Pyrroloiminoquinone and related metabolites from marine sponges
Edith M.
Antunes
a,
Brent R.
Copp
b,
Michael T.
Davies-Coleman
*a and
Toufiek
Samaai
c
aDepartment of Chemistry, Rhodes University, Grahamstown, South Africa. E-mail: M.Davies-Coleman@ru.ac.za
bDepartment of Chemistry, University of Auckland, Auckland, New Zealand
cSchool of Biology, University of Kwazulu-Natal, Westville Campus, Durban, South Africa
First published on 4th January 2005
Abstract
Covering: the literature from the first reported isolation of a pyrroloiminoquinone metabolite from a marine sponge in 1986 up to October 1, 2004.
This review presents the structure, biological activity, biosynthetic studies and, where applicable, references to syntheses of 81 marine alkaloids containing either tetra-, hexa- or octa-hydrogenated variants of pyrrolo[4,3,2-de]quinoline, pyrrolo[4,3,2-de]pyrrolo[2,3-h]quinoline and pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline core skeletons. The literature describing the isolation of pyrroloiminoquinones, and related metabolites, from marine sponges is littered with taxonomic inconsistencies and recent efforts to clarify the taxonomy of the sponges that produce this group of metabolites are discussed.
 Edith M. Antunes Edith M. Antunes | Edith Antunes received her B.Sc. (Hons.), M.Sc. and Ph.D. degrees from Rhodes University. Her Ph.D. dissertation entitled “Pyrroloiminoquinone metabolites from South African Latrunculid Sponges” was supervised by Professor Michael Davies-Coleman. She is presently a post-doctoral researcher in Professor Bill Gerwick's laboratory at Oregon State University. |
 Brent R. Copp Brent R. Copp | Brent Copp received his B.Sc. (Hons.) and Ph.D. degrees from the University of Canterbury, where he studied the isolation, structure elucidation and structure–activity relationships of biologically active marine natural products under the guidance of Professors John Blunt and Murray Munro. He undertook postdoctoral research with Jon Clardy at Cornell and Chris Ireland at The University of Utah. 1992 and 1993 were spent working in industry as a isolation chemist with Xenova Plc., before returning to New Zealand to take a lectureship at the University of Auckland, where he is currently a Senior Lecturer. His research interests revolve around the discovery of novel natural products and their use as drug leads against a variety of disease states or as molecular probes in chemical genomics studies. |
 Michael T. Davies-Coleman Michael T. Davies-Coleman | Michael Davies-Coleman received his B.Sc. (Hons.) and Ph.D. degrees from Rhodes University in South Africa. After completing his Ph.D. research in the field of plant natural products chemistry under the supervision of Professor Douglas Rivett he was appointed to a lectureship in the School of Pharmacy at Rhodes University. In 1991 he was a post-doctoral research fellow with Professor John Faulkner at Scripps Institution of Oceanography in La Jolla, California and in 1999 he was an NIH visiting scientist in Dr Mike Boyd's drug discovery laboratory at the National Cancer Institute in Frederick, Maryland. He is currently a Professor in the Department of Chemistry at Rhodes University and has published widely on the natural products chemistry of southern African marine invertebrates. |
 Toufiek Samaai Toufiek Samaai | Toufiek Samaai received his M.Sc. from Imperial College (London) and his Ph.D. from the University of the Western Cape in 2002, where he studied the systematics of the sponge family Latrunculiidae under the supervision of Professor M. J. Gibbons and Dr M. Kelly (formerly Kelly-Borges). After graduating in 2002, he took up a lecture position at the former University of Durban Westville (now the University of KwaZulu-Natal) in Durban, South Africa. His research interests revolve around sponge systematics, biodiversity and reef ecology of subtropical and tropical areas on the east coast of South Africa. |
1 Introduction
For nearly two decades marine sponges of the families Latrunculiidae (order Poecilosclerida, sub-order Latrunculina incertae sedis) and Acarnidae (order Poecilosclerida, sub-order Microciona) have proved to be a prolific source of biologically active alkaloid metabolites containing either tetra-, hexa- or octa-hydrogenated variants of pyrrolo[4,3,2-de]quinoline 1, pyrrolo[4,3,2-de]pyrrolo[2,3-h]quinoline 2 and pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline 3 core skeletons.1,2 The characteristic pyrroloiminoquinone 4 structural motif dominates in all three core skeletons and provides the name, along with pyrroloquinoline, commonly used to describe the members of this group of secondary metabolites. Marine pyrroloquinoline alkaloids have been previously reviewed as part of a larger review of bioactive marine alkaloids3 and in tandem with pyridoacridine alkaloids from marine sources.4 A recent taxonomic revision of a number of the original voucher specimens of the sponges that have yielded some of the published chemistry of pyrroloiminoquinone metabolites5–10 and some confusion surrounding the alphabetical ordering of discorhabdins have precipitated this review of the structures, bioactivity and, where applicable, biosynthesis and synthesis of this group of marine natural products.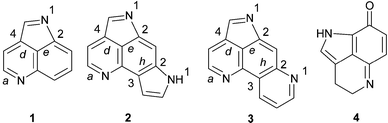
This review begins with a brief overview of the systematics of the sponge families Latrunculiidae, Acarnidae and Podospongiidae where the information provided by marine natural product chemical studies has helped to partly clarify the phylogenetic relationship between the sponges allocated to these families.3,5–10 Following on from the taxonomic discussion, the isolation, structures, biological activity and relevant syntheses of the pyrroloiminoquinone metabolites from the core structural classes, pyrrolo[4,3,2-de]quinoline and pyrrolo[4,3,2-de]pyrrolo[2,3-h]quinoline are presented here in order of increasing structural complexity. Conversely, the pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline core structural class, incorporating the discorhabdins A–V and the closely related prianosins A–D and epinardins A–D, is introduced in a chronological sequence reflecting the reported isolation of these alkaloids. The order of presentation of the structures of the discorhabdin, prianosin and epinardin series is an attempt to clarify some of the structural overlap and anomalies that surround these metabolites. Syntheses, biosynthetic studies and reports of the subsequent isolation of the ubiquitous discorhabdins, and less common structurally related analogues, follow the historically biased review of the discorhabdin/prianosin/epinardin literature.
