A straightforward approach towards cyclic peptides via ring-closing metathesis—scope and limitations†‡
Uli
Kazmaier
*,
Christina
Hebach
,
Anja
Watzke
,
Sabine
Maier
,
Heike
Mues
and
Volker
Huch
Institut für Organische Chemie, Universität des Saarlandes, Saarbrücken, D-66123, Germany. E-mail: u.kazmaier@mx.uni-saarland.de; Fax: +49 681 302 2409; Tel: +49 681 302 3409
First published on 26th November 2004
Abstract
N- and C-terminal diallylated peptides are obtained by several approaches, such as peptide Claisen rearrangement, N- and O- allylation, and the Ugi reaction of allyl-protected components. These diallylated peptides are suitable substrates for ring-closing metathesis and the success of this cyclisation was investigated with respect to the ring size, the position of the allyl moieties and the reaction parameters. In general, excellent yields are obtained for cyclisation of allyl glycine subunits and N-allylated amides, while allyl esters and allyl carbamates often presented serious problems. However, yields of up to 73% were obtained under optimised conditions, and the new generated double bond is formed with excellent trans-selectivity.
Introduction
Cyclic peptides are found in a wide variety of marine organisms and fungi.1 Many of these peptides show significant biological activity,2 and are therefore highly interesting from a pharmaceutical point of view.3 In higher organisms, cyclic structures can be formed by the oxidation of two cysteine subunits, a process which is generally used to stabilise the secondary and tertiary structure of peptides. Cysteine-containing peptides are preferentially found in peptide hormones and a number of redox active proteins, such as glutaredoxin 1.4 Here, the disulfide bridge locks a tetrapeptide fragment of the protein chain into a β-turn type structure.5 Frequently, loops and turns in peptides and proteins are responsible for their biological activity, making these structures are highly interesting from both a pharmaceutical point of view and as targets for peptidomimetics.6 In general, cyclisation of peptides results in an increased stability towards proteases. Since in cysteine peptides the disulfide bonds are sensitive to reduction, the metabolic stability of these compounds can be dramatically increased by replacing the sensitive disulfide bond by a non-cleavable C–C bond.7 Therefore, a lot of investigation has been carried out on the synthesis of carba analogues of cysteine, such as 2,7-diaminosuberic acid.8 Very recently, Williams et al. and Grubbs et al. described syntheses based on the dimerisation of tethered allyl glycines via ring-closing metathesis.9–11 As shown by Grubbs et al. this ring-closing approach can be directly used for the synthesis of cyclic peptides, such as 2, a carba analogue of the glutaredoxin active site 1 (Fig. 1).12 This is an extremely straightforward approach towards β-turn mimetics, and since the publication of this pioneering work, several examples of the synthesis of smaller and larger ring systems were reported, many of which featured interesting pharmaceutical properties.13–15 | ||
| Fig. 1 Natural and artificial peptide loops. | ||
Ring-closing metathesis was not only used for the cyclisation of simple peptides to form cyclic peptidomimetics, but also for the fixation of helix structures and the interconnection of cyclic peptides.16–17
Peptides containing allyl glycines are very often used as precursors,18 thus leading to surrogates for cysteine, as illustrated in Fig. 1. However, other unsaturated substrates can be cyclised, such as allylic ethers, esters or amides.19–21 The yields in the cyclisation step depend on the ring size as well as on the distance between the double bonds and other functionalities. Liskamp et al. investigated the ring-closing metathesis of a wide range of peptides bearing the unsaturation on the amide functionality.22 In this case, the introduced loops connect two amide nitrogens, leaving the amino acid sequence unaffected. Importantly, these macrocycles may be formed by connecting any two amide nitrogens, as long as the length of the alkene substituents is adjusted appropriately. It was established that the ring-closing metathesis can be conducted using N-alkylamides for a loop bridging two amides, N-pentenylamides for a loop spanning three amides and N-homoallylamides when four or more amides are involved in the ring. Libraries of cyclic peptides were obtained by this approach.
For quite some time we have been investigating syntheses of γ,δ-unsaturated amino acids.23 Besides Pd-catalysed allylic alkylations of chelated amino acid ester enolates,24 especially the Claisen rearrangement, proceeding via those chelated amino acid ester enolates is suitable for this purpose.25 If esters of chiral allylic alcohols are used, the corresponding enantiomerically pure amino acids are obtained.26 This protocol is not limited to the rearrangement of amino acid esters, but can also be applied to peptides.27 In particular, allylic esters of tosylated peptides are suitable substrates for the Claisen rearrangement, yielding the products in a highly diastereoselective fashion, while the configuration of the newly formed stereogenic centre is controlled by the peptide chain.28 Tosyl-protected amino acids are also appropriate substrates for palladium-catalysed allylic alkylations, giving rise to N-allylated derivatives under very mild conditions.29 Therefore, these protocols open up an easy access to fully functionalised diallylic peptides, suitable precursors for ring-closing metathesis.30
Results and discussion
We started our investigations with the rearrangement of peptide ester 3 (Scheme 1). In the presence of tin chloride, used for chelation, the rearranged product 4 was obtained with excellent yield and in a highly stereoselective manner. An induced diastereoselectivity of 84% was remarkable, especially with respect to the fact that the only chiral centre in the peptide 3 was 7 atoms away from the newly formed chiral centre. The (R)-configured amino acid was formed preferentially with high syn selectivity (>95%). Peptide 4 was subsequently subjected to a palladium-catalysed N-allylation, giving rise to the product 5 without any racemisation. Unfortunately, all attempts to cyclise this diallylated substrate 5 under different reaction conditions and various amounts of Grubbs' catalyst were unsuccessful.31 | ||
Scheme 1 Peptide Claisen rearrangement of crotyl ester 3; (a) 5.0 equiv LHMDS, 2.0 equiv SnCl2, THF, −78 °C → rt, 16 h; (b) CH2N2, ether, 10 min; (c) 1 mol% (allyl PdCl)2, 4.5 mol% PPh3, allyl carbonate, THF, rt, 16 h; (d) 10–15 mol% RuCl2(PCy3)2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh), toluene, rt → 90 °C, 8 h. CHPh), toluene, rt → 90 °C, 8 h. | ||
Several factors influence the tendency of ring-closure metathesis, but we focused mainly on the following three parameters: (i) the unsaturated amino acid, (ii) the ring size and (iii) the amino acid sequence. In ring-closing metatheses described in the literature so far, unbranched allylglycines (or related amino acids) were generally used as substrates. Therefore, one might reason that the more steric demanding methyl group in α-position to the terminal olefin could inhibit the ring-closure.
Unfortunately, exchanging this amino acid with allylglycine was just as fruitless as reducing the ring size by replacing the β-amino acid by an α-amino acid such as leucine. Obviously 12- and 13-membered peptide rings were difficult to obtain by this strategy. Most likely, the problems with these medium sized rings result from an unsuitable conformation of the linear peptide chain in solution, with trans amide bonds inhibiting the allyl termini from coming within close proximity. Therefore we decided to enlarge the ring size and to introduce one proline, which is able to form cis amide bonds and turn structures. 32
Rearrangement of the tetrapeptide ester 6 gave the desired allylated tetrapeptide 7 in excellent yield (Scheme 2).33N-allylation provided the substrate 8 for the subsequent ring-closing metathesis, which now resulted in the formation of the desired cyclic peptide 9 (15-membered ring) in high yield.34 This was in good agreement with the results described by Grubbs et al. for comparable peptides such as 2 (14-membered ring).12 The double bond was obtained as an inseparable cis/trans-mixture.
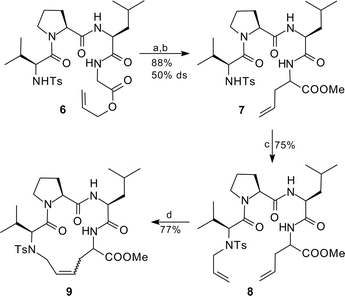 | ||
| Scheme 2 Peptide Claisen rearrangement and subsequent ring-closing metathesis; (a) 5.0 equiv LHMDS, 2.0 equiv SnCl2, THF, −78 °C → rt, 16 h; (b) CH2N2, ether, 10 min; (c) 1 mol% (allyl PdCl)2, 4.5 mol% PPh3, allyl carbonate, THF, rt, 16 h; (d) 10 mol% RuCl2(PCy3)2(CHPh), CH2Cl2, reflux, 14 h. | ||
To determine if the proline plays a significant role in the cyclisation, we came back to our allylated tripeptides such as 10, which could not by cyclised previously after N-allylation. Cleavage of the N-protecting group and coupling with allylated amino acids provided the elongated peptides 11 and 12. Subsequent ring-closing metathesis yielded the corresponding 16-membered cyclic peptides 13 and 14 in even higher yields in comparison to the smaller proline peptide (Scheme 3). Obviously ring size played a major role for the success of this reaction; the amino acid sequence and conformational issues were less important, at least for peptides with more than 14 ring members.
