Synthesis and thermal denaturation studies of novel 2′-O,3′-C-linked bicyclic oligonucleotides with a methoxy or a piperazino group facing the major groove of nucleic acid duplexes†
Michael
Raunkjær
a,
Mads D.
Sørensen
b and
Jesper
Wengel
*a
aNucleic Acid Center‡, Department of Chemistry, University of Southern Denmark, DK-5230, Odense M, Denmark. E-mail: jwe@chem.sdu.dk; Fax: 45 6615 8780; Tel: 45 6550 2510
bDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100, Copenhagen, Denmark
First published on 18th November 2004
Abstract
With the aim of evaluating duplex stabilities of oligonucleotides (ONs) with major groove facing functionalities, two novel 2′-O,3′-C-linked bicyclic nucleoside phosphoramidite building blocks were synthesized by routes involving regioselective O-methylation or piperazine attachment using carbonyldiimidazole coupling chemistry. The novel monomers were incorporated into 9-mer mixed base ONs and the thermal stability toward complementary single stranded DNA and RNA was evaluated by thermal denaturation experiments. O-methylated ONs confirmed the applicability of the functionalized bicylic sugar unit for attachment of groups facing the major groove and satisfactory binding properties towards the RNA complement were observed. For the piperazino modified ONs, experiments were performed in aqueous buffers with low (40 mM) and medium (110 mM) salt concentrations, at pH 5 and pH 7. A change from a medium to a low salt concentration induced a significant relative increase in the thermal stability of modified duplexes toward both DNA and RNA complements, which suggests protonation of the piperazino group under the experimental conditions applied.
Introduction
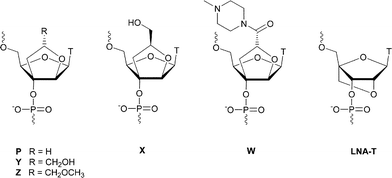
Oligonucleotides (ONs) and analogues have found a widespread application in the fields of therapeutics, biotechnology and nanotechnology.1,2 They form well defined and organized structures due to their unique hybridization properties. Chemical modifications or functionalization of ONs allow modulation of both chemical and physiological properties, which include altered affinity towards complementary strands, nuclease stability and RNase H activity. Successful attempts to modify ONs have been performed by introducing a cationic group linked to the base,3–11 phosphate12–17 or sugar18–26 moieties, aimed at enhancing the affinity towards a complementary strand by net charge reduction of the duplex.We have previously synthesized three 2′,3′-BcNAs (P, X and Y; Fig. 1, T = thymin-1-yl), with each showing interesting binding properties towards complementary RNA when incorporated into a modified ON.27,28 Modification P, constituting a simple bicylic scaffold, showed an enhanced affinity towards complementary RNA when built into an almost fully modified 5′-P13T sequence.27 This result encouraged us to synthesize functionalized bicyclic structures, namely the diastereoisomeric modifications X and Y, with an additional hydroxymethyl group added to the bicyclic sugar unit of nucleoside P. We envisaged the use of the hydroxymethyl group as a major groove facing conjugation site for the attachment of different moieties, such as oligopeptides, oligonucleotides, pyrene units and alkylamines. Very few ONs carrying additional functionalities facing the major groove have been synthesized, but a pyrene conjugate of an ara-uridine monomer showed promising hybridization properties when targeting complementary DNA.29 However, the ara-uridine monomer itself displayed destabilizing behaviour when incorporated into an ON and evaluated against complementary DNA and RNA.