2 The taxonomy of marine sponges that produce pyrroloiminoquinone and related metabolites
Sponges that produce pyrroloiminoquinone and related metabolites generally possess either silicious discorhabd microscleres (Fig. 1A) aligned vertically on the surface of the ectosome (Fig. 1B) or palmate chelae (Fig. 1C) distributed in various parts of their tissues (family Acarnidae).5–9 With the notable exception of the genus Strongylodesma, discorhabd microscleres (which can be further differentiated into aniso, iso, isoconico and isochia forms on closer examination) are found in sponges from the other three genera within the family Latrunculiidae (Scheme 1). Discorhabd microscleres are also superficially similar to the spinorhabd micoscleres found in sponges of the family Podospongiidae (Fig. 1D and E). It is not surprising that the presence of an array of these superficially similar microscleres was traditionally used as an easily recognizable, and often hastily assigned taxonomic identifier to combine diverse sponges into a single genus Latrunculia (family Latrunculiidae).5,7,8 Kelly-Borges and Vacelet11 first drew attention to clear differences in the skeletal architecture and the lack of structural homology in the discorahbd microscleres of sponge genera grouped together in what was then the monophyletic family Latrunculiidae.3 These significant differences necessitated an extensive revision of the family Latrunculiidae5,7–9 and the taxonomic status quo suggests the hypothetical phylogenetic relationship between this family and related families presented in Scheme 1. From an analysis of the plethora of marine natural product investigations of sponges originally loosely grouped together in the sponge family Latrunculiidae, Samaai and Kelly have been able to provide significant chemotaxonomic evidence in support of this phylogenetic revision.3,5–9 For example, Indo-Pacific sponges originally identified as Latrunculia spp. that produce macrocyclic lactones e.g. latrunculin A 5,12 have been re-allocated to the circumtropical genus Negombata in the family Podospongiidae while sponges either originally identified as Latrunculia or Prianos spp., and providing a source of sesterterpene peroxides e.g. trunculin A 6,13 have been re-identified as Dicarnus or Sigmaceptrella species, also in the family Podospongiidae.3,5,6,11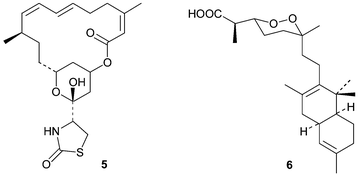
 | ||
| Fig. 1 (A) Anisodiscorhabd from Latrunculia bocagei Ridley and Dendy, 1886. (B) Common arrangement of discorahbds on the surface of the ectosome in Latrunculia species. (C) A palmate chela, characteristic of sponges assigned to the order Microcionina. (D) and (E) Spinorhabds typically found in sponges of the genera Sigmosceptrella and Negombata, respectively. | ||
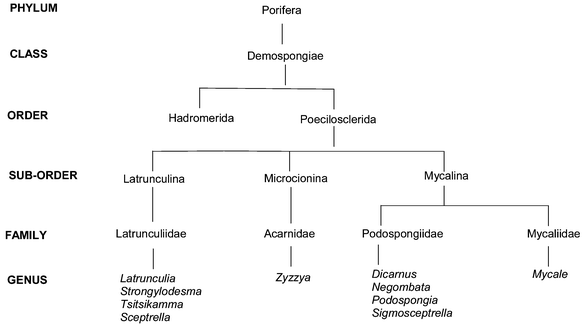 | ||
| Scheme 1 A suggested phylogenetic relationship between the sponge families Latrunculiidae, Acarnidae, Podospongiidae and Mycaliidae. | ||
Sponges of the family Latrunculiidae are ubiquitous in cold water, with diversity centred predominantly in the southern hemisphere.8Latrunculia species are relatively abundant in the sub-tidal (< −50m) benthic communities off New Zealand, Antarctica and South Africa.8,9 The utilization of definitive chemotaxonomic markers within the family Latrunculiidae can be tenuous given the proposed close biosynthetic relationship between pyrroliminoquinone and related metabolites (Scheme 2). Nonetheless, general chemotaxonomic trends are evident, for example the genera Latrunculia (Latrunculiidae) and Strongylodesma (Latrunculiidae) are apparent reservoirs of discorhabdin-type metabolites (see later).
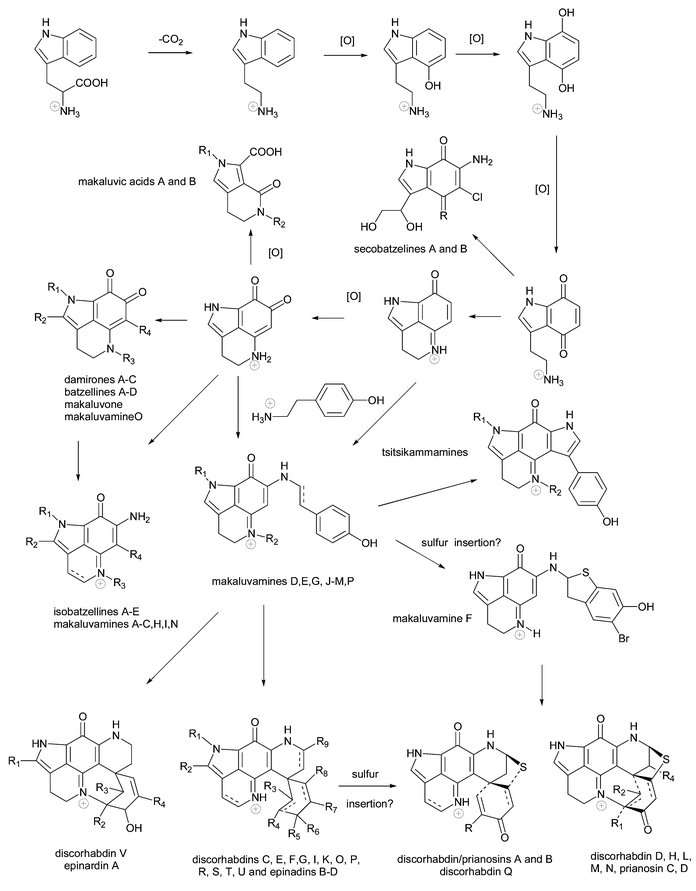 | ||
| Scheme 2 A putative biosynthetic sequence linking marine pyrroloiminoquinone and related metabolites to a phenylalanine precursor (adapted from Urban et al.3 and Lill et al.82). | ||
In a comprehensive study of the taxonomic, environmental and chemical variation within the sponge genus Latrunculia in New Zealand, Miller et al.14 and Alvarez et al.15 concluded the presence of at least eight species rather than the one to four previously believed. The authors found that there were no single diagnostic characters that could be used to distinguish all eight species and a combination of characteristics was required. The characters studied included allozyme analysis using nine-locus genotypes, spicule morphology and chemical analysis. The eight species of Latrunculia displayed fixed or near-fixed genetic differences, indicating that they were reproductively isolated while the discorhabdin alkaloid chemistry produced by the sponges was species specific rather than the result of environmental variation.
