1,3-Alternate calix[4]arenes, selectively functionalized by amino groups
Crenguta
Danila
a,
Michael
Bolte
b and
Volker
Böhmer
*a
aFachbereich Chemie und Pharmazie, Abteilung Lehramt Chemie, Johannes Gutenberg-Universität, Duesbergweg 10-14, D-55099, Mainz, Germany. E-mail: vboehmer@mail.uni-mainz.de
bInstitut für Organische Chemie, Johann Wolfgang von Goethe Universität, Marie Curie-Straße 11, D-60439, Frankfurt/Main, Germany
First published on 26th November 2004
Abstract
General strategies are described to synthesize calix[4]arenes which are fixed in the 1,3-alternate conformation and substituted selectively by amino groups. These derivatives are useful starting materials for the attachment of various groups via amide bonds, as demonstrated by several examples, but may be converted also to ureas, imides or azomethines. Four amino groups may be attached to the narrow rim via (several) methylene groups as spacer by O-alkylation with ω-bromophthalimides or ω-bromonitriles. From the resulting tetraethers the amino functions are obtained by cleavage with hydrazine or by hydrolysis, allowing a selective functionalisation of both sides of the molecule (phenolic units A, C versus B, D). Amino functions at the wide rim are introduced by ipso-nitration of the respective t-butylcalix[4]arene derivatives and subsequent reduction. Selective ipso-nitration of a 1,3-diether, followed by O-alkylation with allylbromide to obtain the tetraether in the 1,3-alternate conformation, hydrogenation of allyl and nitro groups (in one step), protection of the amino functions as phthalimides followed by ipso-nitration of the remaining t-butyl phenol rings, allows again to distinguish both sides of the molecule (units A, C versus B, D). Reaction of a wide rim tetraamine in the 1,3-alternate conformation by Boc-anhydride allows not only to obtain the mono- and tri-Boc derivative, but also in nearly 60% yield the C2-symmetrical di-Boc derivative, in which two adjacent phenolic units are protected (distinction of A, B from C, D). This paves the way for the preparation of chiral derivatives or assemblies. O-Alkylation with ω-bromophthalimides followed by ipso-nitration leads to precursors for octaamines in the 1,3-alternate conformation, in which the potential amino functions on both rims can be selectively “activated” by reduction or hydrazinolysis. The structures of the newly synthesized molecules were mainly confirmed by their 1H NMR spectra, which allow a distinction from isomeric derivatives in the cone and partial cone conformation. Single crystal X-ray analyses were obtained for two analogous derivatives in the 1,3-alternate conformation (27, n = 3,4), an isomeric compound in the cone conformation (27, n = 3,4), and a derivative in the partial cone conformation (11).
Introduction
Calixarenes represent a class of cyclic oligomers, which is readily available in larger quantities and amenable to chemical modifications.1 Thus, they have been frequently used as building blocks for larger molecular structures or assemblies or as platforms on which various kinds of functional groups may be assembled. For these aims calixarenes bearing amino functions may be considered as the most versatile starting material to which further residues can be attached easily via amide,2 imide,3 (thio)urea4 or azomethine5 links. N-Alkylation even allows the attachment of two residues per amino group6 or the formation of cationic sites by quaternisation.7 In general calixarenes can be decorated by amino groups at the narrow rim, attached to the phenolic hydroxyl groups via (CH2)n-spacers, or at the wide rim, directly attached to the para-positions or again via (CH2)n-spacers. In the calix[4]arene series the introduction of ether residues (≥propyl) restricts the conformational mobility and tetraethers can be fixed in one of the four basic conformations (cone, partial cone, 1,2- and 1,3-alternate) among which the cone-conformation is best explored. Thus, CMPO-like functions8 have been attached to the wide9 and to the narrow rim10 to obtain extractants for lanthanides and actinides. Anion receptors were obtained as narrow (or wide) rim di- or tetra-(thio)urea derivatives11 or by the attachment of various amide groups including cobaltocenium.12 The self-assembly of wide rim tetra-urea derivatives to form hydrogen bonded dimeric capsules has been extensively studied,13 and similar capsules have been found for calix[4]arenes bearing dipeptide residues at the wide rim.14 The 1,3-alternate conformation has been less frequently used as platform or building block. Di-urea derivatives as anion receptors15,16 or self-assembled dimers17 may be mentioned as examples, which were derived from the respective amino calix[4]arenes. In general it may be stated that the selective preparation of amino derivatives of calix[4]arenes fixed in the cone conformation is well established, using selective etherification (ω-bromonitriles, ω-bromoalkylphthalimides) for the narrow rim, and selective substitutions (e.g. (ipso)nitration18 followed by hydrogenation) or protection strategies (Boc,19 phthalimide20) for amino groups attached to the wide rim. In this manuscript we discuss general possibilities and describe new strategies for the selective introduction or protection of amino functions in 1,3-alternate calix[4]arenes.Results and discussion
Tetraamines, narrow rim
The introduction of aminoalkyl ether residues can be achieved by O-alkylation of 1 with ω-bromoalkylphthalimides10,21 or ω-bromonitriles.22 Bromopropionitrile (BrCH2CH2 CN, m = 2) cannot be used for the O-alkylation, since elimination to acrylnitrile predominates while only two ether residues are introduced with bromoacetonitrile.23 Elimination cannot be avoided also with bromoethylphthalimide but hydroxy-ethylphthalimide has been used recently to obtain the 1,3-diether from t-butylthiacalix[4]arene under Mitsunobu conditions.24 The O-alkylation with Boc-protected chloroethylamine25 seems to be another alternative to introduce aminoethyl ether groups.26Via the syn-1,3-diether 2 or 3, obtained in excellent yield in the presence of K2CO3, (or under Mitsunobu conditions in the case of 2 (n = 2) a mixed tetraether 4, fixed (mainly) in the 1,3-alternate conformation is available if the second etherification is done with an excess of Cs2CO327 as base. The amount of Cs2CO3 required as template can vary between 8- and 15-fold, with respect to the hydroxyl groups, depending on the nature of the existing ether residue and the p-substituent in the phenolic unit (see below). Partialcone and cone isomers28 are always observed as side products. The formation of tetraethers with four identical ether residues (e.g. alkylphthalimides), can be achieved in one step with Cs2CO3, but two steps, as outlined in Scheme 1, are preferable according to our experience.
 | ||
| Scheme 1 (i) Alkyl bromide, K2CO3; (i′) for n = 2: N-(β-hydroxyethylphthalimide, triphenylphosphine, DIAD, THF; (ii) alkyl bromide, Cs2CO3, DMF, 50 °C; (iii) H2, Raney-Ni, NaOH; (iv) hydrazine, EtOH, reflux; (v) Ac2O; (vi) p-nitrobenzoylchloride. | ||
Compounds of type 4 contain two different precursors for amino functions which are in principle “orthogonal”. The cleavage of the phthalimido groups with hydrazine in refluxing ethanol occurs without reduction of the nitrile functions. As an example, diamine 5 was obtained in 88% yield. After acetylation of the amino functions (92% of 6) the reduction of the nitrile groups is possible by hydrogenation (Raney-Ni, room temperature) under alkaline conditions. In the final step a different residue may be attached to the diamine 7, as shown by the p-nitrobenzamide 8 chosen as an example.
Contrary to our expectations the reduction of the nitrile functions in 4 was not possible without a partial hydrolysis of one of the phthalimido groups. Under various conditions, e.g. lowering the temperature from 50 °C to room temperature, replacing Raney-Ni by Pd/C, using LiOH as weaker base instead of NaOH, the monophthalamide 9 was formed and isolated in yields up to 62%.29 Attempts to hydrogenate the nitrile functions with Raney-Ni in toluene at 60 °C left the starting material unreacted.30
However, the sequence 1 to 8 as outlined in Scheme 1 and realized for one example allows in principle to attach two different residues in alternate sequence to amino groups on the narrow rim of a 1,3-alternate calix[4]arene. Evidently diamines and their derivatives are available analogously.31
Tetraamines, wide rim
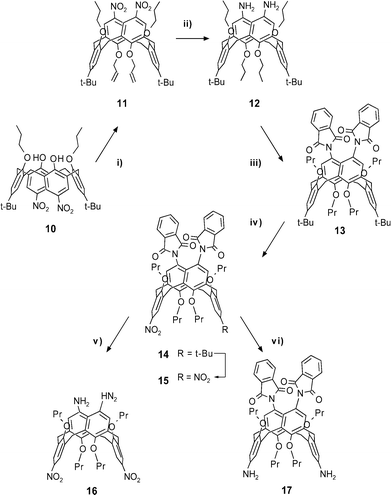
Introduction of amino functions attached to the wide rim is possible by ipso-nitration of a tetraether followed by reduction of the nitro groups. This sequence is possible also with tetraethers in 1,3-alternate conformation32 (and other conformations) and leads to a tetraamine (bearing eventually two different ether residues) where no differentiation of the amino functions is possible.Selective ipso-nitration of syn-1,3-diethers, however, is possible in good yield in the phenolic units.18O-Alkylation of 10, taken as an example for such a dinitro-diether, with allylbromide leads to 11 in the 1,3-alternate conformation (accompanied by the partial cone isomer). Simultaneous reduction of the nitro groups and hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (Raney-Ni, rt) gives the diamine 12, in which the amino groups can be protected as phthalimide (13). Ipso-nitration of the t-butylphenyl ether units in 13 is possible in a clean fashion (without attack on the phthalimide units).20 In the special case we even found conditions (high dilution) to obtain the mono-nitro compound 14 in 79% yield, which could be used to synthesize wide rim triamines. Subsequent ipso-nitration of 14 gave the dinitro compound 15 in 83% yield. Compounds of type 14 and 15 contain two independent (orthogonal) precursors of amino groups. In analogy to similar derivatives in the cone-conformation20,33 either the phthalimide group can be cleaved by hydrazine (16) or the nitro groups can be hydrogenated (17). All steps are summarized in Scheme 2.
C double bonds (Raney-Ni, rt) gives the diamine 12, in which the amino groups can be protected as phthalimide (13). Ipso-nitration of the t-butylphenyl ether units in 13 is possible in a clean fashion (without attack on the phthalimide units).20 In the special case we even found conditions (high dilution) to obtain the mono-nitro compound 14 in 79% yield, which could be used to synthesize wide rim triamines. Subsequent ipso-nitration of 14 gave the dinitro compound 15 in 83% yield. Compounds of type 14 and 15 contain two independent (orthogonal) precursors of amino groups. In analogy to similar derivatives in the cone-conformation20,33 either the phthalimide group can be cleaved by hydrazine (16) or the nitro groups can be hydrogenated (17). All steps are summarized in Scheme 2.
Thus, derivatives with two different amide residues are available from 15 (or 14) by deprotection (or reduction), acylation, reduction (or deprotection) and final acylation. The appropriate reaction sequence may be chosen with respect to the special example.
It should be mentioned that the O-alkylation of 10 requires a reactive bromide, like allylbromide (eventually benzylbromide, ethylbromoacetate), due to the lower nucleophilicity of the p-nitrophenolate units, while usual alkyl bromides are not reactive enough in the presence of Cs2CO3. However, the first ether residue can be chosen more or less freely,34 and the allyl group is easily hydrogenated to the propyl group.