 | ||
| Fig. 1 | ||
Modifications X and Y showed a slight destabilizing effect when incorporated into a mixed 9-mer sequence and measured against a complementary RNA, with a profound destabilizing effect against complementary DNA.28 However, an almost fully modified 5′-X13T sequence displayed enhanced thermal stability towards complementary RNA as observed for the parent modification P. Surprisingly, thermal stability studies showed that modification Y induced a strong self-complexation when incorporated into an almost fully modified 5′-Y13T sequence (ON3, Table 1).28 We suggested that the presence of the 2″-C-hydroxymethyl group pointing towards the thymine moiety plays an important role in this observation. It is likely that the normal Watson–Crick base pairing is disrupted by hydrogen bonding between the hydroxyl group and the C2 carbonyl group. To study this hypothesis, and to evaluate the influence of the stereochemical configuration on the applicability as an attachment site, we decided to synthesize ONs containing the corresponding methylated monomer Z as a probe without the capability of forming a hydrogen bond with the C2 carbonyl group. In addition, the RNA binding observed for ONs containing X prompted us to investigate the applicability of the hydroxymethyl moiety of this monomer for functionalization towards the major groove of nucleic acid duplexes. This is an extension of earlier work focused on utilizing modification Y as a branching point for triple helix forming ONs,30 elaborated upon herein by the introduction of the N-methylpiperazino-modified monomer W, a molecular design stimulated by the opportunity of exploratory N-derivatizations.31,32 The basic N-methylpiperazino moiety allows for studies relating duplex stability with changes in pH value or salt concentration, with the goal of advancing understanding regarding the relation between net charge and duplex stability.
| Entry | ON sequence | DNA target | RNA target | ||
|---|---|---|---|---|---|
| T m | ΔTm | T m | ΔTm | ||
| a Performed with 1.5 µM of the two complementary strands in a medium salt buffer: 0.1 mM EDTA, 100 mM sodium chloride, 10 mM sodium phosphate, pH 7.0. The melting temperature Tm was determined as the maximum of the first derivative of the melting curve (A260vs. temperature). ΔTm = difference in Tm obtained for the modified ON and for Ref A. n.d. = no transition detected above 10 °C. b A value of 61 °C was obtained in an analogous thermal denaturation experiment without the addition of complementary strand. | |||||
| Ref A | 5′-T14 | 31 | — | 29 | — |
| ON1 | 5′-T7YT6 | 27 | −4 | 25 | −4 |
| ON2 | 5′-T5Y4T5 | 10 | −21 | 25 | −4 |
| ON3 | 5′-Y13T | 60 | b | 61 | b |
| ON4 | 5′-T7ZT6 | 27 | −4 | 24 | −5 |
| ON5 | 5′-T5Z4T5 | 10 | −21 | 25 | −4 |
| ON6 | 5′-Z13T | n.d. | — | 38 | +9 |
Results and discussion