Interestingly, a Japanese sponge Prianos melanos,16 a source of the prianosins (synonymous with the discorhabdins – see later) has been re-identified as a species of Strongylodesma (formerly only known from the South Atlantic).5,8 Several deep-water Caribbean Batzella species16 that yielded batzelline, secobatzelline and discorhabdin metabolites (see later), are also now recognized as Strongylodesma species.5,17 Paradoxically, the genera Batzella and Prianos have historically both been used as “taxonomic dustbins” which may account for the handful of unrelated species placed in these genera.3,16,18 Currently, Batzella sensu stricta is placed within the Poecilosclerid family Chondropsidae19 and Prianos sensu strictu is synonymous with the Haplosclerid genus Haliclona (Reniera) (family Chalinidae).20
Makaluvic acids, damirones and makaluvamines are consistently, but not exclusively, reported from sponges assigned to the genus Zyzzya (family Acarnidae, formerly Iophonidae)3 as evidenced by the recent isolation of members of all three of these groups of alkaloids, in addition to discorhabdins, from the South African sponge Latrunculia bellae.21 During the last decade several major changes in familial classifications within Poecilosclerida have occurred, especially in the transfer of genera and in the reorganization of familial assemblages.5–10,19,20 Thus Hooper proposed that the family Iophonidae be renamed Acarnidae as the latter represents the older family name created by Topsent as a family level taxon for Acarnus.10 The original sponge that yielded the damirones, Damiria spp. (see later), and the only reference to this species in the chemistry literature, has been re-identified as Zyzzya fuliginosa.5,16
The discovery of the tsitsikammamine metabolites (see later) in the new South African genus Tsitsikamma (family Latrunculiidae) initially contributed positively to the chemotaxonomic debate as the purported biosynthetic intermediacy of the tsitsikammamine structures between those of the makaluvamines and discorhadins appeared to be paralleled by the mixture of Zyzzya and Latrunculia morphological characteristics evident in T. favus.3,9 However, the absence of tsitsikammamines in a second species of this genus, T. pedunculata, recently discovered off South Africa, negates the chemotaxonomic significance originally assigned to these compounds.21
There have been no reported natural product studies of the fourth genus, Sceptrella, within the family Latrunculiidae and this may be attributed to the difficulty of collecting this sponge from the extreme depths (ca. −500 m) where it is known to exist.8
3 Alkaloids containing a pyrrolo[4,3,2-de]quinoline skeleton
The mono- and bicyclic makaluvic acids and secobatzellines are ring-opened examples of pyrrolo[4,3,2-de]quinolines and are clearly biosynthetically related. Makaluvic acids A 7 and B 8, isolated from Zyzzya fuliginosus (= fuliginosa, species name incorrectly spelt in the original paper) collected at Chuuk Atoll, Micronesia, possibly represent oxidative degradation products of tricyclic alkaloids belonging to the batzelline, isobatzelline or makaluvamine families.22 The structure of 7 was confirmed by an X-ray diffraction study. Makaluvic acid A 7 has also recently isolated from the South African sponge Latrunculia bellae21 and 7 was not cytotoxic in either P388 murine leukaemia22 or HCT-116 human colon tumour cell line screens,21 in accordance with the iminoquinone structural requirement necessary for cytotoxicity in the pyrroloiminoquinone alkaloids (see later). Iminoquinone-containing secobatzelline A 9 and the potentially artifactual quinone analogue secobatzelline B 10 were isolated from deep water specimens of Batzella sp. (original voucher specimen re-identified as a Strongylodesma species by Samaai),5,17 collected in the Caribbean.23 While 9 inhibited the phosphatase activity of calcineurin and the peptidase activity of CPP32 with IC50 values of 0.55 and 0.02 µg mL−1, quinone 10 was less active. In a similar manner, cytotoxicity towards the P388 and A-549 cell lines was more pronounced in the presence of the iminoquinone moiety (9, IC50 0.06, 0.04 µg mL−1) than the quinone (10, IC50 1.2, 2.9 µg mL−1). No syntheses of makaluvic acids or secobatzellines have been reported to date.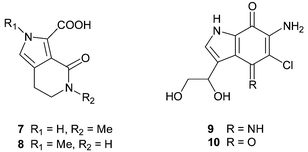
The simplest examples of marine alkaloids that fall within the classification of the tricyclic pyrrolo[4,3,2-de]quinoline class are the ortho-quinones which include batzellines A–D, damirones A–C, makaluvone and makaluvamine O.
The first examples of alkaloids bearing a tricyclic pyrrolo[4,3,2-de]quinoline skeleton isolated from the marine environment were the batzellines A–C 11–13, reported in 1989 from a Batzella sp. sponge (original voucher specimen re-identified as a Strongylodesma species by Samaai),5,17 collected at a depth of 120 m off the Grand Bahama Islands.24 The structure of 11 was secured by an X-ray diffraction study, while those of 12 and 13 were established by chemical modification and comparison with 11. Batzelline D 14, the N-1-desmethyl analogue of batzelline C, was reported more recently from an Abrohlos Island, Indo-West Pacific collection of Zyzzya fuliginosa25 while damirones A 15 and B 16 were isolated from a Palauan collection of Damiria sp.,26 an Indonesian Histodermella sp.27
(both voucher specimens of the Damiria and Histodermella species have been re-identified as Z. fuliginosa by Van Soest28) and more recently from Australian specimens of Z. fuliginosa.29 Investigation of a Pohnpeian specimen of Z. fuliginosa yielded damirone C 17.30 An X-ray diffraction study of damirone A 15 has been reported.31 The most recent examples of ortho-quinoid containing pyrroloquinoline alkaloids are makaluvone 18, isolated, together with damirone B 16, from a Fijian collection of Zyzzya cf. marsailis32
(re-identified as Z. fuliginosa by Kelly5) and makaluvamine O 19, the N-1-desmethyl analogue of makaluvone, purified from the Jamaican sponge Smenospongia aurea.33 The isolation of 19 from S. aurea represents the first report of a pyrroloquinoline metabolite from the sponge family Thorectidae, and the authors, recognizing the chemotaxonomic significance of this discovery, comment that although the voucher specimen is indeed that of S. aurea, the very similar gross morphology of Smenospongia, Zyzzya and Strongylodesma species off the coast of Jamaica cannot rule out the possibility that different sponge species were inadvertently mixed together during the field collection.33
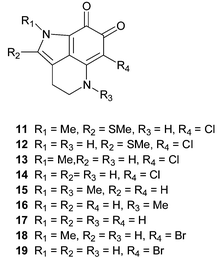
Batzelline A 11 exhibited selective cytotoxicity towards A549 (human lung) cells compared to P388 (murine leukaemia), and showed some selectivity for melanoma cell lines in testing at the NCI.34 No activity was observed in in vivo testing against A549 tumour xenografts in mice. Batzelline D 14 was inactive in an HIV-1 envelope-mediated cell fusion assay,25 damirones A 15 and B 16 exhibited mild cytotoxicity towards the Ehrlich carcinoma cell line (IC50 values of 46.5 and 50 µg mL−1 respectively),31 while damirone B and makaluvone 18 failed to exhibit cytotoxicity towards the HCT-116 cell line and demonstrated no ability to inhibit the function of the DNA processing enzyme topoisomerase II.32 Makaluvamine O 19 demonstrated moderate activity towards Plasmodium falciparum (IC50 0.9 µg mL−1) combined with low VERO cell line toxicity.33