The two different precursors of the amino function in 15 point (in pairs) in one or the other direction. The molecule has C2v-symmetry. An alternative to differentiate between amino functions attached to the wide rim consists in a partial protection by Boc. When 1 mol of tetraamine3518 is treated with 1.9 mol of Boc-anhydride (Scheme 3), 58% of the 1,2-diprotected derivative 20 can be isolated by chromatography, in addition to 23% of the monoprotected compound 19. (The yield of 20 can be increased to 65% and decreased to 18% for 19 using 2 mol of Boc-anhydride). If 3 mol of Boc-anhydride are used, the triprotected compound 22 is available in 63% yield from 18, and the tetra-Boc protected compound was formed as a side product. Similar to the cone-derivative the formation of a 1,3-diprotected 21 compound is not observed.
 | ||
| Scheme 2 (i) Allylbromide, Cs2CO3, DMF, 50 °C; (ii) H2, Raney-Ni, EtOH, rt; (iii) phthalic anhydride, toluene, reflux; (iv) HNO3, CH2Cl2/AcOH; (v) hydrazine, EtOH, reflux; (vi) H2, Raney-Ni, toluene/THF, rt. | ||
Compound 20 is chiral (C2-symmetry), an aspect which is not further discussed in this article and the two protected, as well as the not-protected, amino functions point in different directions which contrasts to 15. Tetraamides with two different amide functions are available from 20 by acylation, deprotection and a second acylation as shown for one example. The acylation of 20 with acetanhydride leads quantitatively to 24 which after deprotection is easily acylated to tetraamide 25.
Tetraamines, narrow/wide rim
If four amino functions attached to a 1,3-alternate calix[4]arene point in one direction, two of them must be attached at the narrow rim (with different length of the alkyl spacer, e.g. two, three or four carbon atoms) and two at the wide rim.A possible reaction sequence to achieve this goal is shown in Scheme 4:
 | ||
| Scheme 3 (i) 2 mol Boc2O; (ii) 3 mol Boc2O; (iii) Ac2O; (iv) CF3COOH, CHCl3, 0 °C; (v) p-nitrobenzoylchloride, CHCl3, NEt3, rt. | ||
Ipso-nitration of the syn-1,3-diether 2, followed by O-alkylation of 26 with allylbromide leads to compounds 27 in a reaction sequence analogous to Scheme 3.
The crucial step of the sequence, the O-alkylation with allylbromide (compare Scheme 2, step (i)), was tried under different conditions in the presence of Cs2CO3. While the reaction is too slow at room-temperature, an increase of the temperature favours the formation of the partial cone conformation over the desired 1,3-alternate. The best results in our hands were found for a concentrated reaction mixture (base and calixarene) in DMF at 40–50 °C for 5–7 days. A higher yield of 27 was obtained for n = 4 (53%) than for n = 3 (19%). This may be due to some steric hindrance between the phthalimido groups and the nitro groups of the inverted phenolic units, which is more pronounced for the shorter chains (n = 3 vs.n = 4). This could also explain why the partial cone (19%) and cone (2%) conformers were isolated as side products in the case of 27 (n = 3) while the formation of the partial cone conformer was detected only by NMR for 27 (n = 4).
 | ||
| Scheme 4 (i) HNO3, CH2Cl2/AcOH; (ii) allylbromide, Cs2CO3, DMF, 40–50 °C; (iii) H2, Raney-Ni, THF, rt; (iv) hydrazine, EtOH, reflux; (v) hydrazine, Pd/C, EtOH, reflux. | ||
Compounds 27 again contain two independent precursors of amino functions. Catalytic hydrogenation leads to the wide rim diamines 28 (n = 3, 4) which can be N-acylated at the wide rim, deprotected and acylated again on the narrow rim to furnish a tetraamide with two different amide residues on the same side of the platform. Cleavage of the phthalimide groups by hydrazine in boiling ethanol leads to aliphatic diamines 30, as shown for n = 3 (57%), in which the nitro groups are retained. After a first N-acylation at the narrow rim, reduction of the nitro groups followed by a second acylation at the wide rim will lead to a mixed tetraamide again. The choice of the appropriate reaction sequence will be due to the acyl residues to be introduced.
The example of 30 (n = 3) shows that the allyl ether groups are converted to propyl ether groups during the hydrazinolysis of the phthalimide groups. After shorter reaction times products were isolated, in which all phthaloyl residues were cleaved, while allyl groups were still present. This suggests that conditions might be found under which the allyl groups remain unchanged, if this would be desired. However, up to now we could not obtain a pure compound 29.
Finally, a direct conversion of 27 into tetraamines 31 (R = H) should be also possible if the cleavage reaction with hydrazine is done in the presence of Pd/C (compare the preparation of 36 below). The resulting tetraamines show (in the case of n = 4) very broad 1H-NMR signals under all conditions studied, and also the unambiguous characterization as tetraacetamide 31 (R = COCH3) failed so far.36
Octaamines
As illustrated in Scheme 5, it is possible also to create four amino functions on each side of a 1,3-alternate calix[4]arene. | ||
| Scheme 5 (i) Alkylbromide, Cs2CO3, THF reflux or DMF 50 °C; (ii) HNO3, CH2Cl2/AcOH; (iii) H2, Raney-Ni, THF/DMF, 50°; then Ac2O; (iv) hydrazine, EtOH, reflux; (v) hydrazine, Pd/C, EtOH, reflux. | ||
The tetraether 32 (n = m = 3) can be obtained from the respective syn-1,3-diether 2 or directly in one step from 1. The two step synthesis makes it possible also to have two different spacer lengths n and m, as shown for 32 (n = 4, m = 3), which is obtained in 60% yield starting from 1,3-syn-diether 2 (n = 4), while the yield was lower for 32 (n = m = 3) (compare the synthesis of 27 discussed above). Ipso-nitration yields 33 (64%), a compound again containing two independent precursors for amino functions. The catalytic hydrogenation of the nitro groups (Raney-Ni, H2) was complicated by the low solubility of 33 (n = m = 3) in usual solvents or solvent mixtures, and 34 (R = H) was directly acylated and characterised as the tetraacetamide 34 (R = COCH3). Cleavage of the phthalimido groups in 33 led to the narrow rim tetraamine 35 and simultaneous reduction of the nitro groups (hydrazine, Raney-Ni) gave octaamine 36
As already mentioned, the formation of tetraethers in the 1,3-alternate conformation starting with syn-1,3-diethers 2 is usually accompanied by partial cone and cone isomers as side products.37 With calixarenes (e.g.1) as starting material the 1,2-alternate conformer must be considered additionally as a potential product which was isolated in the case of 32 (n = m = 3) in 31%.
Characterization by NMR
1H NMR spectroscopy is the most valuable tool to distinguish these different isomers, especially during their separation, while the 13C signals of the methylene bridges are of limited value.38 The attribution of the structure is mainly based on the well known pattern of the Ar–CH2–Ar bridges, and of the aromatic protons.39 Substituents R in p-position and ether residues Y may furnish additional information.In the following we will discuss typical examples proceeding mainly in the sequence given by Schemes 1 to 5. In many cases the molecular structure was additionally proved by X-ray analysis, for which some characteristic examples are presented the next section.
Due to the different ether residues in 4 the methylene protons are not identical and appear as a pair of doublets (AB system, J = 13.8 Hz) at δ = 3.86 and 3.78 ppm. The 1,3-alternate conformation follows nevertheless unambiguously from the small difference in their chemical shift (Δδ = 0.08 ppm), since a cone conformation would require a difference of about 1.2 ppm, and a partial cone conformer would not show a single pair of doublets. For ArH protons, the 1H-NMR displays two close singlets at 6.97 and 7.00 ppm, respectively. Two singlets are also found for t-Bu protons at 1.20 and 1.28 ppm. More pronounced are the differences in the chemical shift for the methylene protons in compound 11 (Δδ = 0.13 ppm), while two strongly separated singlets at 6.96 and 7.95 ppm for aromatic protons are due to the different substituents at the wide rim (t-Bu vs. NO2).
Two 1H-NMR spectra measured in CDCl3 and C6D6 are required for the complete identification of the chiral C2-symmetry of compound 20, where a two-fold axis intersects the carbon atoms of two opposite methylene bridges. Two singlets (3.28, 3.41 ppm) and a pair of doublets (3.32, 3.37 ppm) for the methylene protons are found in CDCl3. Two AB systems for the aromatic protons of 20 (doublets at 6.45/6.47 and 7.23/7.27 ppm) are shown only in C6D6, while this clear splitting cannot be seen in CDCl3. The compounds 19 and 22 show a similar pattern of signals reflecting the same Cs-symmetry in both cases, with the symmetry plane intersecting two opposite phenolic units (protected and unprotected).
Even a small difference in the length of the ether chains is reflected in the 1H-NMR spectrum of 32 (n = 3, m = 4). While the D2d-symmetrical compound 32 (n = m = 3) shows one singlet for the protons of the methylene bridges (3.67 ppm) and one for the aromatic protons (6.92 ppm), two doublets with geminal coupling (∼3.67, 3.70 ppm) for methylene protons and two close singlets (6.91, 6.96 ppm) for the aromatic protons are found for 32 (n = 3, m = 4), where the symmetry is reduced to C2v
The proton signals displayed in 1H-NMR spectrum show that all further amines (34, 35 and 36) generated from the 33 precursor have the same D2d-symmetry indicated by the presence of two singlets, one for methylene protons and the other one for the aromatic protons.
Single crystal X-ray analyses
Four compounds, 27 (1,3-alt) with n = 3, 4, 27 (cone, n = 3) and 11 (paco) have been additionally confirmed by X-ray analysis. The results will be briefly discussed, using the general numbering scheme presented in Fig. 1. The molecular conformation and shape is shown in Fig. 2a–d, while Fig. 3 contains selected packing diagrams. | ||
| Fig. 1 General atomic numbering scheme illustrated by 27 (n = 3). | ||
![Molecular conformation of four calix[4]arenes: (a)
27
(1,3-alt, n = 3); (b)
27
(cone, n = 3); (c)
27
(1,3-alt, n = 4); (d)
11
(paco).](/image/article/2005/OB/b414173c/b414173c-f2.gif) | ||
| Fig. 2 Molecular conformation of four calix[4]arenes: (a) 27 (1,3-alt, n = 3); (b) 27 (cone, n = 3); (c) 27 (1,3-alt, n = 4); (d) 11 (paco). | ||
 | ||
| Fig. 3 The packing diagram of (a) 27 (1,3-alt, n = 3); (b) 27 (cone, n = 3); (c) 27 (1,3-alt, n = 4). | ||
For all compounds bond lengths and bond angles are in the usual range, and the following discussion refers mainly to the shape of the calixarene skeleton. A general and unambiguous description of the conformation of calixarenes is possible, using the torsion angles around the σ-bonds connecting the methylene bridges and the aromatic units (Table 1). The conformation of a given calix[4]arene is characterised by a typical sequence of the signs of these torsion angles, and these sequences are found for all compounds: (+,−)(+,−)(+,−)(+,−) for a cone conformation (27 (cone, n = 3)), (−,−)(+,+)(−,−)(+,+) for 1,3-alternate (27 (1,3-alt)), and (+,−)(+,−)(−,−)(+,+) for a partial cone (11 (paco)).40 While all torsions are in the range of 60° to 69° (absolute values) for 27 (1,3-alt, n = 3), smaller values down to 37° are found for 27 (1,3-alt, n = 4). To visualize the consequences for the shape of these two molecules in the 1,3-alternate conformation, and in general for a more vivid description of the conformation of calix[4]arenes the inclination of the single phenolic units with respect to a reference plane, the best plane through the four methylene carbon atoms (C1–C4), may be used. These δ values41 are also included in Table 1. Surprisingly the strongest deviation from an ideal regular square for C1–C4 is observed for the compound in the cone conformation. The two diagonals differ by about 0.74 Å, and pairs of opposite sides differ by 0.14 Å. Also the average deviation from the best plane (0.23 Å) is significantly higher than for the other three compounds (≤ 0.06 Å). Small but still significant differences are even found for the two molecules in the 1,3-alternate conformation, where the average side length for n = 4 is larger by 0.04 Å, compared with the analogue with n = 3.