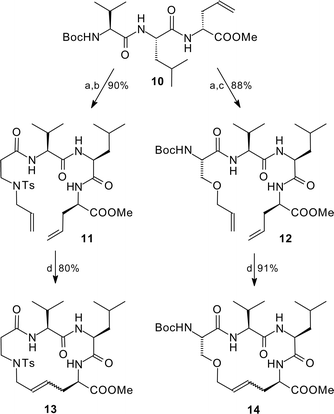 | ||
| Scheme 3 Cyclisation of 16-membered rings; (a) HCl, dioxane, 0 °C, 30 min; (b) 1 equiv N-tosyl-N-allyl-β-Ala, 1.1 equiv TBTU,35 5 equiv, NEt3, CH2Cl2, 10 h; (c) 1 equiv Boc-Ser(OAll), 1.1 equiv TBTU, 5 equiv, NEt3, CH2Cl2, 8 h; (d) 10 mol% RuCl2(PCy3)2(CHPh), CH2Cl2, reflux, 15 h. | ||
We further enlarged the ring size by replacing the allylglycine by an O-allylserine (Scheme 4, 15). For comparison, the proline-containing peptide 17 was also prepared by conventional peptide synthesis. Both peptides 15 and 17 gave the expected 18-membered cyclisation products 16 and 18 in excellent yield, comparable to the 16-membered analogues. Product 18 was obtained as a separable 1 : 1 mixture of the cis/trans-isomers, while in the case of 16 the isomers could not be separated. An interesting observation was made after the C-terminal O-allylserine unit was replaced by the serine allyl ester 19. In this case, the yield of the ring-closing metathesis dropped to 21%, although the ring size was the same as in 16 and 18.
 | ||
| Scheme 4 Cyclisation of 18-membered rings; (a) 10 mol% RuCl2(PCy3)2(CHPh), CH2Cl2, reflux, 15 h. | ||
Ring strain should not be the problem, and therefore one might not expect higher yields at higher temperatures. Indeed, when the cyclisation is carried out in toluene at 60 °C, only unreacted starting material 19 was recovered. Obviously, allylic esters are much less suited for ring-closing metathesis in comparison to allylic ethers or amides. This might be explained by a coordination of the carbonyl group to the ruthenium carbon complex formed in the first step of the metathesis, giving rise to a stable 6-membered chelate ring, which does not undergo further reactions.36 This chelate formation can, in principle, be suppressed by addition of Ti(OiPr)4, which coordinates more strongly to the carbonyl group.37 Unfortunately, the addition of 5 equiv. of Ti(OiPr)4 had no effect on the cyclisation. Another reason for the low yield might result from an interaction of the free hydroxy group on the C-terminal serine with the catalyst. We therefore protected this functionality by benzoylation, but the cyclisation yield was lower than with the free hydroxy group.
Because of our ongoing interest in stereoselective modifications of peptides we wanted to use cyclic peptides as templates for the stereoselective introduction of side chains, since stereoselective modifications of linear peptides are not a trivial issue.38 The advantage of cyclisation via an allylic ester is that the latter can easily be cleaved after the modification under palladium catalysis. Most interesting from this point of view should be a fixation of peptide conformation by metathesis of two allylic protecting groups; an allyl ester on the C-terminus and an allyloxycarbonyl (Alloc)-protecting group on the N-terminus of the peptide. Sequential palladium-catalysed cleavage of both protecting groups in one step should provide fully unprotected modified peptides. We intensively investigated a ring-closure approach with these two allylic protecting groups. As substrates we chose N-allyl protected tripeptide allylic esters, since they lead to 16-membered rings, which has previously been reported to be a suitable size. To get rapid access to our substrates with a high potency of variation, the Passerini and Ugi multi-component reactions have been used, providing the required substrates in one step (Scheme 5).39–40 With Alloc-protected valine as the acid component and the allylic ester of isocyanoacetic acid the depsipeptide, 21 was obtained as diastereomeric mixture in acceptable yield. In the presence of (R)-p-methoxy-1-phenylethylamine the corresponding peptide 22 could be obtained in a 3 : 1 diastereomeric ratio at rt. The selectivity could be increased to nearly 9 : 1 if the reaction was carried out at −30 °C. In this case the diastereomers could be separated by flash chromatography. The substrates were then subjected to ring-closing metathesis. No reaction was observed using depsipeptide 21 even with 10% catalyst, while the major diastereomer of 22 gave cyclisation product 23 in only 13% yield (Table 1, Entry 1).
| Entry | Peptide | Product | Catalyst | Reaction conditions | Yield/% | Comments |
|---|---|---|---|---|---|---|
| 1 | 22 | 23 | 10% A | CH2Cl2, rt | 13 | Only trans double bond |
| 2 | 25 | 28 | 5% A | CH2Cl2, rt | 37 | 21% starting material recovered |
| 3 | 26 | 29 | 10% A | Toluene, 60 °C | 33 | 34% starting material recovered |
| 4 | 22 | 23 | 10% B | CH2Cl2, rt | — | Polymerisation |
| 5 | 22 | 23 | 5% C | Toluene, rt | — | Polymerisation |
| 6 | 25 | 28 | 10% B | CH2Cl2, rt | — | Polymerisation |
| 7 | 25 | 28 | 5% C | CH2Cl2, rt | 5 | Polymerisation |
| 8 | 24 | 27 | 10% A | CH2Cl2, rt | 15 | 21% starting material recovered |
| 9 | 24 | 27 | 10% A | Toluene, 30 °C | 16 | — |
| 10 | 24 | 27 | 10% A | Toluene, 90 °C or N2 stream | 27 | — |
| 11 | 22 | 23 | 10% A | CH2Cl2, rt, 0.3 eq. Ti(OiPr)4 | 34 | — |
| 12 | 22 | 23 | 10% A | CH2Cl2, rt, 1 eq. Ti(OiPr)4 | 26 | — |
| 13 | 22 | 23 | 10% A | CH2Cl2, rt, 4 eq. Ti(OiPr)4 | — | — |
| 14 | 22 | 23 | 10% B | CH2Cl2, rt, 0.3 eq. Ti(OiPr)4 | — | Polymerisation |
| 15 | 22 | 23 | 10% A | Catalyst added in 30 min to a 0.05 M solution of 25 in CH2Cl2 at rt | < 5 | — |
| 16 | 25 | 28 | 5% A | Catalyst added in 30 min to a 0.05 M solution of 28 in CH2Cl2 at rt | < 5 | — |
| 17 | 22 | 23 | 10% A | Catalyst added in 2 h to a 0.05 M solution of 25 in CH2Cl2 at rt | 23 | — |
| 18 | 25 | 28 | 5% A | Catalyst added in 30 min to a 0.05 M solution (CH2Cl2) of 28 under N2 stream at rt | 34 | — |
| 19 | 22 | 23 | 5% A | Catalyst added in 30 min to a 0.05 M solution (CH2Cl2) of 28 under reflux–N2 stream | 38 | — |
| 20 | 25 | 28 | 10% A | Peptide (0.01 M solution) added to catalyst in CH2Cl2 under reflux–N2 stream | 51 | Only trans double bond |
| 21 | 25 | 26 | 10% A | Peptide (0.01 M solution) added to catalyst in CH2Cl2 under reflux–N2 stream | 73 | Only trans double bond |
 | ||
| Scheme 5 Multi component reactions and ring-closing metathesis; (a) 10 mol% RuCl2(PCy3)2(CHPh), CH2Cl2, reflux, 15 h. | ||
Interestingly, only the trans-configured product was obtained (>95%) as determined by HPLC. The olefin geometry as well as the configuration of the newly formed stereogenic centre could be determined by X-ray structure analysis.41§ All amide and ester bonds showed trans configuration, even the N-alkylated amide bond. Therefore it is not obvious, why depsipeptide 21 did not undergo cyclisation.
Since the Ugi reaction turned out to be a valuable approach to the required substrates, we investigated the influence of several parameters on the outcome of the ring-closing metathesis (Scheme 6). One might expect an influence from the peptide sequence, especially if loop inducing amino acids such as prolines or N-alkylated amino acids are incorporated. Therefore we also varied the N-terminal amino acid using Alloc-protected alanine, proline and sterically demanding N-methyl valine as carbocyclic acids. With (S)-m-methoxy-1-phenylethylamine all peptides were obtained in high yield and especially with Alloc-alanine (24) in excellent diastereoselectivity. Unfortunately for the peptides 25 and 26 the diastereomeric ratio could not be determined because of the occurrence of several rotamers. Therefore the diastereomeric mixture was subjected to ring-closing metathesis and the yields are given in Table 1. None of the reactions ran to completion and in all cases the starting material was recovered. In agreement with the cyclisation of 22 only the all-(S)-substrates underwent cyclisation, while the other diastereomer was the major component of the recovered starting material. In all examples only the trans configured double bond (>95 trans) was obtained, as determined by X-ray structure analysis of 28 and comparison of the NMR spectra of the other cyclic peptides.†
 | ||
| Scheme 6 Ugi reactions and ring-closing metathesis. | ||
As expected, the best results were obtained with the proline peptide 25. The isolated yield was better than 26, for which more starting material was recovered.
With this substrate harsher reaction conditions were necessary. In comparison to 24 and 25 the reaction was rather slow at rt, but could be accelerated significantly by heating the reaction mixture to 60 °C. Since the pioneering work of Grubbs et al. using the 1st generation ruthenium catalyst A, several new catalysts have been developed such as the more reactive 2nd generation catalyst B, or the “boomerang catalyst” C, which allows regeneration of the metal complex (Fig. 2).42–44
 | ||
| Fig. 2 Metathesis catalysts investigated. | ||
We chose catalyst A for our investigations, because it is commercially available at a reasonable price and shows high tolerance towards functional groups. With respect to higher yields for cyclisations of our critical substrates, we also investigated the other catalysts, albeit without the desired effect. Both catalysts failed in the cyclisation of peptide 25 and 26. Mainly, polymerisation was observed with catalyst B, while with catalyst C only traces of product (ca. 5%) were obtained in the reaction of 26, and 50–60% of starting material could be recovered. Therefore, catalyst A was used for the further investigation of the influence of reaction conditions.
As illustrated with peptide 26, higher temperatures are sometimes necessary for satisfying results. The combined influence of the reaction temperature and solvent was also investigated (entries 8–10). In the cyclisation of 24 the yield could be increased by switching from CH2Cl2 to toluene at higher temperatures (entry 10), while changing the solvent alone had no influence. An increase of yield was also observed if nitrogen was bubbled through the solution, to remove the ethylene that was formed.
Peptide 22 was used to investigate the influence of Lewis acids, such as Ti(OiPr)4, on the outcome of the reaction. As illustrated in entries 11–14, subequimolar ratios of Lewis acids can indeed increase the yield, while in higher ratios the reaction can be suppressed completely. Lewis acids only accelerated the metathesis reaction with catalyst A, but with the more reactive B, only homodimerisation and polymerisation occurred. The best results were obtained in a 0.01 M solution of substrate 22, but the yield could not be increased to more than 35% under all conditions investigated so far; we thus presume that deactivation of the catalyst by the substrate occurs during the reaction.