Alkaloids belonging to the batzelline and damirone families have been the target of synthetic efforts, which typically utilize indole or quinoline precursors. Complete syntheses of batzelline A 11,35 B 1235 and C 1336–38 and damirones A 1536,39 and B 1639 have been reported, while formal syntheses, involving new routes to synthetic intermediates, of batzelline C40,41 and damirones A40,42,43 and B40,42,43 have been disclosed. The obviously close biosynthetic relationship between batzelline and damirone ortho-quinones and the iminoquinone containing isobatzellines and makaluvamines (see later) has been exploited with alkaline hydrolysis of makaluvamines H and C (see later) affording damirones A and B respectively in high yield.31 Diazotization of isobatzelline A (see below) resulted in a low yield of batzelline A 11.44
Isobatzellines A–D 20–23 were isolated as bioactive components of a deep water collection of Batzella sp. (original voucher specimens HBOM003:00050/51 both re-identified as Strongylodesma species by Samaai),5,17 made off the Grand Bahamas.44 Structurally they are closely related to the batzelline alkaloids, also isolated from Batzella sp. (=
Strongylodesma sp.), with replacement of the ortho-quinoid functionality by a 7-amino substituted iminoquinone skeleton. The fifth member of the family, isobatzelline E 24, was isolated from an Indopacific collection of Zyzzya fuliginosa.25 In stark contrast to their ortho-quinoid analogues, the isobatzelline alkaloids exhibit wide ranging biological activities. Isobatzellines A–D were found to be cytotoxic towards the P388 murine leukaemia cell line with IC50 values of 0.42, 2.6, 12.6 and 20 µg mL−1 respectively in addition to anti-Candida albicans activity with MIC values of 3.1, 25, 50 and 25 µg mL−1 respectively.44 Isobatzelline C 22, but not isobatzelline E 24, was active in an HIV-1 envelope mediated cell fusion assay, however, it was speculated that the activity may have been due to the ability of such compounds to interfere with DNA-processing enzymes (see later under makaluvamines).25 Syntheses of isobatzellines A and B35 and C36,38,40,41 have been reported.
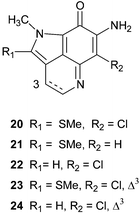
The makaluvamines represent a diverse range of structures based upon a 7-amino substituted pyrroloiminoquinone skeleton. Substitution at positions N-1, N-5 and N-9 are common, with one example having Δ3 unsaturation and one example bearing substitution at C-6. The first examples of the series, makaluvamines A–F 25–30, were reported as cytotoxic pigments of a Fijian collection of the sponge Zyzzya cf. marsailis32
(=
Z. fuliginosa, see earlier).5 More recently, makaluvamine A 25 has also been reported from harvested plasmodial cells of the myxomycete Didymium bahiense, indicating that the biosynthesis of such alkaloids is not limited solely to the marine environment.45 The trisubstituted analogue makaluvamine G 31 was isolated from an Indonesian collection of an unidentified Histodermella species27
(=
Z. fuliginosa, see earlier).28 Investigation of Pohnpei (Micronesia) specimens of Z. fuliginosa afforded six new makaluvamines H–M 32–37,30 the same sponge species collected off Australia's Great Barrier Reef recently yielded makaluvamines C, E, G, H and L29 while Philippines and Vanuatu specimens were found to be a source of makaluvamines N 3846 and P 3947 respectively. Makaluvamine O 19, the only ortho-quinone containing makaluvamine, has been discussed earlier.33
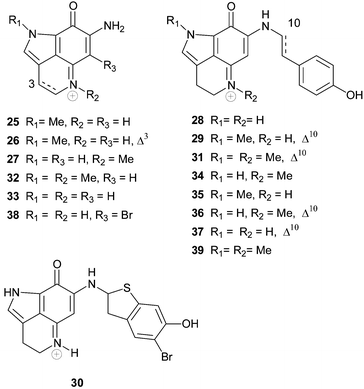
The makaluvamine family of alkaloids typically exhibit cytotoxicity towards tumour cell lines, the mechanism of which is believed to be mediated by inhibition of DNA processing enzymes. Makaluvamines A 25 and C–F 27–30 were found to exhibit cytotoxicity towards the HCT-116 cell line with IC50 values of 1.3, 36.2, 17.1, 1.2 and 0.17 µM respectively.32 The HCT-116 cytotoxicity of makaluvamine C 27 is inconsistent with the general trend observed in this series of analogues and the subsequent screening of this compound, isolated recently from the South African sponge Latrunculia bellae, against HCT-116 (IC50 1.1 µM) would suggest that the HCT-116 cytotoxicity of makaluvamine C is more significant than originally reported.21 Makaluvamines A, C, D, E and F were also shown to inhibit the function of topoisomerase II, to intercalate into DNA and undergo reductive cleavage of DNA.32,48,49 Makaluvamines A and C were found to be more effective in vivo against human ovarian ovcar-3 tumours implanted in nude mice [T/C (tumour volume of the treated group/tumour volume of the control group × 100) 62 and 48% respectively] than P388 murine leukaemia (ILS 0 and 18%).32,48 Interestingly, the Δ3 analogue, makaluvamine B 26, was not cytotoxic.32 Makaluvamine G 31 exhibited general cytotoxicity to a panel of tumour cell lines (IC50 0.4–0.5 µg mL−1), displayed no antifungal or antiviral activity, and while moderate immunomodulatory activity was observed in vitro, no efficacy was observed in in vivo testing.27 In contrast to other makaluvamine alkaloids, makaluvamine G was found to be a moderate inhibitor of topoisomerase I, but did not significantly inhibit topoisomerase II. RNA, DNA (IC50 15 µM) and protein synthesis (IC50 21 µM) were also inhibited by the alkaloid.27 Utilising a panel of genetically engineered yeast strains, it was concluded that makaluvamines D 28, G 31, H 32, J 34, K 35, L 36 and M 37 were inactive against topoisomerase and that makaluvamines I 33 and L 36 were an order of magnitude more cytotoxic towards the HCT-116 cell line than makaluvamines C 27, G 31, H 32 and K 35, and that makaluvamines D 28 and M 37 were the least active.30 Makaluvamines H–M exhibited only mild activity against Bacillus subtilis.30 Makaluvamines A, E and K exhibited cytotoxicity towards P388 murine leukaemia cells (IC50 0.4, 0.6 and 0.7 µg mL−1 respectively)22 while makaluvamines C and H had IC50 values of 26.5 and 12.5 µg mL−1 towards Ehrlich carcinoma cells.31 Makaluvamine N 38 exhibited cytotoxicity towards the HCT-116 cell line (IC50 0.6 µg mL−1) and inhibited the catalytic activity of topoisomerase II,46 while makaluvamine P 39 was also cytotoxic to KB cells (64% inhibition at 3.2 µg mL−1), inhibited xanthine oxidase (IC50 16.5 µM) and was moderately active in a free-radical scavenging (antioxidant) assay.47
Due to their relative structural simplicity and potent biological activities, the makaluvamines represent attractive targets for synthesis. To date there have been reported total or formal syntheses of makaluvamines A,36,40,50–52 B,40,50,51 C,40,42,50,51,53 D,40,41,50–52,54–58 E,51 (racemic) F,59,60 I,52,58 and K.52
There have been surprisingly few reports of the synthesis and biological evaluation of non-natural makaluvamine-type alkaloids. Three new analogues, in addition to makaluvamines D 28, and I 33 were synthesized and found to exhibit moderate cytotoxicity (IC50 0.3–5.3 µM) towards the L1210 cell line.58 Preparation of lexitropsin61 and polypyrroleamide62 pyrroloiminoquinone hybrids have been reported in an effort to increase cellular uptake and cytotoxic potency.