| (a) | (b) | (c) | (d) | |
|---|---|---|---|---|
| I. Torsion angles | ||||
| C12–C11–C1–C43 | −60.5 | 66.8 | −46.6 | −112.7 |
| C11–C1–C43–C42 | −69.3 | −115.4 | −54.5 | 74.8 |
| C22–C21–C2–C13 | 65.5 | 136.6 | 68.4 | 61.3 |
| C21–C2–C13–C12 | 61.8 | −78.4 | 37.2 | 58.9 |
| C32–C31–C3–C23 | −60.2 | 61.3 | −45.2 | −70.0 |
| C31–C3–C23–C22 | −64.1 | −113.6 | −61.0 | −59.9 |
| C42–C41–C4–C33 | 68.5 | 133.2 | 60.3 | −69.9 |
| C41–C4–C33–C32 | 60.3 | −71.1 | 46.6 | 119.6 |
| II. Reference planes C1–C4 | ||||
| rmsd | 0.006 | 0.23 | 0.062 | 0.037 |
| Distance C1–C2 | 5.060 | 4.962 | 5.129 | 5.077 |
| Distance C2–C3 | 5.097 | 5.122 | 5.109 | 5.111 |
| Distance C3–C4 | 5.059 | 4.988 | 5.112 | 5.061 |
| Distance C1–C4 | 5.090 | 5.117 | 5.121 | 5.042 |
| Distance C1–C3 | 7.182 | 7.471 | 7.306 | 7.131 |
| Distance C2–C4 | 7.177 | 6.729 | 7.164 | 7.215 |
| III. Inclination of the aromatic units(δ) | ||||
| C11–C16 | 91.4 | 134.4 | 116.3 | 99.0 |
| C21–C26 | 91.6 | 78.9 | 102.4 | 95.4 |
| C31–C36 | 94.3 | 142.4 | 110.9 | 84.4 |
| C41–C46 | 84.9 | 79.8 | 106.7 | 139.3 |
| (a) 27 (1,3-alt, n = 3) (b) 27 (cone, n = 3) (c) 27 (1,3-alt, n = 4) (d) 11 (paco) | ||||
For 27 (1,3-alt, n = 3) all δ-values are close to 90°, only one nitrophenol unit is bent 5.1° inward and one t-butylphenol unit 4.3° outwards. Rather different is the shape of 27 (1,3-alt, n = 4), where both the nitrophenol units (12.4°, 16.7°) and the t-butylphenol units (26.3°, 20.9°) are strongly bent outwards. This “deformation” of the ideal 1,3-alternate conformation cannot be due to an intramolecular influence of the spacer (consisting of four instead of three carbon atoms). Therefore it must be caused by packing effects (see below) and gives an example of how strong these effects can be.
27 (cone, n = 3) is found in an extremely pinched cone conformation, where both nitrophenol units are bent inwards (11.1°, 10.2°) and both t-butyl phenol units are bent outwards (44.4°, 52.4°). The partial cone compound 11 (paco) can be roughly described as containing structural elements of a 1,3-alternate (aromatic rings 1 to 3) and a pinched cone (aromatic rings 3 to 1) conformer. The deviation of the former rings from an orientation perpendicular to the reference plane is ≤9°. The orientation of the t-butylphenol units is nearly parallel; one of them is bent outwards (9°) and the other one bent inwards (5.6°). Both p-nitrophenol units in-between are outwards oriented by 5.4° and 49.3°, respectively.
Molecular conformations of all four compounds are shown in Fig. 2, while Fig. 3 shows a representative section of the packing for the three phthaloyl substituted compounds.
Intermolecular forces between neighbouring molecules comprise π–π stacking between phthaloyl residues and aromatic units of the calixarenes (mainly between nitrophenol ether units). However, the inspection of the packing of the two 1,3-alternate derivatives 27 (n = 3,4) did not give obvious reasons for the differences found in their conformation.
Conclusions
General strategies have been developed and realized for representative examples to synthesize amino derivatives of calix[4]arenes fixed in the 1,3-alternate conformation. Four amino functions can be attached either to the narrow or to the wide rim, or just to one side of the molecule. Both sides or both rims, respectively, are completely substituted by amino functions in octaamines. The use of protective groups or two independent precursors of amino groups allows a further differentiation, leading to the preparation of mixed amides. Since amino groups are readily substituted by further residues, the compounds and synthetic strategies described will be useful for the synthesis of various selectively substituted calix[4]arenes.Experimental
Materials
Solvents and all other chemicals were purchased from Acros, Aldrich and Lancaster and used without further purification. Silica gel (Merck, 0.040–0.063 mm) was used for column chromatography. 1H-NMR spectra were reordered on a Bruker AC200, AC300 and Brucker DRX400 Avance instrument (400 MHz). FD and ESI mass spectra were measured on a Finningan MAT 8230 spectrometer. Melting points are uncorrected.5,11,17,23-Tetra-t-butyl 26,28-dihydroxy 25,27-diphthalimidoethoxy-calix[4]arene 2 (n = 2) (cone)
A solution of triphenylphosphine (6,0 g, 23 mmol) in THF (40 ml) was cooled down to 0–5 °C (ice bath) and di-iso-propyl azodicarboxylate (DIAD) (23 mmol, 4.5 ml) was added dropwise under nitrogen. After 30 min a white precipitate was formed and a suspension of t-butyl calix[4]arene (5.0 g, 7.7 mmol) and N-(hydroxyethyl)-phthalimide (4.4 g, 23 mmol) in THF (100 ml) and DMF (15 ml) was slowly added. The reaction mixture was stirred 1 h at 0–5 °C and after it was warmed to rt until the suspension became clear (5 h). Then the solvent was partially removed under reduced pressure until a pale yellow precipitate was formed. It was filtered off, washed with water (50 ml) and methanol (30 ml) to give the pure product as a white powder (4.6 g, 60%). mp 297–298 °C (Found: C 77.21, H 7.17, N 12.80. C64H70N2O8 requires C 77.24, H 7.09, N 12.86).1H-NMR (300 MHz, CDCl3) δ 0.86, 1.25 (2s, 36H, t-Bu), 3.25, 4.19 (2d, 8H, 2J = 13.2 Hz, Ar–CH2–Ar), 4.24 (t, 4H, 3J = 6.6 Hz, -CH2–N), 4.42 (t, 4H, 3J = 6.9 Hz, O–CH2-), 6.69 (s, 4H, ArH), 6.77 (s, 2H, OH), 6.98 (s, 4H, ArH), 7.67–7.92 (m, 8H, Phth-H).
5,11,17,23-Tetra-t-butyl-25,27-bis-phthalimidopropoxy-26,28 -bis-cyanopropoxy-calix[4]arene 4 (1,3-alternate)
4 (1,3-alt). mp 241–243 °C, (Found: C 74.55, H 7.16, N 4.48. C74H84N4O8·2H2O requires C 74.47, H 7.43, N 4.69).
1H NMR (300 MHz, CDCl3) δ 1.20, 1.28 (2s, 36H, t-Bu), 1.32–1.42 (m, 8H, -CH2–CH2–CH2-), 1.86 (t, 4H, 3J = 7.5 Hz, -CH2–CN), 3.38 (t, 4H, 3J = 8.0 Hz, -CH2–N), 3.46, 3.52 (2t, 8H, 3J = 7.0, 6.63 Hz, O–CH2-), 3.86, 3.78 (2d, 8H, 2J = 13.8 Hz, Ar–CH2–Ar), 6.97, 7.00 (2s, 8H, ArH), 7.68–7.82 (m, 8H, Phth-H).
4 (paco). mp 233–235 °C after recrystallisation from chloroform–methanol (15 ml, 1 : 2).
1H NMR (300 MHz, CDCl3) δ 1.01, 1.25, 1.27 (3s, 18/9/9H, t-Bu),1.74 (m, 2H, -CH2–CH2–CH2-), 1.89 (m, 2H, -CH2–CH2–CH2-), 2.11 (m, 4H, -CH2–CH2–CH2-), 2.57 (t, 4H, 3J = 7.0 Hz, -CH2–CN), 3.09 (d, 2H, 2J = 12.5 Hz, Ar–CH2–Ar), 3.30 (t, 4H, 3J = 7.3 Hz, -CH2–N), 3.55–3.88 (m, 12H, O–CH2-, Ar–CH2–Ar), 4.00 (d, 2H, 2J = 12.1 Hz, Ar–CH2–Ar), 6.67, 6.86 (2d, 4H, 2J = 2.2 Hz ArH), 7.01, 7.10 (2s, 4H, ArH), 7.68–7.84 (m, 8H, Phth-H).
5,11,17,23-Tetra-t-butyl-25,27-bis-aminopropoxy-26,28-bis-cyanopropoxycalix[4]arene 5 (1,3-alternate)
A suspension of 4 (0.60 g, 0.5 mmol) in ethanol (40 ml) was refluxed with hydrazine (12 ml) for 2 h after which a clear solution was formed. The solvent was evaporated, the organic residue was dissolved in CHCl3 and washed several times with water. The organic solution was dried (MgSO4) and the solvent evaporated again. The final product was purified by crystallization from CHCl3–hexane (20 ml, 1 : 10) to give a yellow powder; yield 0.42 g, 88%; mp 303 °C.1H NMR (300 MHz, CDCl3) δ 1.18–1.37 (m, 8H, -CH2–CH2–CH2-), 1.30, 1.31 (2s, 36H, t-Bu), 1.91 (t, 4H, 3J = 7.7 Hz. CH2–CN), 2.16 (bs, 4H, -NH2), 2.50 (t, 4H, 3J = 6.9 Hz, CH2–NH2), 3.45–3.50 (2t, 8H, -CH2–O), 3.85, 3.86 (2d, 8H, 2J = 17.3 Hz, Ar–CH2–Ar), 7.01, 7.03 (2s, 8H, ArH).
5,11,17,23-Tetra-t-butyl-25,27-bis-acetamidopropoxy-26,28-bis-cyanopropoxy-calix[4]arene 6 (1,3 alternate)
Diamine 5 (0.42 g, 0.46 mmol) was dissolved in acetanhydride (5 ml) containing a few drops of triethylamine. After 12 h stirring at room temperature a white precipitate had formed which was filtered off and washed with water (30 ml). The desired diamide 6 was obtained as a white powder; (0.42 g, 93%); mp 285–286 °C.1H NMR (300 MHz, CDCl3) δ 1.13–1.41 (m, 8H, -CH2–CH2–CH2-), 1.25, 1.30 (2s, 36H, t-Bu), 1.88 (t, 4H, 3J = 7.3 Hz, CH2–CN), 1.94 (s, 6H, -CH3), 2.28–2.94 (m, 4H, -CH2–NH), 3.26 (t, 4H, 3J = 9.0 Hz, -CH2–O), 3.49 (t, 4H, 3J = 6.0 Hz, -CH2–O), 3.84 (s, 8H, Ar–CH2–Ar), 6.4 (bs, 2H, NH), 6.96, 7.01 (2s, 8H, ArH).