The phosphoramidite derivatives 5 and 10 were synthesized from the known bicylic nucleoside 1 (Scheme 1).28 Methylation at the 2″-C-hydroxymethyl position using conditions previously employed26,33 to avoid methylation in the nucleobase moiety gave nucleoside 2 in 55% yield together with 26% recovered starting material. As expected, no N3-methylated product was observed. Oxidation of the 2″-C-hydroxymethyl position in nucleoside 1 by means of the free radical 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and iodobenzene diacetate (BAIB)34,35 afforded the carboxylic acid 6 in a yield of 82%. The synthesis of amide 7 was initially attempted using either dicyclohexylcarbodiimide (DCC) or 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) as coupling reagents, but formation of by-products was observed in both cases. However, activation of the carboxylic acid by 1,1′-carbonyldiimidazole (CDI) in anhydrous THF, followed by addition of N-methylpiperazine smoothly converted nucleoside 6 into amide 7 in a satisfactory yield of 85%. The presence of an amine moiety in nucleoside 7 greatly enhanced the affinity for hydrophilic solvents and a strong basic aqueous solution had to be applied during work-up in order to prevent the product entering the water phase. Hydrogenolysis of nucleosides 2 and 7, using H2 and Pd(OH)2 on carbon as a catalyst, afforded the debenzylated nucleosides 3 and 8, respectively. The use of MeOH or EtOH as solvents only resulted in partial debenzylation of nucleoside 7, whereas the use of acetone gave the fully debenzylated product. Selective protection of the primary hydroxyl group of crude derivatives 3 and 8 was accomplished using 4,4′-dimethoxytrityl chloride (DMTCl) in anhydrous pyridine, giving nucleosides 4 and 9 in overall yields of 74% and 72% from 2 and 7, respectively. 4-(N,N-Dimethylamino)pyridine (DMAP) was added to the reaction mixture of nucleoside 8 in order to accelerate the reaction. Reacting nucleosides 4 and 9 with 2-cyanoethyl N,N-diisopropylphosphoramidochloridite and DIPEA in anhydrous DCM afforded the phosphoramidites 5 and 10 in yields of 65% and 75%, respectively.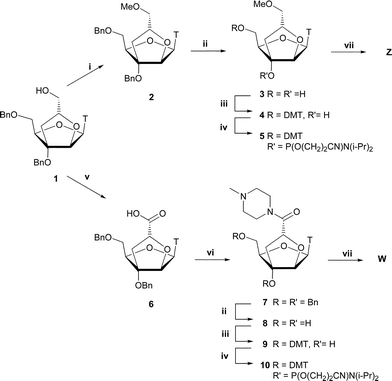 | ||
| Scheme 1 Reagents and conditions: i) CH3I, NaH, anhydrous THF, 0 °C, 55% (+26% 1); ii) 20% Pd(OH)2/C, H2, abs. EtOH (for 3) or acetone (for 7), rt; iii) DMTCl, anhydrous pyridine, DMAP (synthesis of 9), rt, 4: 74% and 9: 72% (two steps); iv) 2-cyanoethyl N,N-diisopropylphosphoramidochloridite, DIPEA, CH2Cl2, 5: 65% and 10: 75%; v) TEMPO, BAIB, CH2Cl2, H2O, 5 °C, 82%; vi) 1-methylpiperazine, CDI, anhydrous THF, rt, 85%; vii) DNA synthesizer. T = thymin-1-yl. | ||
Phosphoramidite 5 was used as a building block for the incorporation of monomer Z into modified 14-mer thymidylate sequences (ON4–ON6, Table 1; see Experimental section for details). As anticipated, and opposed to that observed for monomer Y,28 no self-complexation was observed for 5′-Z13T (ON6). In fact, this modified ON displayed similar hybridization properties to 5′-P13T27 and 5′-X13T28 with an increased affinity towards complementary RNA (ΔTm = +9 °C) and RNA selective hybridization. One or four consecutive modifications of Z induced a moderate decrease in the thermal stability toward RNA (ON4 and ON5, Table 1), which is in agreement with results reported for ONs containing modification Y (ON1 and ON2, Table 1)28 and also corresponds well with the behaviour of ONs modified with the parent compound P.27 The thermal stability studies of ONs modified with Z indicate that the RNA binding capability is preserved when the C2″-hydroxylmethyl moiety is chemically functionalized, which underlines the potential of this group as a conjugation site independent of stereochemical configurations at C2″.