Veiutamine 40 is an unusual member of the tricyclic pyrroloiminoquinone alkaloid family in that it bears carbon substitution at C-6.63 Veiutamine, isolated from a Fijian collection of Zyzzya fuliginosa, was found to exhibit cytotoxicity towards a panel of human tumour cell lines with a mean IC50 value of 0.1 µg mL−1. The alkaloid was seven times more potent than the structurally related makaluvamine D (IC50 2.0 ug mL−1) against the HCT-116 human colon tumour cell line. Veiutamine has been synthesized using methodology that relies upon N-Boc-directed lithiation for introduction of the C-6 substituent.64
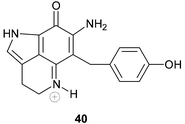
4 Alkaloids containing a pyrrolo[4,3,2-de]pyrrolo[2,3-h]quinoline skeleton
Tsitsikammamines A 41 and B 42 were isolated from the South African latrunculid sponge Tsitsikamma favus.65 However, no tsitsikammamines were found in a second South African member of this genus, T. pedunculata.9,21 A more detailed investigation of the minor metabolites occurring in extracts of T. favus yielded the two novel N-18 oxime analogues 43 and 44 of the tsitsikammamines.21 Natural products containing N-oxide or N-oxime functionalities are uncommon in the marine environment. Several attempts to oxidize 41 and 42 to 43 and 44 were unsuccessful, suggesting that this conversion is of enzymatic origin and is not a result of the isolation protocol.21 Interestingly, the N-18 oximes 43 and 44 exhibited significantly reduced cytotoxicity against human colon tumour (HCT-116) cells (IC50 128.2 and 16.5 µM respectively) when compared with their parent compounds, 41 and 42 (IC50 1.4 and 2.4 µM respectively).21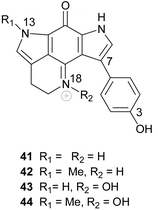
Recently, a biosynthetically related seco-analogue of the tsitsikammamines, zyzzyanone A 45, possessing a pyrrolo[3,2-f]indole-4,8(1H, 7H)-dione core skeleton, was isolated from an Australian sponge, Zyzzya fuliginosa.29 Zyzzyanone A was only moderately cytotoxic (25 µg mL−1) to mouse Ehrlich carcinoma cells and fertilized sea urchin eggs, further highlighting the propensity of the intact pyrroloiminoquinone structural motif to enhance cytotoxicity.29
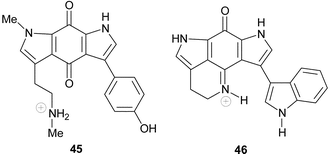
Ironically, the first example of pyrroloiminoquinone metabolites with a pyrrolo[4,3,2-de]pyrrolo[2,3-h]quinoline core skeleton was not obtained from a marine sponge. Wakayin 46 was isolated from a Fijian ascidian, Clavelina spp., by Copp et al.66 and provided the first indication that pyrroloiminoquinone metabolites are not confined to marine sponges, tentatively suggesting a possible symbiont source for these bis-pyrroloiminoquinone metabolites.