5,11,17,23-Tetra-t-butyl-25,27-bis-acetamidopropoxy-26,28-bis-aminobutyloxy-calix[4]arene 7 (1,3 alternate)
A suspension of 6 (0.25 g, 0.25 mmol) in ethanol–THF (1 : 4, 8 ml) and aqueous NaOH (6%, 8 ml) was stirred with Raney-Ni under hydrogen atmosphere. After the hydrogen uptake was complete the catalyst was removed by filtration through sea sand and the solvent removed under reduced pressure. The white residue was extracted with chloroform, the solution washed several times with water, dried (MgSO4) and evaporated. The residue was triturated with CH2Cl2–hexane (1 : 10, 10 ml) to give a yellow oil, yield 0.2 g, 50%.1H NMR (300 MHz, CDCl3) while most of the signals are broad signals characteristic signals for aromatic protons, -NH, and terminal methyl groups can be distinguished: δ 1.25 (bm, 48H, -CH2–CH2–CH2-, t-Bu), 1.91 (s, 6H, -CH3), 2.50–3.81 (bm, 28H, -CH2–NH2, -NH2, -CH2–O, Ar–CH2–Ar), 6.43 (bs, 2H, NH), 6.96 (bs, 8H, ArH).
5,11,17,23-Tetra-t-butyl-25,27-bis-acetamidopropoxy-26,28-bis-(p-nitrobenzoylamino)-butyloxy-calix[4]arene 8 (1,3-alternate)
A solution of p-nitrobenzoylchloride (93 mg, 0.5 mmol) and triethylamine (0.07 ml, 0.5 mmol) were added to a stirred solution of 7 (0.247 g, 0.25 mmol) in chloroform (30 ml). After 12 h at room temperature the solvent was removed under reduced pressure, and the residue was purified by column chromatography (chloroform), to give a yellow powder; yield 0.15 g, 50%; mp 123 °C.1H NMR (300 MHz, CDCl3) δ 1.13–1.41 (m, 8H, -CH2–CH2–CH2-), 1.25, 1.30 (2s, 36H, t-Bu), 1.46 (m, 4H, -CH2–CH2–CH2-), 1.92 (s, 6H, -CH3), 2.28–2.94 (m, 4H, CH2–CH2–CH2), 3.19–3.33 (m, 12H, -CH2–O, -CH2–N), 3.82 (s, 8H, Ar–CH2–Ar), 6.2 (bs, 2H, NH), 6.89 (bs, 2H, NH), 6.94, 6.97 (2s, 8H, ArH), 7.94, 8.23 (2d, 8H, 4J = 8.8 Hz, ArH).
5,11,17,23-Tetra-t-butyl-25-phthalimidopropoxy-27-phthaloylaminopropoxy-26,28-bis-aminobutyloxy-calix[4]arene 9 (1,3 alternate)
The dinitrile 4 (0.63 g, 0.54 mmol) was dissolved in ethanol–THF (1 : 4, 10 ml). A suspension of Pd/C (300 mg) in NaOH solution (10 ml, 6%) was added, and the reaction mixture was stirred under hydrogen atmosphere at 50 °C for 3 h. After the catalyst was filtered off, the solvents were removed under reduced pressure and the residue was treated with a mixture of CH2Cl2 (25 ml) and water (50 ml). The aqueous phase was extracted with CH2Cl2 (3 × 15 ml) and the combined organic phase was dried (MgSO4) and evaporated. The diamine 9 was obtained as a white powder, yield 0.39 g, 62%; mp 152–154 °C.1H NMR (300 MHz, DMSO-d6) δ 1.13, 1.27 (2s + m, 48H, t-Bu, -CH2–CH2–CH2-), 1.97 (t, 4H, 3J = 7.3 Hz, -CH2–NH2), 3.05 (bs, 4H, N–CH2-), 3.40 (bm, 12H, -O–CH2-, -NH2), 3.78, 3.95 (2d, 8H, 2J = 16.5 Hz, Ar–CH2–Ar), 7.03, 7.09 (2s, 8H, ArH), 7.28–7.35 (m, 4H, Phth-H), 7.50, 7.62 (2d, 4H, 2J = 7.0 Hz, PhthCOOH-H), 10.29 (bs, 1H, -COOH).
5,17-Di-t-butyl-11,23-dinitro-25,27-diallyloxy-26,28-dipropoxy-calix[4]arene 11 (1,3-alternate) and 11 (partial cone)
A stirred suspension of dinitro-calixarene 10 (2.0 g, 2.8 mmol) and Cs2CO3 (13.6 g, 42 mmol) in dry DMF (50 ml) was heated to 40 °C under nitrogen. After 1 h allylbromide (3.6 ml, 42 mmol) was added and the reaction was continued for 7 days. The DMF was removed under reduced pressure and the residue was treated with chloroform (25 ml) and water (75 ml). The organic phase was washed with water (2 × 75 ml), dried (MgSO4) and the solvent was evaporated to give a yellow oil. Analysis by TLC showed the presence of two compounds, which were separated and purified by column chromatography (CH2Cl2–hexane 2 : 3) and identified by NMR as the 1,3-alternate and the partial cone isomers.
1H-NMR (300 MHz, CDCl3)
δ 0.79 (t, 6H, 3J
= 7.7 Hz, -CH2–CH3), 1.21 (s, 18H, t-Bu), 1.45 (m, 4H, -CH2–CH2–CH3), 3.56 (t, 4H, 3J
= 7.7 Hz, O–CH2), 3.68, 3.81 (2d, 8H, 2J
= 15.0 Hz, Ar–CH2–Ar), 3.94 (d, 4H, 4J
= 5.1 Hz, CH2CH–CH2–O-), 5.05 (m, 4H, CH2CH-), 5.70 (m, 2H, CH2CH-), 6.96, 7.95 (2s, 8H, ArH).
1H-NMR (300 MHz, CDCl3)
δ 0.98 (m, 24H, t-Bu, -CH2–CH3), 1.89 (m, 4H, -CH2–CH3), 3.19 (d, 2H, 2J
= 12.9 Hz, Ar–CH2–Ar), 3.52 (m, 2H, O–CH2-), 3.64 (d, 2H, 2J
= 13.6 Hz, Ar–CH2–Ar), 3.77 (m, 2H, O–CH2), 3.83 (d, 2H, 2J
= 13.6 Hz, Ar–CH2–Ar), 4.08 (d, 2H, 2J
= 13.2 Hz, Ar–CH2–Ar), 4.16, 4.31 (2d, 4H, 4J
= 5.9 Hz, -O–CH2–CHCH2), 4.90, 5.29 (2m, 4H, -CHCH2), 5.65, 6.07 (2m, 2H, -CHCH2), 6.50, 6.87 (2d, 4H, 4J
= 2.2, 2.6 Hz, ArH), 8.02, 8.23 (2s, 4H, ArH)
5,17-Di-t-butyl-11,23-diamino-25,26,27,28-tetrapropoxy calix[4]arene 12 (1,3-alternate)
Raney-Ni (0.5 g) was added to a solution of the dinitro compound 11 (0.7 g, 0.88 mmol) in THF (20 ml) and the suspension was stirred under hydrogen atmosphere at rt. After the hydrogen uptake was complete, the catalyst was filtered off and the solvent was evaporated. The residue was dissolved in chloroform (10 ml) and reprecipitated by hexane (25 ml) to give the pure diamine as a pink powder, yield 0.28 g, 45%; mp 198 °C.1H-NMR (300 MHz, CDCl3) δ 0.68, 0.76 (2t, 12H, 3J = 7.3, 7.7 Hz, -CH2–CH3), 1.13–1.38 (m, 26H, t-Bu, -CH2–CH3), 3.23–3.71 (m, 12H, O–CH2-, -NH2), 3.68, 3.80 (2d, 8H, 2J = 15.4 Hz, Ar–CH2–Ar), 6.64, 6.91 (2s, 8H, ArH).
5,17-Di-t-butyl-11,23-diphthalimido-25,26,27,28-tetrapropoxy-calix[4]arene 13 (1,3-alternate)
A solution of diamine 12 (0.7 g, 1.3 mmol), phthalic acid anhydride (0.45 g, 3 mmol) and triethylamine (1 ml) in toluene (25 ml) was refluxed for 12 h. The solvent was removed under reduced pressure to give a red sticky mass, which was dissolved in chloroform (5 ml) and passed through a silica column (chloroform) to remove the excess anhydride. 13 was isolated as a pink powder, yield 0.53 g, 42%; mp 384–385 °C; (Found: C 74.63, H 7.10, N 2.66 C64H70N2O8 requires C 77.24, H 7.09, N 2.81).1H-NMR (300MHz, CDCl3) δ 0.58–0.67 (m, 12H, -CH2–CH3), 1.04 (m, 4H, CH3–CH2-), 1.28 (m, 22H, t-Bu, -CH2–CH3), 3.39 (m, 8H, O–CH2-), 3.84, 3.87 (2d, 8H, 2J = 16.5 Hz, Ar–CH2–Ar), 6.97, 7.12 (2s, 8H, ArH), 7.70 (m, 8H, Phth-H).
5-Nitro-17-t-butyl-11,23-diphthalimido-25,27,26,28-tetrapropoxy-calix[4]arene 14 (1,3-alternate)
Glacial acetic acid (3 ml) and fuming nitric acid (0.22 ml) were added to a vigorously stirred, clear solution of diphthalimido 13 (0.5 g, 0.5 mmol) in dry CH2Cl2 (50 ml) at rt. After a certain time (around 7–15 min) when the color of the solution had changed from black-indigo to yellow, water (50 ml) was added and the reaction mixture was stirred for further 30 min. The organic solution was washed several times with water, dried (MgSO4) and the solvent was removed under reduced pressure. The residue was purified by precipitation from CH2Cl2–CH3OH (20 ml, 1 : 1) to give a yellow powder, yield 0.38 g, 78%; mp 313–315 °C.1H NMR (300 MHz, CDCl3) δ 0.80, 0.88 (2t, 6H, 3J = 7.3 Hz, -CH2–CH3), 1.03 (t, 6H, 3J = 7.3 Hz, -CH2–CH3), 1.17 (s, 9H, t-Bu), 1.66–1.91 (m, 8H, -CH2–CH2–CH3), 3.55 (d, 2H, 2J = 14.0 Hz, Ar–CH2–Ar), 3.61–3.83 (m, 12H, Ar–CH2–Ar, O–CH2-), 3.81 (t, 2H, 3J = 7.7 Hz, O–CH2-), 6.98 (s, 2H, ArH), 7.15, 7.20 (2d, 4H, 4J = 2.6 Hz, ArH), 7.47–7.57 (m, 8H, Phth-H), 7.94 (s, 2H, ArH).