The piperazino-functionalized monomer W was successfully incorporated into mixed 9-mer ONs but attempts to produce fully or partly substituted 14-mer oligothymidylates failed as the ONs obtained proved unstable under the conditions of deprotection and cleavage from the solid support (ON7–ON9, Table 2; see Experimental section for details). Thus, MALDI-MS and CE showed the removal of several piperazino moieties during the treatment with aqueous ammonia. The observed lack of stability was somewhat of a surprise to us since alkyl amide linkages are generally known to be compatible with the conditions of ON cleavage from solid supports,36,37 and it is therefore likely that the observed amide hydrolysis occurs under the influence of the nearby thymine moiety. The thermal stability studies of the mixed 9-mers ONs were conducted in medium salt concentration (pH 5 and pH 7, Table 2) and in low salt concentration (pH 5 and pH 7, Table 3). For the reference ONs, Ref B and Ref C (Table 2 and 3), a destabilizing effect was observed when the salt concentration was reduced from 110 mM [Na+] to 40 mM [Na+], as expected owing to a less efficient shielding of the negatively charged backbones of the two hybridizing strands. Likewise, the reference ON was destabilized when the pH was lowered, which confirms that Watson–Crick base-pairing is disturbed at a low pH value,38,39 and no thermal transition was observed for Ref B at a low salt concentration and pH 5. When a single W monomer was introduced in the mixed 9-mer (ON7, Table 2) we observed a slight decrease in Tm value when measuring against complementary DNA and RNA at pH 7. The effect was even more pronounced with three W monomers incorporated (ON8, Table 2). Lowering the pH appeared to change the situation as ON7 no longer showed any destabilization compared to the reference, and ON8 also showed a slight improvement (ΔTm = +1 °C) when measured against RNA at pH 5. This data suggests that the tertiary amine functionality of the piperazino group of monomer W experiences a higher degree of protonation at pH 5 than at pH 7.
| Entry | ON sequence | pH 7b | pH 5c | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DNA target | RNA target | DNA target | RNA target | ||||||
| T m | ΔTm | T m | ΔTm | T m | ΔTm | T m | ΔTm | ||
| a See caption below Table 1. | |||||||||
| b 0.1 mM EDTA, 100 mM sodium chloride, 10 mM sodium phosphate, pH 7.0. c 0.1 mM EDTA, 100 mM sodium chloride, 10 mM sodium acetate, pH 5. | |||||||||
| Ref B | 5′-d(GTG-ATA-TGC) | 29 | 26 | 24 | 21 | ||||
| ON7 | 5′-d(GTG-AWA-TGC) | 27 | −2 | 24 | −2 | 24 | 0 | 21 | 0 |
| ON8 | 5′-d(GWG-AWA-WGC) | 19 | −10 | 21 | −5 | 20 | −4 | 22 | +1 |
| Ref C | 5′-d(GTLG-ATA-TLGC) | 36 | 41 | 32 | 38 | ||||
| ON9 | 5′-d(GTLG-AWA-TLGC) | 34 | −2 | 39 | −2 | 32 | 0 | 38 | 0 |
The influence of the salt concentration was investigated by thermal stability studies as depicted in Table 3. Duplexes of ON7 and ON8 with complementary DNA and RNA targets were, relative to those involving Ref B, more stable at 40 mM [Na+]/pH 7 than at 110 mM [Na+]/pH 7 (see Table 2). In agreement, for ON7 there is no decrease in Tm value towards complementary DNA and only a slight decrease (ΔTm = −1 °C) towards RNA. This observation suggests that the tertiary amine functionality of the piperazino group is at least partly protonated under the experimental conditions applied, and that this protonation plays a role in duplex stabilization. Clearly, the effect of lowering the pH value is even more pronounced at a low salt concentration than at a medium salt concentration. We established this by thermal denaturation studies at 40 mM [Na+]/pH 5 which showed no thermal transition for Ref B but a clear transition for ON7 and ON8.