5 Alkaloids containing a pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline skeleton
The name discorhabdin is derived from the characteristic discorhabd microsleres (Fig. 1A) used to identify sponges from three of the four genera of the family Latrunculiidae. Pyrroloiminoquinone compounds containing multi-hydrogenated forms of the tetracyclic pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline skeleton (also initially referred to as a pyrrolo[1,7]phenanthroline ring system)67,68 all possess one of the following additional spiro-substituents at C-6; cyclohexanone, cyclohexenol, cyclohexenone and cyclohexadienone. A plethora of different substitution patterns around the six-membered spiro-ring, and further cyclization, in the form of a bond linking C-2 and N-18 and a thioether bridge between C-5 and C-8, are not uncommon.The structure of discorhabdin C 47 was established by X-ray diffraction analysis and was the first structure of the discorhabdin series of compounds to be published.67 Discorhabdin C was isolated from the New Zealand latrunculid sponge Latrunculia du Bocage (later re-identified as Latrunculia sp.)5 and at the time (1986) the potently cytotoxic 47
(murine leukaemia L1210 tumour cell line ED50 < 100 ng mL−1) represented a novel class of anti-tumour agents with apparently significant medicinal potential.67 Unfortunately, the relatively potent yet non-specific cytotoxicity of the discorhabdins and related metabolites has precluded their further clinical development. The publication of the fully assigned NMR data for 47 in addition to the structures and bioactivity of discorhabdins A 48 and B 49
(P388 ED50 0.03–0.1 µg mL−1) and details of the bioassay guided isolation of these compounds from the Latrunculia sponge, followed two years later.68 The structure and absolute stereochemistry of prianosin A, isolated from the Okinawan sponge Prianos melanos,69
(re-identified as a Strongylodesma species by Samaai and Kelly)5,7 was published shortly before the structure of discorhabdin A 48 and ironically, although the structures of prianosin A and discorhabdin A proved to be identical, the latter name has subsequently taken precedence in the literature. Significantly, the X-ray structure of prianosin A 48 has subsequently provided a configurational model for the stereochemical assignment of related members of the discorhabdin series.3 Not surprisingly, prianosin A exhibited similar cytotoxicity data to those reported for discorhabdins A–C (L1210 and L5178Y IC50 37 and 14 ng mL−1 respectively).69 Prianosin A also induced a ten-fold increase in the release of Ca2+ ions from sarcoplasmic reticulum when compared with caffeine in the same assay.69
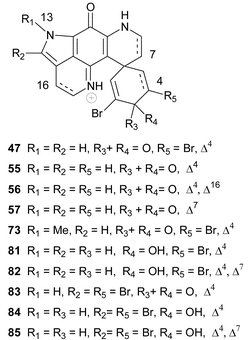
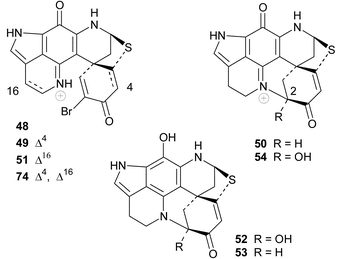
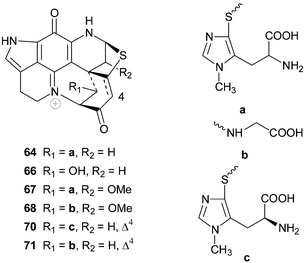
The structures of discorhabdin D 5070 and prianosins B–D 51–53,71 isolated from the same source sponges that provided the earlier members in their respective series, followed in quick succession. NOE difference spectroscopy provided the relative stereochemistry of 50 while comparison of the CD data of 51–53 with those of discorhabdin/prianosin A 48 provided the absolute stereochemistry of the latter three compounds. The p-aminophenol structure initially proposed for ring C of prianosins C 52 and D 53 was inconsistent with the emerging dominance of the ring C iminoquinone structural motif, evident in discorhabdins A–D and prianosins A and B, and the structures of 52 and 53 were later revised to 2-hydroxydiscorhabdin D 54 and discorhabdin D 50 respectively.72 Prianosin B 51 is effectively Δ16-discorhabdin A and further exemplified the structural homology between the discorhabdins and prianosins. Although discorhabdin D 50 exhibited lower in vitro activity than discorhabdins A–C 47–49 against P388 cancer cells, the in vivo activity of the former compound (T/C 132% at 20 mg kg−1) was reportedly significant.70 Prianosins B–D were cytotoxic against murine lymphomas L1210 (0.15–2.0 µg mL−1), L5178Y (0.024–1.8 µg mL−1) and human epidermoid carcinoma KB cell lines (0.46–5 µg mL−1).71
The structures of discorhabdins E 55 and F 56 first appeared (without spectral data) in a 1990 general review of the natural products chemistry of southern Pacific shallow water benthic marine invertebrates by Blunt et al.73 These structures, along with those of discorhabdins G–M, were presented by Munro et al. at the New Zealand Institute of Chemistry Conference (December, 1993) and again later at the 37th meeting of the American Society of Pharmacognosy in 1996.3 Spectral data for discorhabdins F–M originally isolated from New Zealand sponges have as yet not been published in the mainstream chemical literature, leading to some ambiguities in the discorhabdin nomenclature. The spectral data of discorhabdin E 55 (although 55 has a single chiral centre at C-6 the compound was found to be present as a racemate) from Latrunculia cf. bocagei (= Latrunculia sp., see earlier) were later reported, together with the structures of a number of semi-synthetic derivatives of discorhabdin C 47, prepared as part of a discorhabdin structure–activity relationship study.74 In this pioneering study the centrality of the pyrroloiminoquinone functionality to cytotoxicity and the significance of a spiro-2,6-dibromocyclohexadienone substituent in enhancing cytotoxicity was established. The apparent selective toxicity of discorhabdin C 47 to colon and leukaemia cell lines was also noted.74
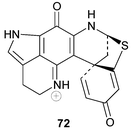
The relative inaccessibility of the original structures and NMR data of discorhabdins G–M has led to alphabetical duplication within discorhabdin nomenclature. In 1995, as part of their ongoing extensive investigations of the natural products chemistry of Antarctic marine invertebrates, Baker and co-workers isolated discorhabdin C 47 from Latrunculia apicalis
(the ubiquitous Antarctic distribution of L. biformis suggests that this sponge may have been incorrectly identified)5 and a new optically active discorhabdin, which they named discorhabdin G 57.75 The structure of this latter compound differed from the structure 58 originally assigned to discorhabdin G by Munro et al. and a compound with the structure 58, recently isolated from a South African latrunculid sponge L. bellae, gave 1H NMR data identical with those of the original discorhabdin G, supplied to Davies-Coleman and co-workers by Munro in 1996.21 Compound 58 was accordingly named discorhabdin G*.21 The configuration at C-8 in 58 was not established.