5,17-Dinitro-11,23-diphthalimido-25,27,26,28-tetrapropoxy-calix[4]arene 15 (1,3alternate)
Glacial acetic acid (0.32 ml) and fuming nitric acid (0.02 ml) were added to a vigorously stirred, clear solution of diphthalimide 13 (0.03 g, 0.03 mmol) in dry CH2Cl2 (6 ml) at rt. The reaction was monitored by tlc. After 5 h the color of the solution had changed from black-indigo to yellow. Water (50 ml) was added and the reaction mixture was stirred for further 30 min. The organic solution was washed twice with water (2 × 10 ml), dried (MgSO4) and the solvent was removed under reduced pressure. The residue was purified by precipitation from CH2Cl2–CH3OH (20 ml, 1 : 1) to give the dinitro compound 15 as yellow powder, yield 0.013 g, 45% (Found: C 69.10, H 5.87, N 5.41. C56H52N4O12 requires C 69.12, H 5.39, N 5.76). mp. 338–340 °C.1H NMR (300 MHz, CDCl3) δ 0.89, 1.07 (2t, 12H, 3J = 7.3 Hz, -CH2–CH3), 1.82 (m, 8H, -CH2–CH2–CH3), 3.68 (s, 8H, Ar–CH2–Ar), 3.74 (m, 8H, O–CH2-), 7.19 (s, 4H, ArH), 7.47–7.57 (m, 8H, Phth-H), 7.96 (s, 4H, ArH).
Dinitro compound 15 was obtained also from the mononitro compound 14 using similar conditions. Glacial acetic acid (1.97 ml) and fuming nitric acid (0.11 ml) were added to a stirred solution of 14 (0.37 g, 0.375 mmol) in CH2Cl2 (25 ml). The reaction was followed by tlc and stopped (the color of the solution had changed from black to yellow) by adding water (50 ml). The organic solution was washed several times with water, dried (MgSO4) and the solvent was removed under reduced pressure. Precipitation from CH2Cl2–CH3OH (20 ml, 1 : 1) gave 15 as yellow powder (0.3 g, 83%).
5,17-Dinitro-11,23-diamino-25,27,26,28-tetrapropoxy-calix[4]arene 16 (1,3-alternate)
A solution of 15 (0.2 g, 0.2 mmol) in EtOH (10 ml) was refluxed with hydrazine (3 ml). After 2 h the solvent was removed under reduced pressure. The residue was dissolved in CHCl3 (10 ml), washed with water (2 × 25 ml), dried (MgSO4) and the solvent was evaporated. The formed powder was dissolved in chloroform (5 ml) and precipitated with hexane (15 ml) to give the pure diamine 15 as a yellow powder, yield 0.11 g, 79%; mp 216 °C.1H NMR (300 MHz, CDCl3) δ 0.69, 0.78 (2t, 12H, 3J = 7.3 Hz, -CH2–CH3), 1.30–1.43 (m, 8H, -CH2–CH2–CH3), 2.69 (bs, 4H, -NH2), 3.27, 3.35 (2t, 8H, 3J = 7.3 Hz, O–CH2), 3.65, 3.66 (2d, 8H, 2J = 15.4 Hz, Ar–CH2–Ar), 6.4, 6.93 (2s, 8H, ArH).
5,17-Diamino-11,23-diphthalimido-25,27,26,28-tetrapropoxy-calix[4]arene 17 (1,3-alternate)
The dinitro compound 15 (0.11 g, 0.11 mmol) was dissolved in toluene (2 ml) and THF (8 ml) and hydrogenated under atmospheric pressure in the presence of Raney-Ni at rt. After the hydrogen uptake was complete, the catalyst was filtered off and the solvent was removed under reduced pressure. The residue was dissolved in chloroform (5 ml) and reprecipitated with hexane (15 ml) to give the pure diamine 17 as a yellow powder, yield; 0.05 g, 50%; mp. 278–280 °C. FD-MS, (M+ + H) m/z = 914.4.1H NMR (400 MHz, DMSO-d6) δ: 0.52, 0.71 (2t, 12H, 3J = 7.4 Hz, -CH2–CH3), 1.17–1.24 (m, 8H, -CH2–CH2–CH3), 3.12 (bt, 4H, O–CH2-), 3.20 (bs, 4H, O–CH2-), 3.64, 3.72 (2d, 8H, 2J = 15.6 Hz, Ar–CH2–Ar), 4.33 (bs, 4H, -NH2), 6.28, 7.07 (2s, 8H, ArH), 7.88, 7.91 (2bs, 8H, Phth-H).
Mono- and di-protection by Boc; calix[4]arenes 19 and 20 (1,3-alternate)
A solution of Boc-anhydride (0.59 g, 2.7 mmol) in dichloromethane (10 ml) was added dropwise to a stirred solution of the tetraamine 18 (1.0 g, 1.5 mmol) in dichloromethane (100 ml). After 24 h of stirring at ambient temperature the solvent was evaporated. The mono- and di-Boc protected compounds 19 and 20 were isolated by column chromatography (EtOAc–hexane = 1 : 1) in 23% (0.25 g), 58% (0.65 g) respectively.Mono-Boc derivative 19: pink powder, mp 138–140 °C (Found: C 71.34, H 7.90, N 6.53 C44H58N4O6 requires C 71.52, H 7.91, N 7.58)
1H NMR (300 MHz, CDCl3) δ 0.87–1.24 (m, 12H, -CH2–CH3), 1.50 (s, 9H, t-Bu), 1.86 (m, 8H, -CH2–CH2–CH3), 3.11–3.6 (m, 22H, Ar–CH2–Ar, O–CH2-, Ar–NH2), 6.34, 6.44 (2s, 4H, ArH), 6.98 (bs, 4H, ArH).
Di-Boc derivative 20: yellow powder, mp 251 °C (Found: C 70.21, H 7.71, N 6.56 C49H66N4O8 requires C 70.14, H 7.93, N 6.68)
1H NMR (400 MHz, C6D6) δ 0.99 (t, 12H, 3J = 7.3 Hz, -CH2–CH3), 1.66 (s, 18H, t-Bu), 1.71 (m, 8H, -CH2–CH2–CH3), 2.94 (bs, 4H, -NH2), 3.42–3.55 (m, 16H, Ar–CH2–Ar, O–CH2-), 6.45, 6.47 (2d, 4H, 4J = 2.9 Hz, ArH), 7.23, 7.27 (2d, 4H, 4J = 2.4 Hz, ArH), 7.5 (bs, 2H, -NH).
1H NMR (400 MHz, CDCl3) δ 1.10–1.29 (bm, 12H, -CH2–CH3), 1.48 (s, 18H, t-Bu), 1.87 (m, 8H, -CH2–CH2–CH3), 3.28 (s, 2H, Ar–CH2–Ar), 3.32 (d, 2H, 2J = 12.5 Hz, Ar–CH2–Ar), 3.37 (d, 2H, 2J = 13.0 Hz, Ar–CH2–Ar), 3.41 (s, 2H, Ar–CH2–Ar), 3.57–3.65 (m, 12H, O–CH2-, -NH2), 6.44, 6.93, 6.95 (3s, 4/2/2H, ArH), 7.82 (bs, 2H, -NH).
Tri- and tetra protection by Boc; calix[4]arenes 22 and 23 (1,3-alternate)
The tri-Boc compound was prepared in the same way, starting from a solution of tetraamino-calixarene 18 (1.0 g, 1.5 mmol) in CH2Cl2 (100 ml) and Boc-anhydride (0.948 g, 4.35 mmol). Isolation and purification by column chromatography (EtOAc–hexane = 1 : 1) gave pure tri-Boc derivative 22 (0.8 g, 60%). mp 242–244 °C (Found: C 66.90, H 7.68, N 5.05 C54H74N4O10·0.5 CH2Cl2 requires C 66.68, H 7.70, N 5.71)1H NMR (300 MHz, CDCl3) δ 0.98, 1.12 (2t, 12H, 3J = 7.3 Hz. -CH2–CH3), 1.50, 1.52 (2s, 18/9H, t-Bu), 1.59, 1.73 (2m, 4H, -CH2–CH2–CH3), 1.91 (m, 4H, -CH2–CH2–CH3), 3.33–3.69 (m, 18H, O–CH2, Ar–CH2–Ar, NH2), 6.14, 6.56 (2s, 1/2H, NH), 6.95, 6.98 (2s, 6/2H, ArH).
The tetra-Boc derivative 23 is formed as side product, but can be obtained in nearly quantitative yield, using 20–30% excess of Boc-anhydride. E.g., starting from a solution of tetraamino-calixarene 18 (1.0 g, 1.5 mmol) in CH2Cl2 (100 ml), pure tetra-Boc compound 23 (1.35 g, 90%) was obtained after purification by reprecipitation from CHCl3–hexane (1 : 5) as a white powder, mp 268–270 °C.
1H NMR (300 MHz, CDCl3) δ 0.80 (t, 12H, 3J = 7.3 Hz, -CH2–CH3), 1.44–1.53 (m, 8H, -CH2–CH2–CH3), 1.50 (s, 36H, t-Bu) 3.41 (t, 8H, 3J = 7.3 Hz, -CH2–CH2–CH3), 3.60 (s, 8H, Ar–CH2–Ar), 6.18 (s, 4H, -NH), 6.99 (s, 8H, ArH).
5,11-Bis-t-butyloxycarbonylamino-17,23-bis-acetamido-25,26,27,28-tetrapropoxy-calix[4]arene 24 (1,3-alternate)
Di-Boc compound 20 (70 mg, 0.084 mmol) was stirred in acetic anhydride (5 ml) with a few drops of triethylamine at rt. After 12 h the reaction mixture was poured on ice and the desired diacetamide precipitated as white solid to give 79 mg of pure 24 (85% yield). mp 262–263 °C. (Found: C 68.58, H 7.86, N 5.71. C54H72N4O10·0.5H2O requires C 68.55, H 7.78, N 5.92)1H NMR (300 MHz, DMSO) δ 0.62 (bs, 12H, -CH2–CH3), 1.26 (bm, 26H, t-Bu, -CH2–CH2–CH3), 1.97 (s, 6H, -CO–CH3), 3.06 (bt, 8H, O–CH2-), 3.58 (bs, 8H, Ar–CH2–Ar), 7.16, 7.25 (2s, 8H, ArH), 8.93, 9.51 (2s, 4H, -NH-).
5,11-Bis-p-nitrobenzoylamino-17,23-bis-acetamido-25,26,27,28-tetrapropoxy-calix[4]arene 25 (1,3-alternate)
To a stirred solution of 24 (67 mg, 0.07 mmol) in CH2Cl2 (7 ml) was added TFA (7 ml). The reaction mixture was stirred at rt for 2 h, then diluted with toluene (15 ml). The solvents were evaporated, the remaining yellow oil was dissolved in chloroform (15 ml) and triethylamine (0.05 ml, 0.35 mmol) and p-nitro-benzoyl chloride (27 mg, 0.15 mmol) were added with stirring. After 12 h the solvent was removed under reduced pressure. The dry residue was purified by column chromatography (chloroform) to obtain 25 as an orange powder (50 mg, 70% yield). mp 223 °C. FD-MS, (M+ + H) m/z = 1036.3.1H NMR (300 MHz, DMSO) δ 0.64 (m, 12H, -CH2–CH3), 1.31 (bm, 8H, -CH2–CH2–CH3), 1.95 (s, 6H, -CO–CH3), 3.2 (bt, 8H, O–CH2-), 3.66 (m, 8H, Ar–CH2–Ar), 7.37, 7.57 (2d, 8H, 4J = 3.7, 3.3 Hz, ArH), 8.11, 8.37 (2d, 8H, 4J = 8.8, 8.4 Hz, ArH), 9.55, 10.26 (2s, 4H, -NH-).