Therefore, the mode of addition was thoroughly investigated with peptides 22 and 25 (entries 15–21). In both cases the yield of cyclisation product was below 5% if a solution of the catalyst was added over 30 min to a solution of the peptide in CH2Cl2 at rt (entries 15,16). If the catalyst solution was added more slowly, over 2 h via a syringe pump, a significant increase in the yield was observed (entry 17). The same results were obtained when the reaction mixture was degassed with N2 to remove the ethylene liberated in the ring-closing step (entries 18–19). But by far the best results were obtained if the peptide solution was added to the catalyst solution in refluxing CH2Cl2 under an N2 stream. Under these conditions the yield could be increased to 51 and 73%, respectively (entries 20 and 21). In both cases, only the product with the trans-configured double bond was obtained, no cis product could be determined by HPLC and NMR.
Conclusion
In conclusion, we have shown that not only the length and sequence of a peptide chain is responsible for the success of a ring-closing metathesis but also the synthetic protocol. While peptides containing amino acids with allylic side chains or with allylether side chains can be cyclised under “standard conditions”, allylic esters and carbamates require optimised reaction conditions.Experimental
All reactions were carried out in oven-dried glassware (100 °C) under argon. All solvents were dried before use. THF was distilled from sodium benzophenone, dichloromethane and diisopropylamine from calcium hydride. LHMDS solutions were prepared from freshly distilled hexamethyldisilazane and commercially available n-butyllithium solution (15% in hexane) in THF at −20 °C directly before use. The starting materials and the products were purified by flash chromatography on silica gel (32–63 µm). 1H and 13C NMR: Bruker AC 300, Bruker Advance 300, Bruker 400 MHz or Bruker DRX-500 spectrometer, respectively.Diastereomeric ratios were determined by analytical HPLC using a Shimadzu workstation with an SPD-M10A diodearray detector and a Lichrosorb silicagel column (250 × 4 mm, 5 µm) or a Daicel “Chiralcel OD–H” column (250 × 4 mm, 5 µm). Optical rotations were measured using a Perkin-Elmer 241 polarimeter.
N-(4-Toluolsulfonyl)-(S)-valinyl-β-alaninyl-glycine crotylester (3)
To a solution of Tos-Val-β-Ala-OH (1.03 g, 3.0 mmol) in THF (15 ml) carbonyldiimidazole (0.49 g, 3.0 mmol) was added at rt. After evolution of CO2 the reaction mixture was stirred for 10 min, before Gly-OAll·HCl (0.50 g, 3.0 mmol) was added as solid. After stirring for 10 h, the reaction mixture was diluted with Et2O and washed with sat. NaHCO3 and 1 N HCl. The aqueous layer was extracted with Et2O. The combined organic layers were dried (Na2SO4) and evaporated in vacuo giving rise to a pale yellow solid. Crystallisation from MeOH–EtOAc–Et2O gave 1.01 g (2.3 mmol, 75%) of 3 as colourless crystals, mp 171–172 °C. 1H NMR (300 MHz, CDCl3): δ 0.73 (3H, d, J 6.8), 0.76 (3H, d, J 6.8), 1.67 (3H, dd, J 6.5 and 1.0), 1.94 (1H, m), 2.32 (2H, dt, J 6.7 and 4.9), 2.40 (3H, s), 3.37 (1H, dd, J 8.3 and 5.9), 3.43 (2H, m), 3.84 (1H, dd, J 18.0 and 5.5), 4.03 (1H, dd, J 18.0 and 6.0), 4.54 (2H, d, J 6.7), 5.54 (1H, dtq, J 15.2 and 6.7, 1.6), 5.62 (1H, d, J 8.5), 5.78 (1H, dq, J 15.2 and 6.6), 6.64 (1H, t, J 5.5), 7.18 (1H, t, J 6.0), 7.23 (2H, d, J 8.9), 7.66 (2H, d, J 8.3). 13C NMR (75 MHz, CDCl3): δ 17.54, 17.74, 19.06, 21.50, 31.05, 36.04, 36.13, 41.48, 62.45, 66.36, 124.34, 127.36, 129.55, 132.38, 136.38, 143.74, 170.46, 171.06, 172.45. C21H31N3O6S (453.56) calcd.: C 55.61 H 6.89 N 9.26 S 7.07; found: C 55.52 H 6.87 N 9.22 S 7.18.N-(4-Toluolsulfonyl)-(S)-valinyl-β-alaninyl-(R)-γ,δ-dehydro-allo-isoleucine methylester (4)
Peptide 3 (227 mg, 0.5 mmol) and tin chloride (190 mg, 1.0 mmol) were placed in a Schlenk flask under argon. Abs. THF (10 ml) was added and the clear solution was cooled to −78 °C. A freshly prepared solution of LHMDS (from 3.13 mmol HMDS and 2.50 mmol BuLi) in THF (3 ml) was slowly added. The reaction mixture was warmed to rt overnight, diluted with Et2O (20 ml) and hydrolysed with 1 N HCl (20 ml). Stirring was continued to destroy the peptide–metal complex. The layers were separated, the organic layer was washed again with 1 N HCl, dried (Na2SO4) and evaporated in vacuo. The crude product was esterified with diazomethane and purified by flash chromatography (hexane–EtOAc–EtOH 6 : 4 : 1) giving rise to 4 (190 mg, 0.44 mmol, 89%) as a colourless solid, mp 215–216 °C. Diastereomeric ratio: 16 : 84. HPLC (Daicel OD–H, hexane–isopropanol 90 : 10, 0.5 ml min−1): tR1 = 28.17 min, tR2 = 35.35 min. 1H NMR (300 MHz, CD3OD): δ 0.81 (3H, d, J 6.4), 0.86 (3H, d, J 6.5), 1.03 (3H, bs), 1.84 (1H, m), 2.27 (2H, m), 2.40 (3H, s), 2.62 (1H, m), 3.15 (2H, m), 3.37 (1H, bs), 3.67 (3H, s), 4.42 (1H, bs), 5.03–5.09 (2H, m), 5.73 (1H, m), 7.33 (2H, d, J 8.4), 7.69 (2H, d, J 7.6). 13C NMR (75 MHz, CD3OD): δ 16.11, 18.56, 19.56, 21.45, 32.46, 35.92, 36.69, 41.33, 52.38, 58.17, 63.81, 116.14, 128.40, 130.53, 138.95, 140.36, 144.77, 173.17, 173.52. C22H33N3O6S (467.58) calcd.: C 56.51 H 7.11 N 8.99; found: C 56.24 H 7.08 N 8.72.[N-Allyl-N-(4-toluolsulfonyl)]-(S)-valinyl-β-alaninyl-(R)-γ,δ-dehydro-allo-isoleucin methylester (5)
A solution of allylpalladium chloride dimer (1.3 mg, 3.4 µmol, 1 mol%), triphenylphosphine (4 mg, 15.2 mmol, 4.5 mol%) and allyl ethyl carbonate (91 mg, 0.7 mmol) in abs. THF (2 ml) was added to a solution of 4 (165 mg, 0.35 mmol) in THF (5 ml). The reaction mixture was stirred overnight and the solvent was evaporated in vacuo. The crude product was purified by flash chromatography on silica (hexane–EtOAc 1 : 1) giving 5 (132 mg, 0.26 mmol, 73%) as a colourless oil. 1H NMR (300 MHz, CDCl3): δ 0.62 (3H, d, J 6.6), 0.81 (3H, d, J 6.5), 1.04 (3H, d, J 7.0), 2.15 (1H, m), 2.34–2.47 (2H, m), 2.38 (3H, s), 2.65 (1H, m), 3.33 (1H, m), 3.45 (1H, m), 3.64 (1H, d, J 10.8), 3.71 (3H, s), 3.86 (1H, dd, J 16.2 and 6.0), 4.13 (1H, dd, J 16.3 and 7.0), 4.60 (1H, dd, J 8.6 and 5.2), 5.01–5.18 (4H, m), 5.62–5.84 (2H, m), 6.03 (1H, d, J 8.2), 6.63 (1H, bs), 7.23 (2H, d, J 8.1), 7.67 (2H, d, J 8.2). 13C NMR (75 MHz, CDCl3): δ 15.54, 19.03, 19.45, 21.26, 27.00, 35.12, 35.26, 40.26, 47.08, 51.92, 55.79, 66.04, 116.23, 117.31, 127.23, 129.25, 134.86, 137.43, 138.21, 143.16, 169.49, 170.72, 171.51. C25H37N3O6S (507.65) calcd.: C 59.15 H 7.35 N 8.28 S 6.32; found: C 59.11 H 7.44 N 8.13 S 6.54.N-(4-Toluolsulfonyl)-(S)-valinyl-(S)-prolinyl-(S)-leucinyl-glycine allylester (6)