| Entry | ON sequence | pH 7b | pH 5c | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DNA target | RNA target | DNA target | RNA target | ||||||
| T m | ΔTm | T m | ΔTm | T m | ΔTm | T m | ΔTm | ||
| a See caption below Table 1. b 0.1 mM EDTA, 30 mM sodium chloride, 10 mM sodium phosphate, pH 7.0. c 0.1 mM EDTA, 30 mM sodium chloride, 10 mM sodium acetate, pH 5. | |||||||||
| Ref B | 5′-d(GTG-ATA-TGC) | 22 | 19 | n.d. | n.d. | ||||
| ON7 | 5′-d(GTG-AWA-TGC) | 22 | 0 | 18 | −1 | 16 | > +6 | n.d. | |
| ON8 | 5′-d(GWG-AWA-WGC) | 18 | −4 | 19 | 0 | 13 | > +3 | 15 | > +5 |
| Ref C | 5′-d(GTLG-ATA-TLGC) | 30 | 35 | 23 | 30 | ||||
| ON9 | 5′-d(GTLG-AWA-TLGC) | 30 | 0 | 33 | −2 | 24 | +1 | 30 | 0 |
To study the compatibility of monomer W with LNA monomers40,41 with the aim of enhancing the thermal stability of duplexes involving the modified 9-mer sequences, we incorporated two LNA-T monomers (Fig. 1) into ON9. As anticipated, the favorable hybridization properties of LNA significantly improved the thermal stability of duplexes formed with both DNA and RNA target strands (Ref C, Table 2 and Table 3). ON9, containing one W monomer and two LNA-T monomers, displayed the same tendency as ON7 under both medium and low salt conditions (pH 7). However, at pH 5, only a negligible positive effect of the piperazino group was observed. These results indicate that under all of the conditions studied, the dominating affinity enhancing effect of the LNA monomers renders the effect of protonation of the basic piperazino group to be of less importance.
Conclusion
The applicability of a 2″-C-hydroxymethyl group of a 2′,3′-bicyclic nucleic acid monomer as a site for conjugation has been demonstrated by satisfactory RNA-binding of the corresponding O-methylated derivative Z. Attachment of a piperazino group to the 2′,3′-bicyclic nucleic acid monomer via an amide bond influences the stability of a duplex formed between both complementary DNA and RNA. Especially at low salt concentrations, the pH value plays a significant role in determining the stability of duplexes involving the modified ONs, indicating that protonation of the tertiary amine plays a duplex-stabilizing role by partial charge reduction of the duplex. The binding affinity of an ON containing a single piperazino monomer could be further improved by incorporation of LNA-T monomers. These results demonstrate that functionalized 2′-O,3′-C-linked bicyclic nucleic acid monomers, e.g.Y (O-conjugation) and W (N-conjugation), are promising scaffolds for attachment of groups facing the major groove of nucleic acid duplexes for modulation of hybridization properties or for systematic mapping of the major groove.Experimental
General
All reagents were obtained from commercial suppliers and used without further purification. THF was distilled from Na and benzophenone. Reactions were carried out under an atmosphere of nitrogen when anhydrous solvents were used. Column chromatography was performed using silica gel 60 (0.040–0.063 mm). Dichloromethane (DCM) used for column chromatography was distilled prior to use. NMR spectra were recorded on a Varian Gemini 200 NMR spectrometer or a Varian Unity 400 with chemical shift values relative to tetramethylsilane as an internal standard (1H and 13C NMR) and relative to 85% H3PO4 as external reference (31P NMR). 2D NMR techniques were used to assign 1H and 13C spectra. Coupling constants (J values) are given in Hz. Microanalyses were performed at the Microanalytical Laboratory, Department of Chemistry, University of Copenhagen. Matrix-assisted laser-desorption ionization-mass spectrometry (MALDI-MS) was performed on a 4.7 Tesla Ultima (IonSpec, Irvine, CA) Fourier transform ion cyclotron resonance (FTICR) mass spectrometer.Oligonucleotide synthesis
All ONs were prepared on a Biosearch 8700 synthesizer using the phosphoramidite approach. Amidite 5 or 10 were used as building blocks for incorporation of monomers Z and W into ONs using a 10 min coupling time with pyridinium hydrochloride as an activator and tert-butyl hydroperoxide as an oxidant in step-wise coupling yields of 98%. The DNA and LNA-T nucleotides were incorporated in step-wise coupling yields of >99% (2 min and 6 min coupling times, respectively.) The 5′-O-DMT-OFF ONs were deprotected using concentrated NH3 in MeOH for 12 h at 55 °C. Precipitation from EtOH afforded ONs of >80% purity as evaluated by gel capillary electrophoresis. The composition of the ON's was verified by MALDI-MS [M − H] analysis: ON4 calc. 4282; found 4276: ON5 calc. 4539; found 4539: ON6 calc. 5312; found 5313: ON7 calc. 2922; found 2921: ON8 calc. 3258; found 3256: ON9 calc. 2976; found 2978.Acknowledgements
The Danish Research Agency and the Danish National Research Foundation are thanked for financial support. Ms Britta M. Dahl, Department of Chemistry, University of Copenhagen, is thanked for oligonucleotide synthesis and Dr Michael Meldgaard, Exiqon A/S, for MALDI-MS analysis.References
- J. Wengel, Org. Biomol. Chem., 2004, 2, 277 RSC.