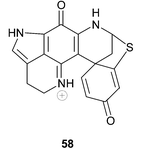
Sub-Antarctic sponges are also a source of discorhabdin-type compounds and the epinardins A–D 59–62 isolated in 1996 from two species of unidentified sponges dredged from a depth of 200 m off the Crozet archipelago (ca. 2600 km SE of South Africa) possess an allylic alcohol functionality as opposed to the enone system in the spiro-six-membered ring common to the discorhabdins.76 The relative stereochemistry of these four compounds was established from NMR data. Epinardin C 61 exhibited strong in vitro cytotoxicity to doxorubicin-resistant L1210/DX tumour cells (IC50 0.3 µg mL−1).76
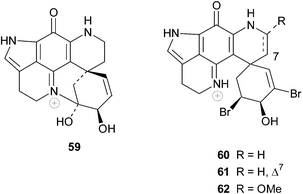
A review of bioactive marine alkaloids by Munro and co-workers published in 2000 has not only shed some light on the original structures of discorhabdins I–M 63–67 but also added two further structures, disorhabdins N and O 68 and 69, to the growing number of discorhabins isolated from New Zealand Latrunculia sponges.3 Paradoxically, the structure of discorhabdin H 70
(Δ4 discorhabdin J) was omitted from Munro and co-workers' review. However, the fully assigned spectral data and structure, including determination of an L-configuration for the thiohistidine residue, of 70
(isolated from the South African sponge Strongylodesma algoaensis) was recently reported by Antunes et al.21 The spectral data of Δ4 discorhabdin N 71, a minor metabolite in the South African sponge L. bellae, were also presented by Antunes et al.21 The relative stereochemistry at the four stereocentres in 71 remains unassigned. The spectral data of discorhabdins “I”
72 and L 66, isolated from deep water Argentian specimens of L. brevis, were published in 2004 by Reyes et al.77 Confusingly, the structure of the former compound possesses a thioether bridge linking ring C with C-5 of the spiro-ring, which is at variance with the structure of discorhabdin I 63 that appears in Munro and co-workers’ review. Discorhabdins “I” and L were both cytotoxic to human colon tumour cells HT-29 (GI50 0.35 and 0.12 µM respectively).77
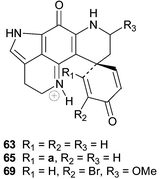
Discorhabdin P 73 (N-13-methyldiscorhabdin C), was isolated from a Batzella sponge (original voucher specimen re-identified as a Strongylodesma species by Samaai5,17) collected using a manned submersible from a depth of 141 m off the Great Bahamas bank.78 The structure of 73 followed from X-ray diffraction analysis. In common with other discorhabdins, 73 was shown to be cytotoxic to P-338 and A-549 tumour cell lines (IC50 0.03 and 0.4 µg mL−1 respectively). Interestingly, 73 was the first discorhabdin alkaloid to be screened for inhibition of calcineurin (an immune system signal transduction enzyme) and the pepitidase activity of CPP32 (caspase 3 proteases are an important group of enzymes implicated in apoptosis).78 Ironically, the nanomolar inhibition of CaN (IC50 0.55 µg mL−1) and CPP32 (IC50 0.55 µg mL−1) by 73 was not shared by discorhabdin C 47, which was inactive in these assays at a concentration of 5 µg mL−1.78
Discorhabdin Q 74 is effectively 16,17-dehydrodiscorhabdin B and was the dominant discorhabdin isolated from cytotoxic extracts of Australian specimens of Latrunculia purpurea, Zyzzya massalis, Z. fulinginosa and an unidentified Zyzzya. spp.79 Discorhabdin Q exhibited only moderate activity in the NCI's 60 cell line screen (mean panel GI50 0.5 µg mL−1) and Boyd and co-workers postulated that Δ16 unsaturation reduces cytoxicity relative to the more common 16,17-saturated members of the discorhabdin series.79
Extracts of a deep water Antarctic Latrunculia sponge and a south Australian Negombata sp. both yielded discorhabdin B 49 and its closely related cis-1,2-epoxy analogue, discorhabdin R 75.80 In light of the clear chemotaxonomic divisions between Latrunculia and Negombata species (see earlier), it is probable that the latter species was incorrectly identified and is possibly Latrunculia purpurea from the widespread distribution of this species off the southern coast of Australia.5 Spectroscopic examination of the two sponge extracts that yielded 75 confirmed that this compound was a natural product and not an oxidation artifact of the chromatographic work-up of the extract.80 No bioactivity data was reported for this compound.
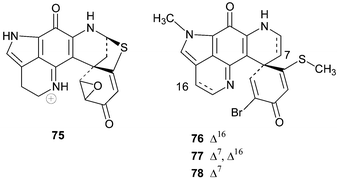
Three cytotoxic discorhabdins, S, T and U 76–78 were isolated from an unidentified Batzella sponge (probably a Strongylodesma species, see earlier) collected by a manned submersible from a depth of 141 m off the coast of the Bahamas.81 NOE data was used to establish the relative stereochemistry at the spiro-carbon in 76 and this configuration was extrapolated to the closely related discorhabdin T 77 (Δ7 discorhabdin S) and discorhabdin U 78 (16,17-dihydrodiscorhabdin S). Discorhabdins S–U 76–78 exhibited in vitro cytotoxicity to murine P-388 tumour cells (IC50 3.1, >5, and 0.17 µM respectively), A-549 human lung adenocarcinoma cells (IC50 >5, >5 and 0.17 µM respectively) and PANC-1 human pancreatic tumour cells (IC50 2.6, 0.7 and 0.07 µM respectively).81 The enhanced cytotoxicity of 78 relative to 76 and 77 against these three cell tumour lines further supports the hypothesis of Boyd and co-workers that cytotoxicity is diminished by Δ16-unsaturation in discorhabdins.79
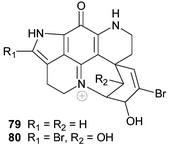
Discorhabdin V 79, and the closely related 14-bromo-1-hydroxydiscorhabdin V 80, recently isolated from a South African sponge Tsitsikamma pedunculata, exhibited cytotoxicity to HCT-116 human colon tumour cells (1.2 and 12.5 µM respectively).21 The relative configurations of the spiro-carbon, the oxymethine carbon(s) in the spiro-ring and the ring junction linking this ring to N-18 were not established in either compound.
Investigations of the natural product chemistry of latrunculid sponges, containing discorhabdin-type alkaloids, usually inadvertently leads to the re-isolation of known members of the discorhabdin ensemble. Reports of the re-isolation of ubiquitous discorhabdins in addition to new records of the isolation of closely related analogues of these compounds are presented below. Details of the single discorhabdin biosynthetic study are also discussed.
Discorhabdin A 48 has been identified in extracts of Zyzzya cf. marsailis32 (= Z. fuliginosa, see earlier) and more recently, the South African sponge Strongylodesma algoaensis21 and the Argentinian sponge Latrunculia brevis.76 The latter sponge also yielded discorhabdin B 49.76 Interestingly, in a comparative screening of 20 pyrroloiminoquinone and related metabolites against the human colon tumour cell line HCT116, 48 was the most cytotoxic (IC50 0.08 µM).21
The biosynthesis of discorhabdin B 49 has been investigated utilizing tissue slices of New Zealand collections of Latrunculia sp. B.82 [U-14C]-L-phenylalanine was found to be incorporated into the alkaloid, with similar levels of incorporation being experienced for sponge tissue that had been pre-treated with antibiotics and tissue that had not, leading to the conclusion that alkaloid biogenesis was probably a product of the sponge cells and not microbial symbionts. The putative biosynthetic sequence of discorhabdin B from phenylalanine by Munro et al. forms the basis of the expanded biosynthetic sequence outlined in Scheme 2 in which recently isolated pyrroloiminoquinone structural classes have been included.