5,17-Di-t-butyl-11,23-dinitro-26,28-diphthalimidoethoxy-calix[4]arene 26 (n = 2)
Nitric acid (65%, 7.5 ml) was added with stirring to a cold (0 °C) solution of 2 (n = 2) (2.5 g, 2.5 mmol) in dry CH2Cl2 (75 ml). After 10 min the color changed from black-indigo to yellow and the reaction was complete. Water was added (100 ml) and the mixture was stirred for 30 min. After phase separation the organic phase was washed with water (3 × 100 ml) until a neutral pH was reached, dried (MgSO4) and the solvent was evaporated. The residue was dissolved in chloroform (10 ml) and the pure product was precipitated with methanol (25 ml) as yellow powder (1.6 g, 67%), m.p 298–300 °C.1H-NMR (300MHz, CDCl3) δ 0.9 (s, 18H, t-Bu), 3.41, 4.15 (2d, 8H, 2J = 13.2 Hz, Ar–CH2–Ar), 4.29 (t, 4H, 3J = 6.2 Hz, -CH2–N), 4.47 (t, 4H, 3J = 6.3 Hz, O–CH2-), 6.73 (s, 4H, ArH), 7.69–7.93 (m, 8H, Phth-H), 7.98 (s, 4H, ArH), 8.20 (s, 2H, OH).
5,17-Di-t-butyl-11,23-dinitro-26,28-diphthalimidoproxy-calix[4]arene 26 (n = 3)
Reaction and work up as described above. 2.0 g (1.95 mmol) 2 (n = 3) finally gave 1.5 g, (75%) of pure product as yellow powder; m.p 253.5–254 °C.1H NMR (200 MHz, CDCl3) δ 1.00 (s, 18H, t-Bu), 2.45 (m, 4H, -CH2–CH2–CH2), 3.48 (d, 4H 2J = 13.8 Hz, Ar–CH2–Ar), 4.11 (m, 8H, O–CH2-, -CH2–N), 4.30 (d, 4H, 2J = 13.8 Hz, Ar–CH2–Ar), 6.88 (s, 4H, ArH), 7.62–7.78 (m, 8H, Phth-H), 8.04 (s, 4H, ArH), 8.94 (s, 2H, -OH).
5,17-Di-t-butyl-11,23-dinitro-26,28-diphthalimidobutoxy-calix[4]arene 26 (n = 4)
Reaction and work up as described above. 3.0 g (2.85 mmol) 2 (n = 4) finally gave 2.0 g, (70%) of pure product as yellow powder; m.p 229–231 °C; (Found: C 68.82, H 6.30, N 5.35 C60H60N4O10·H2O requires C 68.82, H 5.97, N 5.35).1H NMR (300 MHz, CDCl3) δ 1.01 (s, 18H, t-Bu), 2.10 (bs, 8H, -CH2–CH2–CH2), 3.44 (d, 4H 2J = 13.2 Hz, Ar-CH2–Ar), 3.89 (bt, 4H, -CH2–N), 4.06 (bt, 4H, O–CH2-) 4.21 (d, 4H, 2J = 13.2 Hz, Ar-CH2–Ar), 6.88 (s, 4H, ArH), 7.66–7.82 (m, 8H, Phth-H), 8.00 (s, 4H, ArH), 9.07 (s, 2H, OH).
5,17-Di-t-butyl-11,23-dinitro-26,28-diphthalimidopropoxy-25,27-diallyloxy-calix[4]arene 27 (n = 3) (1,3-alternate, partial cone, cone)
A stirred suspension of dinitro calixarene 26 (n = 3) (0.5 g, 0.5 mmol) and Cs2CO3 (1.3 g, 4 mmol) in dry DMF (12.5 ml) was heated to 40 °C under nitrogen. After 1 h allylbromide (0.34 ml, 4 mmol) was added and the reaction mixture was kept under these conditions for 7 days. The DMF was removed at reduced pressure and the residue was treated with chloroform (15 ml) and water (50 ml). The organic phase was washed twice with water (2 × 50 ml), dried (MgSO4) and evaporated. TLC analysis of the residue showed the presence of two isomers (1,3-alt and paco). After column chromatography (dichloromethane–ethylacetate, 95 : 5) three different fractions were isolated which were identified as 1,3-alternate, partial cone and cone conformers. Recrystallization from chloroform–methanol (10 ml, 1:4) gave 27 (n = 3, 1,3-alt) as colorless crystals (0.1 g, 19%), 27 (n = 3, paco) as yellow powder (0.1 g, 19%) and 27a (n = 3, cone) as colorless crystals (10 mg, 2%).
1H-NMR (400 MHz, CDCl3)
δ 1.20 (s, 18H, t-Bu), 1.88 (m, 4H, -CH2–CH2–CH2-), 3.69 (m, 8H, O–CH2-, -CH2–N), 3.69, 3.81 (2d, 8H, 2J
= 15.6 Hz, Ar–CH2–Ar), 3.97 (d, 4H, 2J
= 9.7 Hz, CH2CH–CH2–O-), 5.03 (m, 4H, CH2CH-), 5.70 (m, 2H, CH2CH-), 6.95 (s, 4H, ArH), 7.74 (m, 8H, Phth-H), 7.99 (s, 4H, ArH).
1H-NMR (400 MHz, CDCl3)
δ 1.00, 1.24 (2s, 18H, t-Bu), 2.21 (m, 4H, -CH2–CH2–CH2-), 3.19, 3.68 (2d, 4H, 2J
= 12.9, 13.7 Hz, Ar–CH2–Ar), 3.88 (m, 8H, O–CH2-, -CH2–N), 3.90, 4.07 (2d, 4H, 2J
= 13.3, 12.9 Hz, Ar–CH2–Ar), 4.19, 4.24 (2d, 4H, 2J
= 6.3 Hz, -O–CH2–CHCH2), 4.89, 5.28 (2m, 4H, CH2CH-), 5.54, 6.02 (2m, 2H, CH2CH-), 6.35, 6.87 (2d, 4H, 4J
= 1.9 Hz, ArH), 7.83 (m, 8H, Phth-H), 8.05, 8.35 (2s, 4H, ArH).
1H-NMR (200 MHz, CDCl3)
δ 1.28 (s, 18H, t-Bu), 2.28 (m, 4H, -CH2–CH2–CH2-), 3.19 (d, 4H, 2J
= 13.6 Hz, Ar–CH2–Ar), 3.81 (t, 4H, 3J
= 7.3 Hz, -CH2–N), 4.07 (t, 4H, 3J
= 7.5 Hz, -CH2–O-), 4.38 (d, 4H, 2J
= 5.9 Hz, -O–CH2–CHCH2), 4.41 (d, 4H, 2J
= 13.6 Hz, Ar–CH2–Ar), 5.11 (m, 4H, CH2CH-), 6.13 (m, 2H, CH2CH-), 7.03, 7.12 (2s, 8H, ArH), 7.78 (m, 8H, Phth-H).
5,17-Di-t-butyl-11,23-dinitro-26,28-diphthalimidobutyloxy-25,27-diallyloxy-calix[4]arene 27 (n = 4) (1,3-alternate)
Compound 27 (n = 4) was obtained in an analogous way starting from the dinitro compound 26 (n = 4) (1.6 g, 1.55 mmol) in dry DMF (40 ml) and Cs2CO3 (5 g, 15.5 mmol). The desired 1,3-alternate isomer of 27 (n = 4) was isolated by crystallization from chloroform–methanol (25 ml, 1 : 4) as colorless crystals (0.9 g, 53%). mp 199 °C (Found: C 71.30, H 6.41, N 4.96. C66H68N4O12 requires C 71.46, H 6.18, N 5.05).
1H-NMR (300 MHz, CDCl3)
δ 1.20 (s, 18H, t-Bu), 1.55, 1.72 (2m, 8H, -CH2–CH2–CH2-), 3.69 (m, 8H, O–CH2-, -CH2–N), 3.62, 3.66 (2d, 8H, J
= 15.6 Hz, Ar–CH2–Ar), 3.97 (d, 4H, J
= 5.1 Hz, CH2CH–CH2–O-), 5.12 (m, 4H, CH2CH-), 5.74 (m, 2H, CH2CH-), 6.95 (s, 4H, ArH), 7.67 (m, 8H, Phth-H), 7.99 (s, 4H, ArH).
5,17-Di-t-butyl-11,23-diamino-26,28-diphthalimidopropoxy-25,27-dipropoxy-calix[4]arene 28 (n = 3) (1,3-alternate)
A clear solution of the dinitrocompound 27 (n = 3) (0.6 g, 0.55 mmol) in THF (20 ml) was hydrogenated under atmospheric pressure in the presence of Raney-Ni at rt. After the hydrogen uptake was complete the catalyst was filtered off and the solvent was evaporated. The dry residue was dissolved in chloroform (5 ml) and the diamine was reprecipitated with hexane (10 ml) as a yellow powder (0.4 g, 80%). mp 133 °C. FD-MS, (M+ + H) m/z = 1026.3.1H-NMR (200 MHz, CDCl3) δ 0.58 (t, 6H, 3J = 7.3 Hz, CH3–CH2-), 1.04–1.18 (m, 22H, CH3–CH2–CH2-, t–Bu), 1.55 (m, 4H, -CH2–CH2–CH2-), 3.18 (bt, 4H, 3J = 6.8 Hz, -CH2–N), 3.33–3.3.45 (bm, 12H, O–CH2-, -NH2), 3.57, 3.68 (2d, 8H, 2J = 15.5 Hz Ar–CH2–Ar), 6.38 (s, 4H, ArH), 6.88 (s, 4H, ArH), 7.59–7.76 (bm, 8H, Phth-H)
5,17-Di-t-butyl-11,23-diamino-26,28-diphthalimidobutoxy-25,27-dipropoxy-calix[4]arene 28 (n = 4) (1,3-alternate)
The diamine 28 (n = 4) was obtained as described above for the analogous compound 28 (n = 3) starting from a clear solution of 27 (n = 4) (0.2 g, 0.18 mmol) in THF (15 ml). Analogous work up gave a beige powder (0.16 g, 90%); mp 129 °C.1H-NMR (300MHz, CDCl3) δ 0.65 (t, 6H, 3J = 7.7 Hz, CH3–CH2-), 1.03–1.46 (m, 26H, -CH2-, t-Bu), 1.81 (m, 4H, -CH2-), 3.18–3.69 (m, 24H, -CH2–N, O–CH2-, -NH2, Ar–CH2–Ar), 6.40, 6.92 (2s, 8H, ArH), 7.67–7.80 (bm, 8H, Phth-H).
5,17-Di-t-butyl-11,23-dinitro-26,28-diaminopropoxy-25,27-dipropoxy-calix[4]arene 30 (n = 3) (1,3-alternate)
A solution of 27 (n = 3) (0.58 g, 0.53 mmol) in EtOH (60 ml) was refluxed with hydrazine hydrate (4.5 ml) for 3 h. Then solvent was evaporated til dry and the residue was dissolved in CH2Cl2 (10 ml) and washed with water (2 × 20 ml). The organic phase was dried (MgSO4) and evaporated again. The resulting powder was dissolved in CH2Cl2 (5 ml) and the desired diamine was reprecipitated with hexane (10 ml) as a yellow powder (0.25 g, 57%), mp 155–157 °C. (Found: C 67.99, H 7.48, N 6.29. C48H66N4O8·0.25CHCl3 requires C 67.63, H 7.79, N 6.54) FD-MS, (M+) m/z = 825.05.1H-NMR (300 MHz, CDCl3) δ 0.79 (t, 6H, 3J = 7.3 Hz, -CH2–CH3), 1.23 (s, 18H, t-Bu) 1.39–1.58 (m, 8H, -CH2-), 2.64 (t, 4H,3J = 7.0 Hz -CH2–NH2), 3.50 (t, 4H, 3J = 7.3 Hz, O–CH2-) 3.66–3.73 (m, 8H, O–CH2-, Ar–CH2–Ar), 3.84 (d, 4H, 2J = 15.0 Hz Ar–CH2–Ar), 6.89, 7.98 (2s, 8H, ArH).