- K. F. Pirollo, A. Rait, L. S. Sleer and E. H. Chang, Pharmacol. Ther., 2003, 99, 55 Search PubMed.
- L. E. Heystek, H. Zhou, P. Dande and B. Gold, J. Am. Chem. Soc., 1998, 120, 12165 CrossRef CAS.
- A. M. Soto, B. I. Kankia, P. Dande, B. Gold and L. A. Marky, Nucleic Acids Res., 2002, 30, 3171 CrossRef CAS.
- T. P. Prakash, D. A. Barawkar, V. Kumar and K. N. Ganesh, Bioorg. Med. Chem. Lett., 1994, 4, 1733 CrossRef CAS.
- K. S. Ramasamy, M. Zounes, C. Gonzales, S. M. Freier, E. A. Lesnik, L. L. Cummins, R. H. Griffey, B. P. Monia and P. D. Cook, Tetrahedron Lett., 1994, 35, 215 CrossRef CAS.
- M. Manoharan, K. S. Ramasamy, V. Mohan and D. P. Cook, Tetrahedron Lett., 1996, 37, 7675 CrossRef CAS.
- M. A. Maier, I. Barber-Peoc'h and M. Manoharan, Tetrahedron Lett., 2002, 43, 7613 CrossRef CAS.
- H. Hashimoto, M. G. Nelson and C. Switser, J. Am. Chem. Soc., 1993, 115, 7128 CrossRef CAS.
- F. Seela and M. Zulauf, Helv. Chim. Acta, 1999, 82, 1878 CrossRef CAS.
- K.-Y. Lin and M. D. Matteucci, J. Am. Chem. Soc., 1998, 120, 8531 CrossRef CAS.
- R. L. Letsinger, C. N. Singman, G. Histand and M. Salunkhe, J. Am. Chem. Soc., 1988, 110, 4470 CrossRef CAS.
- P. M. Jung, G. Histand and R. L. Letsinger, Nucleosides Nucleotides, 1994, 13, 1597 CAS.
- T. Horn, S. Chaturvedi, T. N. Balasubramaniam and R. L. Letsinger, Tetrahedron Lett., 1996, 37, 743 CrossRef CAS.
- R. Fathi, Q. Huang, G. Coppola, W. Delaney, R. Teasdale, A. M. Krieg and A. F. Cook, Nucleic Acids Res., 1994, 22, 5416 CAS.
- A. F. Cook, R. Fathi, Q. Huang, G. Coppola, W. Delaney and J.-L. Syi, Nucleosides Nucleotides, 1995, 14, 1005 CAS.
- T. Michel, F. Debart and J.-J. Vasseur, Tetrahedron Lett., 2003, 44, 6579 CrossRef CAS.
- D. T. Hickman, T. H. S. Tan, J. Morral, P. M. King, M. A. Cooper and J. Micklefield, Org. Biomol. Chem., 2003, 1, 3277 RSC.
- R. Meier, S. Grüschow and C. Leumann, Helv. Chim. Acta, 1999, 82, 1813 CrossRef CAS.
- H. M. Pfundheller, T. Bryld, C. E. Olsen and J. Wengel, Helv. Chim. Acta, 2000, 83, 128 CrossRef CAS.
- H. Aurup, T. Tuschl, F. Benseler, J. Ludwig and F. Eckstein, Nucleic Acids Res., 1994, 22, 20 CAS.