Tetrapeptide 6 was obtained by standard peptide coupling reactions in a 4 mmol scale. Crystallisation from EtOAc–Et2O–hexane gave colourless needles, mp 158–159 °C. 1H NMR (300 MHz, CDCl3): δ 0.82 (3H, d, J 6.1), 0.84 (3H, d, J 6.0), 0.86 (3H, d, J 6.6), 0.91 (3H, d, J 6.8), 1.47–2.14 (8H, m), 2.39 (3H, s), 3.13 (1H, m), 3.38 (1H, m), 3.60 (1H, dd, J 9.9 and 6.5), 3.94 (1H, dd, J 18.2 and 5.4), 4.02 (1H, dd, J 18.2 and 5.3), 4.13 (1H, dd, J 8.4 and 2.6), 4.33 (1H, m), 4.58 (2H, dt, J 6.2 and 1.3), 5.22 (1H, dt, J 10.4 and 1.3), 5.28 (1H, dt, J 17.2 and 1.4), 5.86 (1H, ddt, J 17.1, 10.4 and 6.3), 6.39 (1H, d, J 9.9), 6.76 (1H, t, J 5.4), 7.17 (1H, d, J 7.8), 7.26 (2H, d, J 8.1), 7.69 (2H, d, J 8.3). 13C NMR (75 MHz, CDCl3): δ 17.68, 19.07, 21.47, 21.77, 22.88, 24.57, 24.90, 27.14, 31.68, 40.60, 41.18, 47.24, 51.90, 59.34, 59.67, 65.87, 118.82, 127.41, 129.43, 131.48, 136.01, 143.59, 169.31, 170.92, 171.14, 172.06. C28H42N4O7S (578.73) calcd.: C 58.11 H 7.32 N 9.68 S 5.54; found: C 57.94 H 7.29 N 9.53 S 5.68.N-(4-Toluolsulfonyl)-(S)-valinyl-(S)-prolinyl-(S)-leucinyl-allylglycine methylester (7)
According to the preparation of 4, rearrangement product 7 was obtained from 6 (289 mg, 0.50 mmol) as a colourless foam in a diastereomeric ratio of 40 : 60. Yield: 275 mg (0.46 mmol, 93%). HPLC (Daicel OD–H, hexane–isopropanol 90 : 10, 0.5 ml min−1): tR1 = 24.67 min, tR2 = 30.83 min. 1H NMR (300 MHz, CDCl3): δ 0.82 (3H, d, J 6.1), 0.86 (3H, d, J 6.2), 0.89 (3H, d, J 6.8), 0.94 (3H, d, J 6.7), 1.46–2.09 (8H, m), 2.39 (3H, s), 2.47 (2H, m), 3.12 (1H, m), 3.37 (1H, m), 3.64 (1H, dd, J 10.1 and 6.3), 3.69 (3H, s), 4.04 (1H, dd, J 8.4 and 2.7), 4.30 (1H, m), 4.56 (1H, m), 5.04 (1H, d, J 15.4), 5.05 (1H, d, J 11.6), 5.65 (1H, m), 6.06 (1H, d, J 10.0), 6.59 (1H, d, J 7.7), 7.00 (1H, d, J 7.7), 7.27 (2H, d, J 8.2), 7.69 (2H, d, J 8.3). 13C NMR (75 MHz, CDCl3): δ 17.28, 18.99, 21.27, 21.66, 22.66, 24.42, 25.12, 26.85, 31.40, 36.04, 40.57, 46.99, 51.44, 51.66, 52.07, 59.13, 59.47, 118.79, 127.25, 129.18, 132.03, 136.61, 143.36, 170.47, 170.85, 171.17, 171.60. HMRS: calcd. for C29H45N4O7S ([M+H]+), 593.2993; found, 593.3022. HMRS: calcd. for C29H44N4O7SNa ([M+Na]+), 615.2812; found, 615.2847.[N-(4-Toluolsulfonyl)-N-allyl]-(S)-valinyl-(S)-prolinyl-(S)-leucinyl-allylglycine methylester (8)
According to the preparation of 5, N-allylated product 8 was obtained from 7 (260 mg, 0.440 mmol) as a pale yellow oil. Yield: 210 mg (0.33 mmol, 75%). 1H NMR (300 MHz, CDCl3): δ 0.83 (3H, d, J 6.1), 0.85 (3H, d, J 7.2), 0.88 (3H, d, J 6.7), 0.89 (3H, d, J 6.6), 1.39–1.49 (1H, m), 1.53–1.89 (2H, m), 1.89 (1H, m), 1.98–2.20 (4H, m), 2.38 (3H, s), 2.49 (2H, m), 3.52 (1H, m), 3.65–3.76 (1H, m), 3.69 (3H, s), 3.96 (1H, dd, J 16.6 and 5.3), 4.22–4.32 (3H, m), 4.39 (1H, dd, J 16.5 and 7.7), 4.56 (1H, m), 4.99–5.15 (4H, m), 5.63 (1H, m), 5.87 (1H, m), 6.70 (1H, d, J 7.7), 6.86 (1H, d, J 7.9), 7.23 (2H, d, J 7.9), 7.58 (2H, d, J 8.3). 13C NMR (75 MHz, CDCl3): δ 8.59, 19.75, 21.25, 21.78, 22.57, 24.50, 24.66, 27.32, 28.93, 36.06, 40.49, 47.31, 51.43, 51.70, 51.91, 52.07, 59.89, 62.16, 116.64, 118.78, 127.04, 129.18, 132.05, 135.71, 137.09, 143.30, 170.73, 170.84, 171.19, 171.61. C32H48N4O7S (632.82) calcd.: C 60.74 H 7.64 N 8.85 S 5.07; found: C 60.94 H 7.59 N 8.79 S 5.08. HMRS: calcd. for C32H49N4O7S ([M+H]+), 633.3305; found, 633.3329. HMRS: calcd. for C32H48N4O7SNa ([M+Na]+), 655.3124; found, 655.3154.Cyclopeptide 9
Tetrapeptide 8 (85 mg, 0.134 mmol) was dissolved in CH2Cl2 (10 ml) in a Schlenk tube under argon. A solution of Grubbs’ catalyst A (12 mg, 13.4 µmol, 10 mol%) in CH2Cl2 (15 ml) was added slowly to the peptide solution via a syringe. The mixture was refluxed for 2 h and allowed to stir overnight at rt. The solvent was evaporated in vacuo and the dark brown residue was purified by flash chromatography (hexane–EtOAc 6 : 4) giving rise to 9 (67 mg, 0.103 mmol, 77%) as a pale yellow oil. 1H NMR (300 MHz, CDCl3): δ 0.53 (3H, d, J 6.1), 0.83 (3H, d, J 6.4), 0.89 (3H, d, J 6.6), 0.94 (3H, d, J 6.6), 1.48–2.33 (8H, m), 2.40 (3H, s), 2.82 (2H, m), 3.67 (3H, s), 3.64–3.70 (3H, m), 3.82 (1H, dd, J 16.5 and 10.5), 3.96 (1H, m), 4.09 (1H, m), 4.23 (1H, d, J 10.5), 4.74 (1H, m), 5.19 (1H, t, J 10.5), 5.47 (1H, d, J 8.4), 5.55 (1H, t, J 10.9), 7.27 (2H, d, J 8.3), 7.70 (2H, d, J 8.3), 8.61 (1H, d, J 8.3). 13C NMR (75 MHz, CDCl3): δ 19.11, 20.73, 21.50, 23.17, 24.34, 25.72, 27.68, 28.36, 28.63, 40.66, 40.81, 47.25, 52.36, 52.41, 54.79, 56.94, 61.52, 127.80, 128.56, 128.62, 129.52, 136.97, 143.73, 167.61, 171.35, 171.99, 172.71. HMRS: calcd. for C30H45N4O7S ([M+H]+), 605.2993; found, 605.3029. HMRS: calcd. for C30H44N4O7SNa ([M+Na]+), 627.2812; found, 627.2820.Cyclopeptide 13
According to the preparation of 9, peptide 13 was obtained from 11 (61 mg, 0.10 mmol) as a pale yellow oil. Yield: 46 mg (0.08 mmol, 80%). 1H NMR (500 MHz, d7-DMF): δ 0.86 (3H, d, J 6.3), 0.88 (3H, d, J 6.4), 0.99 (3H, d, J 7.0), 1.00 (3H, d, J 7.0), 1.62–1.70 (2H, m), 1.76 (1H, m), 2.11 (1H, m), 2.43 (3H, s), 2.44–2.52 (3H, m), 2.69 (1H, m), 3.26 (1H, m), 3.51 (1H, m), 3.64 (3H, s), 3.75 (1H, dd, J 14.7 and 6.3), 3.81 (1H, dd, J 14.8 and 7.1), 3.97 (1H, dd, J 6.0 and 5.7), 4.43 (1H, m), 4.59 (1H, m), 5.43 (1H, dt, J 15.4, 6.7), 5.64 (1H, dt, J 15.4, 7.0), 7.47 (2H, d, J 8.0), 7.62 (1H, d, J 8.0), 7.71 (1H, d, J 8.3), 7.75 (2H, d, J 8.0), 8.15 (1H, d, J 6.1). 13C NMR (125 MHz, d7-DMF): δ 18.67, 19.50, 21.23, 21.35, 23.38, 25.13, 30.43, 33.88, 40.62, 43.28, 50.18, 51.94, 52.27, 52.39, 61.86, 127.81, 128.84, 130.42, 131.29, 137.32, 144.18, 171.89, 171.98, 172.50, 172.76. C28H42N4O7S (578.73) calcd.: C 58.11 H 7.32 N 9.68; found: C 58.15 H 7.38 N 9.27.Cyclopeptide 14
According to the preparation of 9, peptide 14 was obtained from 12 (80 mg, 0.14 mmol) as a colourless solid, mp 178–179 °C. Yield: 69 mg (0.13 mmol, 91%). 1H NMR (300 MHz, CD3OD): δ 0.92 (3H, d, J 6.2), 0.94 (3H, d, J 6.9), 0.96 (3H, d, J 6.0), 0.97 (3H, d, J 6.2), 1.42 (9H, s), 1.63 (2H, m), 1.69 (1H, m), 2.10 (1H, m), 2.35 (1H, m), 2.60 (1H, m), 1.57 (2H, m), 3.71 (3H, s), 3.84 (1H, d, J 11.8), 3.93 (1H, d, J 11.5), 4.26 (1H, m), 4.28 (1H, d, J 6.9), 4.37 (1H, m), 4.66 (1H, dd, J 9.8 and 3.3), 5.58 (2H, m). 13C NMR (75 MHz, CD3OD): δ 18.44, 19.75, 22.56, 22.97, 25.99, 28.65, 32.29, 34.88, 41.74, 52.80, 52.85, 53.70, 55.40, 62.07, 70.77, 72.19, 80.76, 128.18, 130.34, 157.42, 172.79, 173.04, 173.29, 173,69. HMRS: calcd. for C26H45N4O8 ([M+H]+), 541.3222; found, 541.3256. HMRS: calcd. for C26H44N4O8Na ([M+Na]+), 563.3041; found: 563.3052.Cyclopeptide 16