5,17,11,23-Tetra-t-butyl-25,27,26,28-tetra-phthalimidopropoxy-calix[4]arene 32 (n = m = 3) (1,3-alternate)
32 (n = m = 3, 1,3-alt). mp 255–257 °C (Found: C 75.60, H 7.02, N 4.05. C88H92N4O12 requires C 75.60, H 6.63, N 4.01)
1H-NMR (200 MHz, CDCl3) δ 1.15 (s, 36H, t-Bu), 1.67 (m, 8H, -CH2–CH2–CH2-), 3.44 (t, 8H, 3J = 7.8 Hz, -CH2–N), 3.62 (t, 8H, 3J = 6.8 Hz, -CH2–O), 3.67 (s, 8H, Ar–CH2–Ar), 6.92 (s, 8H, ArH), 7.65–7.82 (m, 16H, Phth-H).
32 (n = m = 3, 1,2-alt). mp 197–199 °C
1H-NMR (200 MHz, CDCl3) δ 1.05 (m, 4H, -CH2–CH2–CH2-) 1.26 (s, 36H, t-Bu), 1.60 (m, 4H, -CH2–CH2–CH2-), 3.00 (d, 2H, 2J = 12.1 Hz, Ar–CH2–Ar), 3.54–3.87 (m, 16H, -CH2–N, -CH2–O), 3.88 (s, 4H, Ar–CH2–Ar), 4.07 (d, 2H, 2J = 12.1 Hz, Ar–CH2–Ar), 7.05, 7.09 (2d, 8H, 2J = 2.2 Hz, ArH), 7.51–7.66 (m, 16H, Phth-H).
32 (n = m = 3, paco). mp 278–280 °C
1H-NMR (200 MHz, CDCl3) δ 0.95, 1.25, 1.28 (3s, 18/9/9H, t-Bu), 1.87, 2.09, 2.22 (3m, 2/2/4H, -CH2–CH2–CH2-), 2.99 (d, 2H, 2J = 8.5 Hz Ar–CH2–Ar), 3.54–3.87(m, 20H, -CH2–N, -CH2–O, Ar–CH2–Ar), 4.01 (d, 2H, 2J = 8.3 Hz, Ar–CH2–Ar), 6.53, 6.78 (2d, 4H, 4J = 1.4, 2.5 Hz, ArH), 7.02, 7.17 (2s, 4H, ArH), 7.52–7.71 (m, 16H, Phth-H).
5,17,11,23-Tetra-t-butyl-25,27-diphthalimidobutyloxy-26,28-diphthalimidopropoxy-calix[4]arene 32 (n = 4, m = 3) (1,3-alternate)
Compound 32 (n = 4, m = 3) was obtained as described for 32 (n = m = 3), starting from 2 (n = 4) (0.5 g, 0.47 mmol), N-(3-bromopropyl)-phthalimide (1,0 g, 3.76 mmol) and Cs2CO3 (1.2 g, 3.76 mmol) in dry DMF (15 ml). The desired 1,3-alternate isomer of 32 (n = 4, m = 3) was isolated by crystallization from chloroform–methanol (20 ml, 1 : 4) as white powder (0.4 g, 60%) mp 204 °C.1H-NMR (300 MHz, CDCl3) δ 1.18, 1.19 (2s, 36H, t-Bu), 1.32, 1.71 (2m, 16H, -CH2–CH2–CH2-), 3.38–3.66 (m, 20H, O–CH2-, -CH2–N Ar–CH2–Ar), 3.70 (d, 4H, 2J = 16.53 Hz Ar–CH2–Ar), 6.91, 6.96 (2s, 8H, ArH), 7.66–7.83 (m, 16H, Phth-H).
1H-NMR (400 MHz, C6D6) δ 1.39, 1.41 (2s, 36H, t-Bu), 1.59, 1.86 (2m, 8H, -CH2–CH2–CH2-), 3.38 (t, 4H 3J = 7.4 Hz, -CH2–N), 3.57–3.66 (m, 12H, O–CH2-, -CH2–N), 3.87, 3.92 (2d, 8H, 2J = 15.6 Hz Ar–CH2–Ar), 6.86–6.88 (m, 8H, Phth-H), 7.11, 7.26 (2s, 8H, ArH), 7.46–7.49 (m, 8H, Phth-H).
5,11,17,23-Tetra-nitro-25,26,27,28-tetraphthalimidopropoxy-calix[4]arene 33 (1,3-alternate)
Glacial acetic acid (13.5 ml) and fuming nitric acid (8 ml) were added to a vigorously stirred suspension of tetraphthalimido compound 32 (n = m = 3) in dry CH2Cl2 (50 ml) at rt. The reaction was complete when the color of the solution had changed from black-indigo to yellow (about 12 h). Then water (100 ml) was added and the mixture was stirred for 30 min. The organic phase was washed several times with water (3 × 100 ml) and dried (MgSO4). The solvent was removed under reduced pressure and the residue was purified by slow crystallization from CH2Cl2–CH3OH (40 ml, 1 : 2) which gave yellow crystals (1.12 g, 71%). mp. 387–389 °C (Found: C 62.85, H 4.29, N 8.16. C72H56N8O20·H2O requires C 63.06, H 4.26, N 8.17).1H NMR (200 MHz, CDCl3) δ 2.26 (m, 8H, -CH2–CH2–CH2-), 3.84 (s, 8H, Ar–CH2–Ar), 3.95 (m, 16H, -CH2–N, O–CH2-), 7.68–7.86 (m, 16H, Phth-H), 8.06 (s, 8H, ArH).
5,11,17,23-Tetra-acetamido-25,26,27,28-tetraphthalimidopropoxy-calix[4]arene 34 (1,3-alternate)
A clear solution of the tetranitro compound 33 (n = m = 3) (0.75 g, 0.55 mmol) in THF–DMF (50 ml, 1 : 1) at 50 °C was hydrogenated under atmospheric pressure in the presence of Raney-Ni. After the hydrogen uptake was complete the catalyst was filtered off and the solvent was evaporated. The dry residue was dissolved in chloroform (10 ml) and the tetraamine 34 (R = H) was reprecipitated with hexane (20 ml) as pale yellow powder (0.53 g, 85%) which shows a broad 1H-NMR spectrum in CDCl3 as well as in DMSO. Therefore it was characterized as tetraacetamide 34 (R = COCH3). The solution of the tetra amine (0.2 g, 0.16 mmol) in acetic anhydride (5ml) and a few drops of triethylamine was stirred for 12 h at rt. The reaction mixture was poured in ice to form a sticky brown mass which was dissolved in chloroform (10 ml). After separation the organic phase was dried and the solvent was evaporated. The tetra-acetamide 34 (R = COCH3) was obtained after column chromatography using (CHCl3–MeOH; 9.8 : 0.2) as eluent as a yellow powder (0.14 g, 64%). m.p 335 °C. FD-MS, (M+ + H) m/z = 1403.5.1H NMR (400 MHz, CDCl3) δ 2.19 (bm, 20H, CH2–CH2–CH2-, -CH3), 3.45 (s, 8H, Ar–CH2–Ar), 3.79 (t, 8H, 3J = 4.7 Hz, O–CH2-), 4.24 (t, 8H, 3J = 7.8 Hz, -CH2–N), 7.36 (s, 8H, ArH), 7.70–7.81 (m, 16H, Phth-H), 8.09 (s, 4H, NH).
5,11,17,23-Tetra-nitro-25,26,27,28-tetra-aminopropoxy-calix[4]arene 35 (1,3-alternate)
A suspension of 33 (1.12 g, 0.83 mmol) in EtOH (60 ml) was refluxed with hydrazine hydrate (20 ml) for 4 h (the solution became clear). After the solvent was evaporated in vacuo til dry the residue was dissolved in CH2Cl2 (15 ml) and washed twice with water (2 × 30 ml). The organic phase was dried (MgSO4) and the solvent was removed in vacuo. The residue was dissolved in chloroform (10 ml) and the tetraamine 35 was reprecipitated with hexane to give a yellow powder (0.39 g, 57%). mp 249–251 °C. (Found: C 56.34, H 6.30, N 12.41 C40H50N8O12·H2O requires C 56.33, H 6.15, N 13.14). FD-MS, (M+ + H) m/z = 835.21H NMR (200 MHz, CDCl3) δ 1.85 (m, 16H, -NH2, -CH2–CH2–CH2-), 2.79 (t, 8H, 3J = 6.3 Hz, -CH2–N), 3.53 (s, 8H, Ar–CH2–Ar), 3.78 (t, 8H, 3J = 6.5 Hz, O–CH2-), 7.78 (s, 8H, ArH).
5,11,17,23-Tetra-amino-25,26,27,28-tetra-aminopropoxy-calix[4]arene 36 (1,3-alternate)
A suspension of 33 (0.4 g, 0.4 mmol) and Pd/C (100 mg) in EtOH (30 ml) was refluxed with hydrazine hydrate (8 ml) for 4 h. Then, the solvent was evaporated under reduced pressure the residue was dissolved in CHCl3 (10 ml) and washed twice with water (2 × 25 ml). The organic solution was dried (MgSO4) and the solvent was removed in vacuo. The formed powder was dissolved in chloroform (5 ml) and the octaamine was obtained as bright yellow powder by reprecipitation with hexane (0.39 g, 57%). mp 259 °C.1H NMR (300 MHz, CDCl3) δ 1.98–2.06 (m, 8H, -CH2–CH2–CH2-), 3.00 (t, 8H, 3J = 6.6 Hz, -CH2–N), 3.73 (s, 8H, Ar–CH2–Ar), 3.98 (t, 8H, 3J = 6.6 Hz, O–CH2-), 8.00 (s, 8H, ArH).