- M. Kanazaki, Y. Ueno, S. Shuto and A. Matsuda, J. Am. Chem. Soc., 2000, 122, 2422 CrossRef CAS.
- R. H. Griffey, B. P. Monia, L. L. Cummins, S. Freier, M. J. Greig, C. J. Guinosso, E. Lesnik, S. M. Manalili, V. Mohan, S. Owens, B. R. Ross, H. Sasmor, E. Wancewicz, K. Weiler, P. D. Wheeler and P. D. Cook, J. Med. Chem., 1996, 39, 5100 CrossRef CAS.
- T. P. Prakash, M. Manoharan, A. S. Fraser, A. M. Kawasaki, E. L. Lesnik and S. R. Owens, Tetrahedron Lett., 2000, 41, 4855 CrossRef CAS.
- M. Prhavc, T. P. Prakash, G. Minasov, P. D. Cook, M. Egli and M. Manoharan, Org. Lett., 2003, 5, 2017 CrossRef CAS.
- G. Wang and W. E. Seifert, Tetrahedron Lett., 1996, 37, 6515 CrossRef CAS.
- P. Nielsen, H. M. Pfundheller, C. E. Olsen and J. Wengel, J. Chem. Soc., Perkin Trans. 1, 1997, 3423 RSC.
- M. Raunkjær, C. E. Olsen and J. Wengel, J. Chem. Soc., Perkin Trans. 1, 1999, 2543 RSC.
- N. N. Dioubankova, A. D. Malakhov, D. A. Stetsenko, M. J. Gait, P. E. Volynsky, R. G. Efremov and V. A. Korshun, ChemBioChem, 2003, 4, 841 CrossRef CAS.
- M. D. Sørensen, M. Meldgaard, V. K. Rajwanshi and J. Wengel, Bioorg. Med. Chem. Lett., 2000, 10, 1853; CrossRef CAS; M. Meldgaard, M. Raunkjær, V. K. Rajwanshi, J. Wengel and M. D. Sørensen, Bioorg. Med. Chem. Lett., 2001, 11, 751 CrossRef.
- A preliminary report has been published: M. Raunkjær, T. Bryld and J. Wengel, Chem. Commun., 2003, 1604 Search PubMed.
- T. Bryld, T. Højland and J. Wengel, Chem. Commun., 2004, 1064 RSC.
- H. M. Pfundheller, A. A. Koshkin, C. E. Olsen and J. Wengel, Nucleosides Nucleotides, 1999, 18, 2017 CAS.
- A. D. Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem, 1997, 62, 6974 CrossRef CAS.
- J. B. Epp and T. S. Widlanski, J. Org. Chem., 1999, 64, 293 CrossRef CAS.
- G. V. Petersen and J. Wengel, Tetrahedron, 1995, 51, 2145 CrossRef CAS.
- J. Lebreton, A. D. Mesmaeker, A. Waldner, V. Fritsch, R. M. Wolf and S. M. Freier, Tetrahedron Lett., 1993, 34, 6383 CrossRef CAS.
- N. Sugimoto, P. Wu, H. Hara and Y. Kawamoto, Biochemistry, 2001, 40, 9396 CrossRef CAS.
- I. Rouzina and V. A. Bloomfield, Biophys. J., 2001, 80, 894 CAS.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607 CrossRef CAS.
- S. Obika, D. Nanbu, Y. Hari, J.-I. Andoh, K.-I. Morio, T. Doi and T. Imanishi, Tetrahedron Lett., 1998, 39, 5401 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: copies of 13C NMR spectra of compounds 6–9 and copies of 31P NMR spectra of compounds 5 and 10. See http://www.rsc.org/suppdata/ob/b4/b414454f/ |
| ‡ A research center funded by the Danish National Research Foundation for studies on nucleic acid chemical biology. |
| This journal is © The Royal Society of Chemistry 2005 |