According to the preparation of 9, peptide 16 was obtained from 15 (80 mg, 0.134 mmol) using 5% catalyst (6 mg, 0.067 mmol). Purification of the crude product by flash chromatography (hexane–EtOAc 6 : 4 → 1 : 1) gave 16 as a colourless solid, mp 184–185 °C. Yield: 65 mg (0.144 mmol, 85%). The cis/trans ratio could not be determined. [α]20D = −3.5 (c 1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 0.89–0.97 (12H, m), 1.43 (9H, s), 1.50–1.57 (3H, m), 2.20 (1H, m), 3.60–3.63 (2H, m), 3.75 (3H, s), 3.78–3.81 (2H, m), 4.10–4.15 (4H, m), 4.20–4.25 (2H, m), 4.44 (1H, m), 4.65 (1H, m), 5.30–5.43 (2H, m), 5.86 (1H, m), 6.06 (1H, dbr), 7.25 (1H, dbr), 7.35 (1H, dbr). 13C NMR (75 MHz, CDCl3): δ 16.47, 19.49, 21.84, 22.81, 24.83, 28.23, 31.05, 39.05, 52.46, 52.59, 52.75, 57.95, 69.63, 70.32, 71.29, 80.27, 127.81, 129.44, 155.73, 170.25, 170.95, 171.16, 171.32. C27H46O9N4 (570.684) calcd.: C 56.83 H 8.12 N 9.82; found: C 55.69 H 8.07 N 8.91. HMRS: calcd. for C27H47N4O9 ([M+H]+), 571.3343; found, 571.3359. HMRS: calcd. for C27H46N4O9Na ([M+Na]+), 593.3163; found, 593.3127.Cyclopeptide 18
According to the preparation of 9, peptide 18 was obtained from 17 (134 mg, 0.23 mmol) using 5% catalyst A (9 mg, 0.01 mmol). To remove traces of the catalyst, the products were dissolved in methanol and stirred vigorously after addition of 3% H2O2 solution. The mixture was diluted with CH2Cl2 and the phases were separated. The organic layer was nearly colourless. In this case the cis/trans isomers could be separated by flash chromatography (hexane–EtOAc 2 : 8 → EtOAc) giving 66 mg (0.12 mmol, 52%) trans-21 and 52 mg (0.09 mmol, 41%) cis-21. Overall yield: 118 mg (0.21 mmol, 93%). HPLC (Daicel OD-H, hexane–isopropanol 85 : 15, 0.5 ml min−1): trans-18: tR1 = 28.14 min, cis-18: tR2 = 31.17 min. trans-18: [α]22D = +11.7° (c 0.5, CHCl3). 1H NMR (300 MHz, CDCl3): δ 0.70 (3H, d, J 6.8), 0.94 (3H, d, J 6.8), 1.40 (9H, s), 1.86–1.99 (2H, m), 2.06–2.13 (2H, m), 2.38 (1H, m), 3.46–3.65 (5H, m), 3.71 (3H, s), 3.72 (1H, m), 3.80 (1H, m), 3.96 (1H, m), 4.13–4.20 (2H, m), 4.29 (1H, sbr), 4.52 (1H, m), 4.59–4.66 (2H, m), 5.49 (1H, d, J 4.8), 5.90 (1H, d, J 15.5), 6.09 (1H, d, J 15.5), 7.81 (1H, d, J 7.9), 7.87 (1H, d, J 8.5). 13C NMR (75 MHz, CDCl3): δ 15.81, 20.16, 25.79, 26.31, 28.33, 30.15, 47.51, 52.31, 52.52, 52.91, 55.97, 60.31, 70.04, 70.46, 70.67, 71.42, 79.82, 127.10, 128.67, 154.94, 170.52, 170.70, 170.98, 171.15. cis-18: [α]22D = −44.4° (c = 0.5, CHCl3). 1H NMR (300 MHz, CDCl3): δ 0.84 (3H, d, J 6.7), 0.96 (3H, d, J 6.7), 1.43 (9H, s), 1.79 (1H, m), 1.94 (1H, m), 2.00 (1H, m), 2.11 (1H, m), 2.39 (1H, m), 3.51 (1H, m), 3.56–3.58 (2H, m), 3.63–3.69 (3H, m), 3.72 (3H, s), 3.90 (1H, m), 3.94–3.96 (2H, m), 3.99 (1H, m), 4.22 (1H, m), 4.49 (1H, d, J 7.7), 4.54 (1H, m), 4.76 (1H, dd, J 9.2 and 4.2), 5.53 (1H, d, J 5.7), 5.67 (1H, m), 5.75 (1H, m), 7.16 (1H, d, J 9.2), 7.70 (1H, d, J 7.4). 13C NMR (75 MHz, CDCl3): δ 16.45, 19.59, 24.61, 27.20, 28.04, 31.30, 47.06, 52.28, 52.67, 54.78, 59.91, 67.52, 69.51, 69.84, 70.69, 80.12, 126.85, 129.39, 154.94, 170.20, 170.51, 170.55, 170.85. HMRS: calcd. for C26H43N4O9 ([M+H]+), 555.3030; found, 555.3036. HMRS: calcd. for C26H42N4O9Na ([M+Na]+), 577.2849; found, 577.2839.Cyclopeptide 20
According to the preparation of 9, peptide 20 was obtained from 19 (88 mg, 0.15 mmol) using 5% catalyst A (7 mg, 0.008 mmol). Yield: 18 mg (32.4 µmol, 21%) of a grey–brown oil. 1H NMR (300 MHz, CDCl3): δ 0.88–0.94 (12H, m), 1.44 (9H, s), 1.50–1.60 (3H, m), 1.88 (1H, m), 3.74 (2H, m), 3.91–4.00 (2H, m), 4.09 (1H, m), 4.60–4.65 (7H, m), 5.57 (1H, dbr), 5.68–5.75 (2H, m), 6.75 (1H, dbr), 6.90 (1H, dbr), 7.05 (1H, dbr). 13C NMR (75 MHz, CDCl3): δ 18.41, 19.28, 20.89, 23.19, 24.83, 28.26, 30.51, 40.05, 51.57, 54.98, 63.41, 63.95, 69.95, 80.30, 127.74, 128.45, 155.71, 169.77, 171.21, 171.60, 172.34. HMRS: calcd. for C26H45N4O9 ([M+H]+), 557.3187; found, 557.3173. HMRS: calcd. for C26H44N4O9Na ([M+Na]+), 579.3006; found, 579.2990.Depsipeptide 21
A solution of (S)-Aloc-ValOH (1.94 g, 5.00 mmol) in trifluoroethanol (5 ml) was added to a solution of pivalaldehyde (0.54 ml, 5.00 mmol) in the same solvent (5 ml) at 0 °C. Isocyanoacetic acid allylester (625 mg, 5.00 mmol) was added via a syringe over 20 min and the mixture was allowed to warm to rt overnight. Stirring at rt was continued for 24 h, before the solvent was removed in vacuo. The residue was dissolved in EtOAc (30 ml) and the organic layer was washed twice with sat. NaHCO3 (30 ml) and 1 N KHSO4 (30 ml). After drying (Na2SO4) and evaporation of the solvent the crude product was purified by flash chromatography (hexane–EtOAc 7 : 3 → 6 : 4). Subsequent crystallisation from ether gave 21 (1.10 g, 2.55 mmol, 53%) as a single diastereomer, mp 173–174 °C. The absolute configuration of the newly formed stereogenic centre was not determined. 1H NMR (500 MHz, CDCl3, HH-COSY): δ 0.88–0.98 (15H, m), 2.12 (1H, m), 3.82 (1H, dd, J 18.0 and 4.4), 4.11 (1H, m), 4.19 (1H, dd, J 18.0 and 5.8), 4.48–4.62 (5H, m), 5.15–5.32 (4H, m), 5.71 (1H, d, J 8.5), 5.84–5.88 (2H, m), 6.73 (1H, d, J 8.5). 13C NMR (125 MHz, CDCl3, HMBC, DEPT): δ 16.4, 18.4, 25.5, 25.6, 30.5, 33.7, 40.1, 59.1, 59.2, 64.7, 65.0, 116.8, 117.9, 130.5, 131.1, 156.8, 168.5, 169.7, 170.4. C20H32N2O7 (412.48) calcd.: C 58.24 H 7.82 N 6.79; found: C 58.17 H 7.75 N 6.83.Allyloxycarbonyl-(S)-valyl-N-[(R)-1-methyl-p-methoxy-benzyl]-(R,S)-valyl-glycine allylester ((S,R,R/S)-22)
Isobutyraldehyde (360 mg, 5.0 mmol) was added to a solution of (R)-1-methyl-p-methoxy-benzylamine (755 mg, 5.0 mmol) in trifluoroethanol (5 ml). The solution was stirred for 15 min and subsequently cooled to −30 °C before (S)-Aloc-ValOH (1120 mg, 5.5 mmol) in trifluoroethanol (5 ml) was added. After a further 15 min, isocyanoacetic acid allylester (750 mg, 5.0 mmol) was added via a syringe over 20 min. The solution was kept at −30 °C for three days before the solvent was evaporated in vacuo. Workup was carried out as described for 21. The diastereomers could be separated by flash chromatography (hexane–EtOAc 7 : 3 → 6 : 4) giving rise to (S,R,R)-22 (196 mg, 0.4 mmol) and (S,R,S)-22 (1589 mg, 3.1 mmol) as viscous aromatic oils. Overall yield: 1.785 g (3.6 mmol, 71%). The diastereomeric ratio was determined to 11 : 89 by HPLC (silica, hexane–EtOAc 73 : 27, 2 ml min−1): tR(S,R,R-22) = 7.40 min; tR(S,R,S-22) = 10.71 min. (S,R,R)-22: [α]22D = +44.5° (c 1, CHCl3, >99% de). 1H NMR (300 MHz, HH-COSY): δ 0.21 (3H, d, J 6.6), 0.70 (3H, d, J 6.6), 1.00 (3H, d, J 6.8,) 1.08 (3H, d, J 6.6), 1.54 (3H, d, J 7.0), 2.28 (1H, m), 2.81 (1H, m), 3.15 (1H, d, J 10.8), 3.75–3.85 (4H, m), 4.20 (1H, dd, J 18.0 and 7.0), 4.52–4.55 (4H, m), 4.80 (1H, m), 5.14–5.38 (5H, m), 5.58 (1H, d, J 9.6), 5.81–5.96 (2H, m), 6.88 (2H, d, J 8.5), 7.28 (2H, d, J 8.8), 8.50 (1H, m). 13C NMR (75 MHz, HMBC): δ 17.0, 17.8, 19.6, 20.2, 20.8, 27.1, 27.1, 31.4, 41.0, 55.3, 57.2, 57.4, 65.8, 68.8, 113.8, 117.5, 118.8, 126.6, 132.8, 130.0, 130.2, 131.7, 156.2, 159.5, 169.6, 173.4, 173.6. HMRS: calcd. for C28H41N3O7 ([M]+), 531.2944; found, 531.2951; calcd. for C28H42N3O7 ([M+H]+), 532.3022; found, 532.2994. (S,R,S)-22, [α]22D = −54.5° (c 1, CHCl3, >98% de). 1H NMR (300 MHz, HH-COSY): δ 0.79 (3H, d, J 6.3), 0.83 (3H, d, J 6.3), 1.04 (3H, d, J 7.0), 1.06 (3H, d, J 7.7), 1.64 (3H, d, J 6.8), 2.19 (1H, m), 2.80–2.94 (2H, m), 3.67 (1H, dd, J 18.2 and 4.7), 3.76 (3H, s), 3.93 (1H, dd, J 18.2 and 6.5), 4.54–4.61 (5H, m), 5.17–5.33 (5H, m), 5.45 (1H, d, J 9.4), 5.83–5.93 (2H, m), 6.78 (2H, d, J 8.8), 7.20 (2H, d, J 8.8), 8.38 (1H, bs). 13C NMR (75 MHz, HMBC): δ17.2, 17.9, 19.9, 20.0, 20.4, 26.5, 31.6, 40.8, 55.2, 55.8, 57.2, 65.6, 65.9, 69.9, 113.9, 117.6, 118.6, 128.9, 129.9, 131.7, 132.6, 156.2, 159.3, 169.5, 171.4, 174.1. HMRS: calcd. for C28H41N3O7 ([M]+), 531.2944; found, 531.2953; calcd. for C28H42N3O7 ([M+H]+), 532.3022; found, 532.3036.Cyclopeptide 26
In a 100 ml Schlenk flask peptide, (S,R,S)-22 (531 mg, 1.00 mmol) was dissolved in CH2Cl2 (80 ml) under reflux and a constant stream of N2 was bubbled through the solution. A solution of Grubbs’ catalyst (81 mg, 0.10 mmol, 10 mol%) in CH2Cl2 (10 ml) was added slowly over a period of 2 h via a syringe. After 4 h when nearly all starting material was consumed (TLC), the solvent was removed in vacuo and the crude product was purified by flash chromatography (hexane–EtOAc 6 : 4 → 1 : 1→ 3 : 7) giving rise to 23 (367 mg, 0.73 mmol, 73%) as a colourless solid, mp 170–172 °C. In addition a small amount of 22 (80 mg, 0.15 mmol, 15%) was recovered. The trans-isomer was obtained with 95% selectivity as determined by HPLC. 1H NMR (500 MHz, CDCl3, HH-COSY): δ 0.28 (3H, d, J 6.6), 0.65 (3H, d, J 6.6), 0.99 (3H, d, J 7.0), 1.13 (3H, d, J 6.6), 1.63 (3H, d, J 6.9), 2.29 (1H, m), 2.74 (1H, m), 3.13 (1H, d, J 11.0), 3.60 (1H, dd, 1H, J 17.8 and 2.5), 3.78 (3H, s), 4.16 (1H, m), 4.47 (1H, dd, J 17.3 and 7.7), 4.51 (1H, dd, J 12.1 and 7.0), 4.68 (1H, dd, J 12.1 and 3.5), 4.80 (1H, dd, J 9.9 and 3.7), 4.95 (1H, dd, J 13.0 and 4.8), 5.31 (1H, q, J 6.6), 5.40 (1H, d, J 9.0,) 5.78–5.96 (2H, m), 6.90 (2H, d, J 8.8), 7.30 (2H, d, J 8.8), 8.42 (1H, d, J 5.9). 13C NMR (75 MHz, HMBC): δ 16.5, 18.7, 19.8, 20.6, 27.0, 31.0, 42.1, 55.3, 56.8, 57.8, 63.2, 64.6, 68.4, 113.9, 127.3, 130.9, 129.8, 130.2, 156.7, 159.6, 168.7, 173.3, 174.7. HMRS: calcd. for C26H38N3O7 ([M+H]+), 504.2621; found, 504.2655; calcd. for C26H37N3O7Na ([M+Na]+), 526.2532; found, 526.2482.Allyloxycarbonyl-(S)-alanyl-N-[(S)-1-methyl-m-methoxy-benzyl]-(R,S)-valyl-glycine allylester ((S,S,R/S)-24)
According to the preparation of 22, peptide 24 was obtained from (S)-Aloc-AlaOH (952 mg, 5.50 mmol), (S)-1-methyl-m-methoxy-benzyl amine (830 mg, 5.00 mmol), isobutyraldehyde (0.45 ml, 5.00 mmol) and isocyanoacetic acid allyl ester (625 mg, 5.00 mmol) in 91% yield. Flash chromatography (hexane–EtOAc 8 : 2 → 7 : 3→ 6 : 4) allowed the separation of the diastereomers giving rise to (S,S,R)-24 (1.50 g, 2.98 mmol, 60%) and (S,S,S)-24 (0.65 g, 1.29 mmol, 28%) as viscous oils. HPLC (silica, hexane–EtOAc 75 : 25, 2 ml min−1): tR(S,S,R-27) = 12.05 min; tR(S,S,S-24) = 14.94 min. (S,S,R)-24: [α]22D = −52.9° (c 0.8, CHCl3). 1H NMR (500 MHz, CDCl3, HH-COSY): δ 0.16 (3H, d, J 6.4), 0.71 (3H, d, J 6.6), 1.42 (3H, d, J 6.7), 1.65 (3H, d, J 7.0), 2.78 (1H, m), 3.10 (1H, m), 3.75–3.80 (4H, m), 4.21 (1H, dd, J 18.1 and 7.0), 4.56–4.60 (4H, m), 5.11 (1H, t, J 7.3), 5.16–5.32 (5H, m), 5.58 (1H, d, J 8.3), 5.83–5.93 (2H, m), 6.84 (1H, dd, J 8.4 and 2.2), 7.05 (1H, d, J 8.6), 7.07 (1H, s), 7.26 (1H, t, J 8.0), 8.39 (1H, bs). 13C NMR (75 MHz, CDCl3): δ 17.0, 18.8, 19.6, 19.8, 27.5, 40.8, 48.4, 55.4, 56.5, 65.7, 68.2, 114.2, 114.4, 117.7, 118.8, 120.6, 129.4, 131.7, 132.6, 139.4, 155.7, 159.9, 169.4, 173.7, 174.5. HMRS: calcd. for C26H37N3O7 ([M]+), 503.2632; found, 503.2632. (S,S,S)-24: [α]22D = −6.9° (c 1.1, CHCl3). 1H NMR (500 MHz, CDCl3, HH-Cosy): δ 0.85 (3H, d, J 6.7), 0.89 (3H, d, J 6.4), 1.35 (3H, d, J 6.7), 1.65 (3H, d, J 7.0), 2.85 (1H, m), 3.20 (1H, m), 3.68–3.79 (5H, m), 4.55–4.60 (4H, m), 4.83 (1H, m), 5.19 (1H, dd, J 10.4 and 1.2), 5.21 (1H, dd, J 10.6 and 1.2), 5.25–5.32 (3H, m), 5.67 (1H, d, J 6.7), 5.83–5.93 (2H, m), 6.78 (1H, dd, J 8.2 and 2.4), 6.83 (1H, s), 6.85 (1H, d, J 7.6), 7.19 (1H, t, J 7.9), 8.05 (1H, bs). 13C NMR (75 MHz, CDCl3): δ 17.4, 19.2, 19.9, 20.4, 26.8, 40.9, 48.3, 55.2, 59.5, 65.7, 65.7, 68.2, 113.2, 114.0, 117.7, 118.7, 119.5, 129.5, 131.7, 132.7, 139.6, 155.6, 159.7, 169.4, 171.1, 174.7. HMRS: calcd. for C26H37N3O7 ([M]+), 503.2632; found, 503.2635. HMRS: calcd. for C26H38N3O7 ([M+H]+), 504.2709; found, 504.2713.Allyloxycarbonyl-(S)-prolinyl-N-[(S)-1-methyl-m-methoxy-benzyl]-(R,S)-valyl-glycine allylester ((S,S,R/S)-25)
According to the preparation of 22, peptide 25 was obtained from (S)-Aloc-ProOH (995 mg, 5.00 mmol), (S)-1-methyl-m-methoxy-benzyl amine (830 mg, 5.50 mmol), isobutyraldehyde (0.45 ml, 5.00 mmol) and isocyanoacetic acid allyl ester (625 mg, 5.00 mmol) in 12 h at 4 °C. Flash chromatography (hexane–EtOAc 7 : 3 → 6 : 4) provided 25 (2.27 g, 4.38 mmol, 89%) as viscous colourless oil as a mixture of diastereomers and rotamers. 1H NMR (500 MHz, CDCl3, HH-COSY): δ 0.21 (1.8H, d, J 6.6), 0.37 (1.8H, d, J 6.6), 1.66 (1.8H, d, J 5.4), 1.93–2.33 (4H, m), 2.82–2.87 (1H, m), 3.04 (1H, m), 3.57–3.87 (6H, m), 4.33 (1H, dd, J 17.9 and 7.9), 4.30–4.78 (4H, m), 5.11–5.35 (6H, m), 6.84 (2H, m), 6.86 (1H, m), 7.15 (0.6H, d, J 7.7), 7.20 (0.6H, s), 7.30 (1H, m), 8.55 (1H, m). Selected signals of the minor isomer: δ = 0.71 (1.2H, d, J 6.3), 0.73 (1.2H, d, J 6.0), 1.70 (1.2H, d, J 5.6), 7.00 (0.4H, s), 7.06 (0.4H, d, J 7.5). 13C NMR (125 MHz, CDCl3, HMBC, DEPT): δ 16.8, 18.8, 18.8, 23.3, 27.8, 29.5, 30.6, 40.7, 46.8, 55.4, 55.9, 58.6, 65.7, 65.9, 69.8, 115.2, 117.2, 118.7, 120.6, 121.0, 129.8, 131.7, 133.0, 139.9, 154.8, 159.8, 169.6, 173.6, 174.2. Selected signals of the minor isomer: δ = 16.6, 24.3, 27.4, 30.6, 47.4, 55.2, 56.0, 58.9, 66.4, 69.3, 112.7, 114.2, 118.3, 121.0, 154.2, 174.3. HMRS: calcd. for C28H40N3O7 ([M+H]+), 530.2801; found, 530.2861.Allyloxycarbonyl-(S)-N-methyl-valyl-N-[(S)-1-methyl-m-methoxy-benzyl]-(R,S)-valyl-glycine allylester ((S,S,R/S)-26)