Crystallographic data
Data were collected on a STOE-IPDS-II two-circle diffractometer employing graphite-monochromated Mo Kα radiation (0.71073 Å). Data reduction was performed with the X-Area software.42 For 27 (1,3-alt, n = 4) an empirical absorption correction was performed using the MULABS43 option in PLATON.44 Structures were solved by direct methods with SHELXS-9045 and refined by full-matrix least-squares techniques with SHELXL-97.46 All non-H atoms were refined with anisotropic displacement parameters. Hydrogens were included at calculated positions and allowed to ride on their parent atoms. Table 2 lists the most important parameters of the X-ray analysis.| Compound | 27 (1,3-alt, n = 3) | 27 (cone, n = 3) | 27 (1,3-alt, n = 3) | 11 (paco) |
|---|---|---|---|---|
| Formula | C64H64N4O12 | C64H64N4O12 | C67H70Cl2N4O12 | C48H58N2O8 |
| M w | 1081.19 | 1081.19 | 1194.17 | 790.96 |
| Crystal system | monoclinic | triclinic | triclinic | triclinic |
| Space group | C2/c (No. 15) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2)
(No. 2) |
P
(No. 2) |
P
(No. 2) |
| T/K | 173 | 173 | 173 | 100 |
| a/Å | 16.7651(15) | 14.1953(7) | 13.4904(6) | 10.3423(7) |
| b/Å | 29.395(3) | 14.6587(7) | 14.1373(7) | 13.1610(9) |
| c/Å | 24.507(2) | 15.3671(7) | 17.7409(8) | 16.5605(7) |
| α/° | 83.459(4) | 101.868(5) | ||
| β/° | 95.993(7) | 92.666(4) | 69.464(4) | 96.988(5) |
| γ/° | 76.454(4) | 91.968(5) | ||
| V/Å | 12011.3(19) | 2923.2(2) | 3078.5(2) | 2185.5(3) |
| Z | 8 | 2 | 2 | 2 |
| µ/mm−1 | 0.083 | 0.085 | 0.171 | 0.081 |
| Unique refins. Measured | 61211 | 62074 | 83961 | 31174 |
| Unique refins.[I > 2σ(I)] | 11107 | 17077 | 14584 | 9295 |
| wR(F2) | 0.3033 | 0.1492 | 0.3124 | 0.1077 |
One of the tert-butyl groups of 27 (cone, n = 3) is disordered over two positions with a ratio of the site occupation factors of 0.471(8) : 0.529(8). For 11 (paco) the tert-butyl groups are disordered over two positions with a ratio of the site occupation factors of 0.461(3) : 0.539(3) and 0.483(6) : 0.517(6), respectively and also, one of the methylene bridges is disordered over two positions with a ratio of the site occupation factors of 0.319(6) : 0.681(6). Residual electron densities: 27 (1,3-alt, n = 3) 1.89 e Å−3 at 0.5000, 0.1015, 0.2500, 27 (cone, n = 3) 1.02 e Å−3 at 0.4964, 0.2884, 0.0518, 27 (1,3-alt, n = 4) 3.00 e Å−3 at 0.8438, 0.0734, 0.2070. Crystallographic data in CIF format have been deposited with the Cambridge Crystallographic Data Centre: CCDC reference numbers 249854–249857. See http://www.rsc.org/suppdata/ob/b4/b414173c/ for crystallographic data in .cif or other electronic format.
Acknowledgements
This work has been financially supported by the European Commission in the framework of the research program on Management and Storage of Radioactive Waste (Contracts No. FI4W-CT96-0022 and No FIKW-CT-2000-00088) and by the Deutsche Forschungsgemeinschaft (SFB 625).References
- For reviews on calixarenes see: (a) Calixarenes 2001, ed. V. Böhmer, Z. Asfari, J. Harrowfield, J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001. Search PubMed; (b) C. D. Gutsche in Calixarenes Revisited, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1998. Search PubMed; (c) V. Böhmer, Angew. Chem., 1995, 107, 785–818 CrossRef; V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef.
- A. Casnati, F. Bonetti, F. Sansone, F. F. Ugozzoli and R. Ungaro, Collect. Czech. Chem. Commun., 2004, 69, 1063–1079 CrossRef CAS.
- (a) J. Y. Kim, Y. H. Kim, J.-I. Choe and S. K. Chang, Bull. Korean Chem. Soc., 2001, 22, 635–637 CAS; (b) M. O. Vysotsky, V. Böhmer, F. Würthner, C.-C. You and K. Rissanen, Org. Lett., 2002, 4, 2901–2904 CrossRef CAS.
- S. J. Kim, M. G. Jo, J. Y. Lee and B. H. Kim, Org. Lett., 2004, 6, 1963–1966 CrossRef CAS.
- J.-A. Pérez-Adelmar, H. Abraham, C. Sánchez, K. Rissanen, P. Prados and J. de Mendoza, Angew. Chem., Int. Ed. Engl., 1996, 9, 1009–1011 CrossRef.
- P. Wang, M. Saadioui, C. Schmidt, V. Böhmer, V. Host, J. F. Desreux and J.-F. Dozol, Tetrahedron, 2004, 60, 2509–2515 CrossRef CAS.
- S. Shinkai, Y. Shirahama, T. Tsubaki and O. Manabe, J. Am. Chem. Soc, 1989, 111, 5477–5478 CrossRef CAS.
- CMPO = carbamoylmethyl phosphine oxide; the (N,N-diisobutyl carbamoylethyl) octylphenyl phosphine oxide is used in the TRUEX process: E. P. Horwitz, D. G. Kalina, H. Diamond, D. G. Vandegrift and W. W. Schultz, Solvent Extr. Ion Exch., 1985, 3, 75 Search PubMed.
- (a) F. Arnaud-Neu, V. Böhmer, J.-F. Dozol, C. Grüttner, R. A. Jakobi, D. Kraft, O. Mauprivez, H. Rouquette, M.-J. Schwing Weill, N. Simon and W. Vogt, J. Chem. Soc., Perkin Trans. 2, 1996, 1175–1182 RSC; (b) C. Schmidt, M. Saadioui, V. Böhmer, V. Host, M.-R. Spirlet, J. F. Desreux, F. Brisach, F. Arnaud-Neu and J.-F. Dozol, Org. Biomol. Chem., 2003, 1, 4089–4096 RSC.
- S. Barboso, A. Garcia Carrera, S. E. Matthews, F. Arnaud-Neu, V. Böhmer, J. F. Dozol, H. Rouquette and M.-J. Schwing-Weill, Perkin Trans. 2, 1999, 719–723 RSC.
- (a) J. Scheerder, M. Fochi, J. F. J. Engbersen and D. N. Reinhoudt, J. Org. Chem., 1994, 59, 7815–7820 CrossRef CAS; (b) K. C. Nam, S. O. Kang, H. S. Jeong and S. Jeon, Tetrahedron Lett., 1999, 40, 7343–7346 CrossRef CAS; (c) A. Casnati, M. Fochi, P. Minari, A. Pochini, M. Reggiani, R. Ungaro and D. N. Reinhoudt, Gazz. Chim. Ital., 1996, 126, 99–106 CAS.
- P. D. Beer, Chem. Commun., 1996, 6, 689–696 RSC.
- J. Rebek Jr., Chem. Commun., 2000, 8, 637–643 RSC.
- (a) A. Rincon, M. P. Prados and J. de Mendoza, J. Am. Chem. Soc., 2001, 123, 3493–3498 CrossRef CAS; (b) F. Sansone, L. Baldini, A. Casnati, E. Chierici, G. Faimani, F. Ugozzoli and R. Ungaro, J. Am. Chem. Soc., 2004, 126, 6204–6205 CrossRef CAS.
- J. Budka, P. Lothak, V. Michlova and I. Stibor, Tetrahedron Lett., 2001, 42, 1583–1586 CrossRef CAS.
- J. Scheerder, J. P. M. van Duynhoven, J. F. G. Engbersen and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1996, 35, 1090–1093 CrossRef.
- J. J. Gonzalez, P. Prados and J. de Mendoza, Angew. Chem., Int. Ed. Engl., 1999, 38, 525–528 CrossRef CAS.
- O. Mogck, V. Böhmer, G. Ferguson and W. Vogt, J. Chem. Soc., Perkin Trans. 1, 1966, 1711–1715 Search PubMed.
- M. Saadioui, A. Shivanyuk, V. Böhmer and W. Vogt, J. Org. Chem., 1999, 64, 3774–3777 CrossRef CAS.
- A. Pop, M. O. Vysotsky and V. Böhmer, Coll. Czech. Chem. Commun., 2004, 69, 1009–1026 CrossRef.
- L. A. J. Chrisstoffels, F. de Jong, D. N. Reinhoudt, S. Sivelli, S. Gazzola, A. Casnati and R. Ungaro, J. Am. Chem. Soc., 1999, 121, 10142–10151 CrossRef CAS.
- J. Scheerder, M. Fochi, J. F. J. Engbersen and D. N. Reinhoudt, J. Org. Chem., 1994, 59, 7815–7820 CrossRef CAS.
- E. M. Collins, M. A. McKervey, E. Madigan, M. B. Moran, M. Owens, G. Ferguson and S. J. Harris, J. Chem. Soc., Perkin Trans. 1, 1991, 3137–3142 RSC.
- I. Bitter and V. Csokai, Tetrahedron Lett., 2003, 44, 2261–2265 CrossRef CAS.
- O. Seneque and O. Reinaud, Tetrahedron, 2003, 59, 5563–5568 CrossRef CAS.
- For the formation of Y = CH2CH2NH2via azides see ref. 10.
- (a) W. Veerboom, S. Datta, Z. Asfari, S. Harkema and D. N. Reinhoudt, J. Org. Chem., 1992, 57, 5394–5398 CrossRef CAS; (b) K. Iwamoto and S. Shinkai, J. Org. Chem., 1992, 57, 7066–7073 CrossRef CAS.
- With Na2CO3 the cone isomer is formed as main product.
- Similar experiences were made with a mixed tetraether in the cone conformation, where both phthalimide groups were hydrolyzed to the monoamide: P. Wang and V. Böhmer, unpublished.
- These conditions were successfully applied for monocyano-hepthyloxy surrounded by tripropyloxy residue attached to calixarenes by Y. Rudzevich and V. Böhmer, unpublished results.
- S. E. Matthews, P. Parzuchowski, A. G. Carrera, C. Grüttner, J. D. Böhmer and V. Böhmer, Chem. Commun., 2001, 5, 417–418 RSC.
- P. Lhoták, J. Svoboda, I. Stibor and J. Sykora, Tetrahedron Lett, 2002, 43, 7413–7417 CrossRef CAS.
- A. Pop, M. O. Vystosky, M. Saadioui and V. Böhmer, Chem. Commun., 2003, 10, 1124–1125 RSC.
- The use of bromoalkyl phthalimides or bromonitriles in the first O-alkylation step leads to hexaamines with two amino groups attached to the narrow rim.
- A. Soi, W. Bauer, H. Mauser, C. Moll, F. Hampel and A. Hirsch, J. Chem. Soc., Perkin Trans. 2, 1998, 1471–1478 RSC.
- A tetra-CMPO derivative could be obtained from 28 (n = 3) in three steps by acylation, cleavage of the phthalimide groups and acylation again: C. Danila and V. Böhmer, unpublished.
- Tetraethers in the cone-conformation can be obtained much more selectively.
- J. Oriol Magrans, J. de Mendoza, M. Pons and P. Prados, J. Org. Chem., 1997, 62, 4518–4520 CrossRef.
- V. Böhmer in Calixarenes, The Chemistry of Phenols, ed. Z. Rappoport, Wiley, Chichester, 2003. Search PubMed.
- Since starting point and direction are in principle arbitrary, the sequence (+,−)(+,−)(−,−)(+,+) is identical to (−,+) (+,+)(−,−)(−,+), listed in Table 1.
- M. Perrin and D. Oehler in Calixarenes, A Versatile Class of Macrocyclic Compounds, ed. J. Vicens and V. Böhmer, Kluwer Academic Publishers, Dordrecht, 1991; values >90° mean that the phenolic unit is bent outwards Search PubMed.
- Stoe & Cie 2001. X-Area. Area-Detector Control and Integration, Software. Stoe & Cie, Darmstadt, Germany Search PubMed.
- R. H. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33–38 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, C-34.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |