Coord. Chem. Rev., 2000, 199, 181 CrossRef CAS; (g) V. Amendola, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Poggi and A. Taglietti, Coord. Chem. Rev., 2001, 219, 821 CrossRef; (h) L. Fabbrizzi, M. Licchelli, G. Rabaioli and A. Taglietti, Coord. Chem. Rev., 2000, 205, 85 CrossRef CAS; (i) P. D. Beer, Chem. Commun., 1996, 689 RSC.
- Supramolecular Chemistry of Anions, eds. E. Bianchi, K. Bowman-James and E. Gracía-Espanña, Wiley-VCH, New York, 1997 Search PubMed.
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS; (b) J. Scheerder, J. F. J. Engersen and D. N. Reinhoudt, Recl. Trav. Chim. Pays-Bas, 1996, 115, 307 CAS.
- (a) T. Gunnlaugsson, A. Harte, J. P. Leonard and M. Nieuwenhuyzen, Supramol. Chem., 2003, 15, 505 CrossRef CAS; (b) T. Gunnlaugsson, A. Harte, J. P. Leonard and M. Nieuwenhuyzen, Chem. Commun., 2002, 2134 RSC.
- (a) T. Gunnlaugsson, A. P. Davis, G. M. Hussey, J. Tierney and M. Glynn, Org. Biomol. Chem., 2004, 2, 1856 RSC; (b) T. Gunnlaugsson, P. E. Kruger, T. Clive Lee, R. Parkesh, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 35, 6575 CrossRef; (c) T. Gunnlaugsson, A. P. Davis and M. Glynn, Chem. Commun., 2001, 2556 RSC.
- (a) Recent examples of luminescent anion sensing include: K. Kim, N. J. Singh, S. J. Kim, H. G. Kim, J. K. Kim, L. W. Lee, K. S. Kim and J. A. Yoon, Org. Lett., 2003, 54, 2083 Search PubMed; (b) N. Marcotte and A. Taglietti, Supramol. Chem., 2003, 15, 617 CrossRef CAS; (c) A. P. de Silva, G. D. McClean and S. Pagliari, Chem. Commun., 2003, 2010 RSC; (d) E. J. Cho, J. W. Moon, S. W. Ko, J. Y. Lee, S. K. Kim, J. Yoon and K. C Nam, J. Am. Chem. Soc., 2003, 125, 12376 CrossRef CAS; (e) Y. Kubo, S. Ishihara, M. Tsukahara and S. Tokita, J. Chem. Soc., Perkin Trans. 2, 2002, 1455 Search PubMed; (f) D. H. Lee, H. Y. Lee and J. P. Hong, Tetrahedron Lett., 2002, 34, 7273 CrossRef CAS; (g) S. O. Kang, S. Jeon and K. C. Nam, Supramol. Chem., 2002, 14, 405 CrossRef CAS; (h) K. Kim and J. T. Yoon, Chem. Commun., 2002, 770 RSC; (i) S. Sasaki, D. Citterio, S. Ozawa and K. S. Suzuki, J. Chem. Soc., Perkin Trans. 2, 2001, 2309 RSC; (j) P. E. Kruger, P. R. Mackie and M. Nieuwenhuysen, J. Chem. Soc., Perkin Trans. 2, 2001, 1079 RSC; (k) Y. Kubo, M. Tsukahara, S. Ishihara and S. Tokita, Chem. Commun., 2000, 653 RSC; (l) P. Anzenbacher Jr., K. Jursíková and J. L. Sessler, J. Am. Chem. Soc., 2000, 122, 9350 CrossRef CAS; (m) H. Miyaji, P. Anzenbacher Jr., J. L. Sessler, E. R. Bleasdale and P. A. Gale, Chem. Commun., 1999, 1723 RSC; (n) H. Xie, S. Yi, X. Yang and S. Wu, New J. Chem., 1999, 23, 1105 RSC; (o) C. B. Blake, B. Andrioletti, A. C. Try, C. Ruiperez and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 10438 CrossRef CAS; (p) C. R. Cooper, N. Spencer and T. D. James, Chem. Commun., 1998, 1365 RSC; (q) R. S. Dickens, T. Gunnlaugsson, D. Parker and R. D. Peacock, Chem. Commun., 1988, 1643 Search PubMed.
- (a) S. Watanabe, N. Higashi, M. Kobayashi, K. Hamanaka, Y. Takata and K. Yoshida, Tetrahedron Lett., 2000, 41, 4583 CrossRef CAS; (b) V. Král, A. Andrievsky and J. L. Sessler, J. Am. Chem. Soc., 1995, 117, 2954.
- (a) Urea receptors have been used to bind bis-anions where the recognition was monitored by NMR, for examples see: B. R. Linton, M. S. Goodman, E. Fan, S. A. van Arman and A. D. Hamilton, J. Org. Chem., 2001, 66, 7313 Search PubMed; (b) E. Fan, S. A. van Arman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369 CrossRef CAS.
- Preliminary communication: T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Lett., 2002, 4, 2449 Search PubMed.
- (a) A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42 CrossRef; (b) A. P. de Silva and K. R. A. S. Sandanayake, Angew. Chem., Int. Ed. Engl., 1990, 29, 1173 CrossRef.
- (a) R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire, C. P. McCoy and K. R. A. S. Sandanayake, Top. Curr. Chem., 1993, 168, 223; (b) R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire and K. R. A. S. Sandanayake, Chem. Soc. Rev., 1992, 21, 187 RSC.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef.
- T. M. Fyles and V. V. Suresh, Can. J. Chem., 1994, 72, 1246 CAS.
- (a) B. Altava, M. I. Burguete, B. Escuder, S. V. Luis, E. García-España and M. C. Muñoz, Tetrahedron, 1997, 53, 2629 CrossRef CAS; (b) B. Alpha, E. Anklam, R. Deschenaux, J. M. Lehn and M. Pietraskiewicz, Helv. Chim. Acta, 1988, 71, 1043.
- R. L. Barnes and J. B. Briks, Proc. R. Soc. London, Ser. A, 1966, 291, 570 CAS.
- (a) T. Gunnlaugsson, T. C. Lee and R. Parkesh, Org. Biomol. Chem., 2003, 1, 3265 RSC; (b) T. Gunnlaugsson, B. Bichell and C. Nolan, Tetrahedron Lett., 2002, 43, 4989 CrossRef CAS; (c) T. Gunnlaugsson, M. Nieuwenhuyzen, L. Richard and V. Thoss, J. Chem. Soc., Perkin Trans. 2, 2002, 141 RSC.
- S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 7831 CrossRef CAS.
- M. Mei and S. Wu, New J. Chem., 2001, 25, 471 RSC.
- S. Nishizawa, R. Kato and N. Teramae, J. Am. Chem. Soc., 1999, 121, 9463 CrossRef CAS.
- C. R. Cooper and T. D. James, J. Chem. Soc., Perkin Trans. 1, 2000, 963 RSC.
| This journal is © The Royal Society of Chemistry 2005 |