According to the preparation of 22, peptide 26 was obtained from N-Aloc-N-Me-ValOH (1.07 g, 5.00 mmol), (S)-1-methyl-m-methoxy-benzyl amine (830 mg, 5.50 mmol), isobutyraldehyde (0.46 ml, 5.00 mmol) and isocyanoacetic acid allyl ester (625 mg, 5.00 mmol) in 12 h at 4 °C. Flash chromatography (hexane–EtOAc 7 : 3 → 6 : 4) provided 26 (1.96 g, 3.53 mmol, 72%) as viscous colourless oil as a mixture of diastereomers and rotamers. The diastereomers could in part be separated by a second flash chromatography. (S,S,R)-26: [α]20D = −137.4° (c 1.1, CHCl3). 1H NMR (500 MHz, CDCl3, HH-COSY, mixture of rotamers): δ 0.16–0.18 (2.1H, d, J 6.5), 0.87 (2.1H, d, J 6.5), 0.82–0.97 (7.8H, m), 1.59 (3H, d, J 7.1), 2.43 (1H, m), 2.79–3.18 (5H, m), 3.63 (3H, s), 4.31–4.63 (2H, m), 4.59–4.63 (4H, m), 4.94 (0.7H, d, J 10.5), 5.06 (0.3H, d, J 10.5), 5.20–5.27 (4H, m), 5.31 (0.3H, q, J 6.9), 5.42 (0.7H, q, J 7.2), 5.56–5.98 (2H, m), 6.79–6.89 (2H, m), 7.19–7.26 (2H, m), 8.32 (0.3H, bs), 8.40 (0.7H, bs). Coalescence was observed for the signals of the rotamers at 398 K in d6-DMSO. 13C NMR (125 MHz, CDCl3, HMBC, DEPT): δ 17.1, 17.3, 17.9, 18.1, 19.4, 19.7, 19.8, 19.9, 20.1, 20.9, 25.8, 27.4, 28.5, 28.7, 29.2, 29.7, 40.8, 40.9, 53.2, 54.7, 55.0, 55.2, 60.6, 62.5, 63.2, 65.4, 65.6, 66.6, 68.5, 111.1, 111.9, 113.3, 113.6, 117.3, 117.7, 118.3, 118.6, 118.9, 120.0, 128.5, 129.5, 131.4, 131.8, 132.1, 132.5, 139.7, 144.7, 156.7, 159.3, 168.7, 171.2, 171.4. (S,S,S)-26: [α]20D = −111.0° (c = 1, CHCl3). 1H NMR (500 MHz, CDCl3, HH-COSY, mixture of rotamers): δ 0.84–1.04 (11.1H, m), 1.11 (0.9H, d, J 6.7), 1.56 (0.9H, d, J 7.1), 1.59 (2.1H, d, J 7.1), 2.59 (1H, m), 2.96–2.97 (2H, m), 3.07 (3H, s), 3.67–3.73 (3.7H, m), 3.80 (0.7H, dd, J 18.4 and 6.6), 3.82 (0.3H, dd, J 18.5 and 5.7), 3.95 (0.3H, dd, J 18.5 and 5.7), 4.46–4.75 (5H, m), 5.07 (0.3H, dd, J 10.3 and 1.0), 5.15 (0.7H, ddd, J 10.5, 1.2 and 1.0), 5.22–5.51 (3H, m), 5.42 (0.3H, q, J 6.8), 5.53 (0.7H, q, J 6.8), 5.74 (0.3H, m), 5.79–5.96 (1.7H, m), 6.59 (1H, m), 6.64 (0.3H, m), 6.65–6.81 (2H, m), 7.07 (0.3H, t, J 8.2), 7.20 (0.7H, t, J 9.0), 8.40 (0.3H, bs), 8.46 (0.7H, bs). 13C NMR (125 MHz, CDCl3, HMBC, DEPT): δ 17.2, 17.3, 17.9, 18.0, 19.4, 19.7, 19.8, 19.9, 20.1, 20.9, 25.9, 27.3, 28.5, 28.8, 29.2, 29.6, 40.85, 40.9, 53.4, 54.9, 55.0, 55.1, 60.7, 62.4, 63.0, 65.5, 65.8, 66.6, 70.2, 111.0, 111.7, 113.3, 113.8, 117.2, 117.5, 118.2, 118.6, 118.9, 119.9, 128.5, 129.3, 131.5, 131.7, 132.1, 132.6, 139.7, 144.6, 156.6, 159.6, 168.9, 171.2, 171.5. HMRS: calcd. for C29H43N3O7 ([M]+), 545.3101; found, 545.3072. HMRS: calcd. for C29H44N3O7 ([M+H]+), 546.3179; found, 546.3177.Cyclopeptide 27
In a 250 ml Schlenk flask, peptide 24 (505 mg, 1.00 mmol) was dissolved in toluene (100 ml) at rt and a constant stream of N2 was bubbled through the solution. A solution of Grubbs’ catalyst (82 mg, 0.10 mmol, 10 mol%) in toluene (8 ml) was added slowly over a period of 2 h via a syringe. After stirring for 16 h, no starting material could be detected and the solution was concentrated to 20 ml. A 1% aqueous solution of H2O2 (to destroy the catalyst) was added and the mixture was stirred vigorously for 1 h, before charcoal was added. After 15 min the mixture was filtered through celite. The solvent was removed in vacuo and the crude product was purified by flash chromatography (hexane–EtOAc 6 : 4 → 3 : 7 → EtOAc) giving rise to 27 (129 mg, 0.27 mmol, 27%) as a colourless solid, mp 164–167 °C. Crystallisation from CH2Cl2–toluene–EtOAc provided crystals suitable for X-ray structure analysis.†1H NMR (500 MHz, CDCl3, HH-COSY): δ 0.81 (3H, d, J 6.3), 0.83 (3H, d, J 6.3), 1.44 (3H, d, J 7.3), 1.60 (3H, d, J 7.3), 2.76 (1H, m), 2.94 (1H, d, J 11.2), 3.50 (1H, dd, J 17.6 and 2.4), 3.70 (3H, s), 3.85 (1H, dd, J 17.6 and 6.4), 4.19 (1H, d, J 13.7), 4.53 (1H, m), 4.66 (1H, m), 4.93–5.01 (2H, m), 5.41 (1H, d, J 8.8), 5.54 (1H, q, J 6.9), 5.90–5.92 (2H, m), 6.79 (1H, dd, J 8.2 and 2.4), 6.92 (1H, d, J 7.8), 6.98 (1H, s), 7.20 (1H, dd, J 8.3 and 7.8), 8.20 (1H, d, J 5.4). 13C NMR (125 MHz, CDCl3, HMBC): δ 16.7, 18.9, 19.8, 20.6, 26.2, 42.3, 48.2, 55.2, 55.8, 63.2, 64.7, 70.0, 113.7, 114.5, 119.5, 126.2, 131.2, 129.2, 139.4, 156.1, 159.7, 168.2, 170.3, 175.9. HMRS: calcd. for C24H33N3O7Na ([M+Na]+), 498.2216; found, 498.2226.Cyclopeptide 28
In a 250 ml Schlenk flask, peptide 25 (257 mg, 0.50 mmol) was dissolved in CH2Cl2 (50 ml) at rt and a constant stream of N2 was bubbled through the solution. A solution of Grubbs’ catalyst (20 mg, 0.025 mmol, 5 mol%) in CH2Cl2 (2.5 ml) was added slowly over a period of 90 min via a syringe. After all starting material was consumed, workup was carried out as described for 27. The solvent was removed in vacuo and the crude product was purified by flash chromatography (hexane–EtOAc 6 : 4 → 1 : 1→ 3 : 7) giving rise to 28 (90 mg, 0.19 mmol, 38%) as a colourless solid. Crystallisation from EtOAc–heptane provided crystals suitable for X-ray structure analysis.† If the reaction was carried out in the presence of 10% Grubbs’ catalyst under reflux according to the preparation of 23, the yield could be increased to 51%. 1H NMR (500 MHz, CDCl3, HH-COSY): δ 0.85 (3H, d, J 6.7), 0.87 (3H, d, J 6.6), 1.60 (3H, d, J 7.1), 2.02 (2H, m,), 2.25 (2H, m,), 2.80 (1H, m), 2.91 (1H, d, J 11.0), 3.56–3.62 (2H, m), 3.76–3.79 (4H, m), 3.84 (1H, dd, J 17.8 and 6.1), 4.31 (1H, d, J 14.2), 4.43 (1H, dd, J 11.1 and 3.8), 4.88 (1H, dd, J 11.2 and 6.8), 4.92 (1H, dd, J 14.2 and 3.8), 5.03 (1H, dd, J 7.7 and 4.0), 5.63 (1H, q, J 6.9), 5.88–5.90 (2H, m), 6.82 (1H, dd, J 8.2 and 2.2), 6.98 (1H, d, J 7.6), 7.16 (1H, s), 7.24 (1H, t, J 8.0), 8.48 (1H, d, J 4.1). 13C NMR (125 MHz, CDCl3, HMQC, HMBC): δ 16.9, 19.7, 20.7, 24.7, 26.2, 30.6, 42.8, 46.5, 55.2, 55.9, 58.0,63.7, 64.7, 70.4, 114.0, 114.4, 119.5, 125.7, 131.7, 129.2, 138.6, 154.2, 159.8, 168.0, 170.4, 175.6. C26H35N3O7 (501.58) calcd.: C 62.26 H 7.03 N 8.38; found: C 62.07 H 6.82 N 8.29. HMRS: calcd. for C26H35N3O7 ([M]+), 501.2481; found, 501.2478.Cyclopeptide 29
In a 100 ml Schlenk flask, peptide 26 (272 mg, 0.50 mmol) was dissolved in toluene (20 ml) at rt and a solution of Grubbs’ catalyst (41 mg, 0.05 mmol, 10 mol%) in toluene was added slowly via a syringe. The solution was heated to 60 °C and the workup was carried out as described for 30. The solvent was removed in vacuo and the crude product was purified by flash chromatography (hexane–EtOAc 6 : 4 → 1 : 1 → 3 : 7) giving rise to 29 (85 mg, 0.17 mmol, 33%) as a colourless solid and recovered 26 (93 mg, 0.18 mmol, 34%). 1H NMR (500 MHz, CDCl3): δ 0.79 (3H, d, J 6.6), 0.94 (3H, d, J 6.4), 1.01 (3H, d, J 6.6), 1.08 (3H, d, J 6.9), 1.59 (3H, d, J 7.1), 2.41 (1H, m), 2.83 (1H, m), 3.02 (1H, d, J 10.1), 3.20 (3H, s), 3.32 (1H, dd, J 16.7 and 3.0), 3.72 (3H, s), 4.25–4.31 (2H, m), 4.41 (1H, m), 4.48 (1H, d, J 10.3), 4.93 (1H, m), 5.09 (1H, dd, J 14.4 and 4.9), 5.89 (1H, m), 6.06 (1H, d, J 15.7), 6.16 (1H, q, J 7.1), 6.65–6.90 (3H, m), 7.13 (1H, t, J 8.2), 8.11 (1H, d, J 6.9). 13C NMR (125 MHz, CDCl3, DEPT, HSQC): δ 18.2, 19.7, 19.8, 20.1, 22.2, 25.5, 28.9, 30.5, 41.1, 55.1, 55.9, 61.1, 63.2, 64.8, 71.0, 111.3, 117.0, 121.1, 126.6, 128.9, 132.0, 139.5, 157.0, 159.5, 169.3, 170.3, 174.4. HMRS: calcd. for C27H40N3O7 ([M+H]+), 518.2865; found, 518.2866.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie is gratefully acknowledged. We also thank the Degussa-Hüls AG for generous gifts of amino acids.References
- Amino Acids, Peptides and Proteins, Specialist Periodical Reports, Chem. Soc., London Search PubMed.
-
(a)
U. Gräfe, Biochemie der Antibiotika, Spektrum, Heidelberg, 1992 Search PubMed
; (b) U. Gräfe, Antibiotics and Antiviral Compounds, ed. H. A. Kirst, K. Krohn and H. Maag, VCH, Weinheim , 1993 Search PubMed
-
E. Mutschler, Arzneimittelwirkungen, Wissenschaftliche Verlagsgesellschaft, Stuttgart, 1991 Search PubMed
-
(a) H.-J. Musiol, F. Siedler, D. Quarzago and L. Moroder, Biopolymers, 1994, 34, 1553–1562 CrossRef CAS
- J. Rizo and L. M. Gierasch, Annu. Rev. Biochem., 1992, 61, 387–418 CrossRef CAS
- Reviews:
(a)
Tetrahedron, 1993, 49, 3433–3690 Search PubMed
- R. F. Nutt, D. F. Veber and R. Saperstein, J. Am. Chem. Soc., 1980, 102, 6539–6545 CrossRef CAS
-
(a) R. F. Nutt, R. G. Strachan, D. F. Veber and F. W. Holly, J. Org. Chem., 1980, 45, 3078–3080 CrossRef CAS
- R. M. Williams and J. Liu, J. Org. Chem., 1998, 63, 2130–2132 CrossRef CAS
- D. J. O'Leary, S. J. Miller and R. H. Grubbs, Tetrahedron Lett., 1998, 39, 1689–1690 CrossRef CAS
- Reviews:
(a) R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446–452 CrossRef CAS
- S. J. Miller, H. E. Blackwell and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 9606–9614 CrossRef CAS
-
(a) S. J. Miller and R. H. Grubbs, J. Am. Chem. Soc., 1995, 117, 5855–5856 CrossRef CAS
-
(a) E. R. Jarvo, G. T. Copeland, N. Papaioannou, P. J. Bonitatebus and S. J. Miller, J. Am. Chem. Soc., 1999, 121, 11638–11643 CrossRef CAS
-
(a) A. S. Ripka, R. S. Bohacek and D. H. Rich, Bioorg. Med. Chem. Lett., 1998, 8, 357–360 CrossRef CAS
-
(a) H. E. Blackwell and R. H. Grubbs, Angew. Chem., 1998, 110, 3469–3472 CrossRef
- T. D. Clark, K. Kobayashi and M. R. Ghadiri, Chem. Eur. J., 1999, 5, 782–792 CrossRef CAS
-
(a) J. Pernerstorfer, M. Schuster and S. Blechert, Chem. Commun., 1997, 1949–1950 RSC
-
(a) S. Sasmal, A. Geyer and M. E. Maier, J. Org. Chem., 2002, 67, 6260–6263 CrossRef CAS
-
(a) S. Rajesh, B. Banerji and J. Iqbal, J. Org. Chem., 2002, 67, 7852–7857 CrossRef CAS
-
(a) E. N. Prabhakaran, I. N. Rao, A. Boruah and J. Iqbal, J. Org. Chem., 2002, 67, 8247–8250 CrossRef CAS
-
(a) J. F. Reichwein, B. Wels, J. A. W. Kruijtzer, C. Versluis and R. M. J. Liskamp, Angew. Chem., 1999, 111, 3906–3910 CrossRef
- Review: U. Kazmaier, Liebigs Ann., Recl., 1997, 285–295 Search PubMed
-
(a) U. Kazmaier and F. L. Zumpe, Angew. Chem., 1999, 111, 1572–1574 CrossRef
- U. Kazmaier, Angew. Chem., 1994, 106, 1046–1047 CAS
-
(a) U. Kazmaier and C. Schneider, Synlett, 1996, 975–977 CrossRef
-
(a) U. Kazmaier, J. Org. Chem, 1994, 59, 6667–6670 CrossRef CAS
- U. Kazmaier and S. Maier, J. Org. Chem., 1999, 64, 4574–4575 CrossRef CAS
-
(a) F. L. Zumpe and U. Kazmaier, Synlett, 1998, 1199–1220 CAS
-
(a) U. Kazmaier and S. Maier, Org. Lett., 1999, 1, 1763–1766 CrossRef CAS
- No reaction was observed, even at temperatures up to 90 °C. These conditions only resulted in decomposition of the catalyst.
- Very recently, Iqbal et al. reported on a ring-closing metathesis of peptides containing a proline and a D-configured amino acid; B. Banerji, M. Bhattacharya, R. B. Madhu, S. K. Das and J. Iqbal, Tetrahedron Lett., 2002, 43, 6473–6477 Search PubMed
- The newly generated amino acid was not formed selectively, because proline containing peptides fail to undergo stereoselective Claisen rearrangements (see ref. 28).
- The double bond was obtained as an inseparable cis/trans-mixture..
- R. Knorr, A. Trzeciak, W. Bannwarth and D. Gillessen, Tetrahedron Lett., 1989, 30, 1927–1930 CrossRef CAS
-
(a) J. Feldman, J. S. Murdzek, W. M. Davis and R. R. Schrock, Organometallics, 1989, 8, 2260 CrossRef CAS
- A. Fürstner and K. Langemann, Synthesis, 1997, 792–803 CrossRef CAS
- Review:
D. Seebach, A. K. Beck, A. Studer, in Modern Synthetic Methods Vol. 7, ed. B. Ernst and C. Leumann, HCA, Basel/VCH, Weinheim, 1995, pp. 1–178 and references cited therein Search PubMed
- M. Passerini, Gazz. Chim. Ital., 1921, 51, 126–181 CAS
-
(a) Reviews: I. Ugi, in Isonitrile Chemistry, Academic, New York, 1971 Search PubMed
- U. Kazmaier and C. Hebach, J. Chem. Soc., Chem. Commun., 2003, 596–597 RSC
- Recent reviews:
(a) S. J. Connon and S. Blechert, Angew. Chem., 2003, 115, 1944–1968 CrossRef
-
(a) A. K. Gosh, J. Cappiello and D. Shin, Tetrahedron Lett., 1998, 39, 4651–4654 CrossRef CAS
-
(a) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation and analytical/spectroscopic data of linear tetrapeptides, X-ray data of cyclopeptides 27–29. See http://www.rsc.org/suppdata/ob/b4/b411228h/ |
| ‡ Dedicated to the memory of Professor U. Schmidt, a pioneer in cyclopeptide chemistry. |
| § CCDC reference numbers 197619, 245782, 245783. See http://www.rsc.org/suppdata/ob/b4/b411228h/ for crystallographic data in .cif format. |
| This journal is © The Royal Society of Chemistry 2005 |