Discorhabdin C 47 is seemingly ubiquitous in New Zealand Latrunculia species14,15,67,68,70,73,74 and a major metabolite in the Antarctic sponge L. apicalis (possibly L. biformis, see earlier).75 Although 47 has not been isolated from South African latrunculid sponges, a plethora of discorhabdin C analogues have been obtained from Tistsikamma favus and T. pedunculata.21,65T. pedunculata yielded 3-dihydrodiscorhabdin C 81 previously synthesized via reduction of discorhabdin C 47,73,74 and 3-dihydro-7,8-dehydrodiscorhabdin C 82.21 Bromination at C-14 in the left hand hemisphere of the pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline skeleton has only been found in discorhabdin-type metabolites isolated from the genus Tsitsikamma and may prove to be a suitable chemotaxonomic marker for this genus. Both 14-bromodiscorhabdin C 83 and 14-bromo-3-dihydrodiscorhabdin C 84 are prevalent in T. favus and T. pedunculata.21,65 A further minor C-14 brominated discorhabdin C metabolite, 14-bromo-3-dihydro-7,8-dehydrodiscorhabdin C 85, was also isolated from T. pedunculata.21 All five discorhabdin C analogues, 81–85, were cyctotoxic to HCT-116 tumour cells (IC50 0.32, 0.20, 0.01, 0.65, 0.22 µM respectively).21
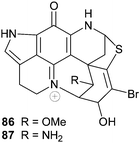
The South African sponge Strongylodesma algoaensis yielded discorhabdin D 50 while two minor discorhabdin D analogues, 1-aminodiscorhabdin D 86 and 1-methoxydiscorhabdin D 87 were isolated from L. bellae, a species that co-exists with S. algoaensis in Algoa Bay, South Africa.21 The stereochemistry at the four chiral carbon atoms in 86 and 87 was not established. Both 86 and 87 exhibited cytotoxicity in an HCT-116 cancer cell line screen (IC50 0.12 and 0.23 µM respectively).21
Discorhabdins serve as both pigments and chemical defence agents in sponges and a recent investigation of the distribution of the green pigment discorhabdin G 57 in the Antarctic sponge Latrunculia apicalis (possibly L. biformis, see earlier) revealed that the majority of this compound (>50%) is confined to the ectoderm of the sponge (0–2 mm).83L. apicalis is rarely preyed upon by the dominant Antarctic seastar spongivore, Perknaster fuscus, and in an elegant series of seastar tube foot retraction response experiments Baker and co-workers were able to confirm the role of discorhabdin G as a chemical deterrent to predation in L. apicalis.83
The complexity of the pyrido[2,3-h]pyrrolo[4,3,2-de]quinoline skeleton and the universal cytotoxicity associated with alkaloids possessing this core structure have made the discorhabdins attractive synthetic targets. The synthetic challenge provided by these complex metabolites is possibly reflected in the paucity of published discorhabdin syntheses with only the total synthesis of discorhabdins A 48,84,85 C 47,37,40,55,86–88 and E 5586 having been reported.
6 Conclusion
Over the last eighteen years pyrroloiminoquinone and related metabolites have been regularly isolated from sponges generally confined, after recent fairly extensive taxonomic revision,5–10 to the two families Latrunculiidae and Acarnidae. The sponges that yielded these metabolites have been collected from both shallow and deep-water habitats stretching from Antarctica to Japan and the Caribbean with the centres of diversity of the family Latrunculiidae, in particular, situated predominantly in the southern hemisphere.8 Although there have been substantial investigations of the chemistry of latrunculid sponges collected from the colder water regions off New Zealand,3,67,68,70,74 Australia,79,80 Antarctica,75,80,83 the sub-Antarctic islands76 and more recently South Africa,21,65 our knowledge of the pyrroloiminoquinone metabolites of South American latrunculid sponges remains limited.77 Therefore, it is likely that latrunculid sponges collected from the cold water areas off South America may yield a cohort of new pyrroloiminoquinone metabolites in future studies. No natural product investigations of the deep-water latrunculid genus Sceptrella have been reported and chemotaxonomic evidence in support of the inclusion of Sceptrella in the family Latrunculiidae is still required. The isolation of makaluvamine A from a terrestrial myxomycete45 and wakayin from an ascidian66 clearly indicates that pyrroloiminoquinone and related metabolites are neither the sole preserve of marine sponges nor restricted to the marine environment. A microbial source for the pyrroloiminoquinone metabolites isolated from a New Zealand Latrunculia species has been tentatively discounted.82 However, it may be premature to extrapolate this result to all pyroloiminoquinone-producing sponges and an investigation of a possible symbiont origin of the bis-pyrroloiminoquinones wakayin and the tsitsikammamines, in particular, would be useful.21 Finally, the complex structures of the discorhabdins and the potent, albeit generally nonspecific, universal cytotoxicity of marine pyrroloiminoquinone metabolites will continue to make these compounds attractive and challenging synthetic targets in the future.7 References
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2004, 21, 1 RSC; J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2003, 20, 1 RSC.
- D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1 RSC (and earlier reviews in this series).
- S. Urban, S. J. H. Hickford, J. W. Blunt and M. H. G. Munro, Curr. Org. Chem., 2000, 4, 765 CAS.
- Q. Ding, K. Chichak and J. W. Lown, Curr. Med. Chem., 1999, 6, 1 CAS.
- T. Samaai, Ph.D. Thesis, 2002, University of the Western Cape, South Africa.
- M. Kelly and T. Samaai in Systema Porifera. Guide to the supraspecific classification of sponges and spongiomorphs (Porifera), eds. J. N. A. Hooper and R. W. M. Van Soest, Kluwer Academic/Plenum, New York, 2002, p. 694 Search PubMed.
- M. Kelly and T. Samaai in Systema Porifera. Guide to the supraspecific classification of sponges and spongiomorphs (Porifera), eds. J. N. A. Hooper and R. W. M. Van Soest, Kluwer Academic/Plenum, New York, 2002, p. 707 Search PubMed.
- T. Samaai and M. Kelly in Systema Porifera. Guide to the supraspecific classification of sponges and spongiomorphs (Porifera), eds. J. N. A. Hooper and R. W. M. Van Soest, Kluwer Academic/Plenum, New York, 2002, p. 708 Search PubMed.
- T. Samaai, M. J. Gibbons, M. Kelly and M. Davies-Coleman, Zootaxa, 2003, 371, 1 Search PubMed.
- J. N. A. Hooper in Systema Porifera. Guide to the supraspecific classification of sponges and spongiomorphs (Porifera), eds. J. N. A. Hooper and R. W. M. Van Soest, Kluwer Academic/Plenum, New York, 2002, p. 412 Search PubMed.
- M. Kelly-Borges and J. Vacelet, Mem. Queensl. Mus., 1995, 38, 477 Search PubMed.
- A. Groweiss, U. Shmueli and Y. Kashman, J. Org. Chem., 1983, 48, 3512 CrossRef CAS.
- R. J. Capon, J. K. MacLeod and A. C. Willis, J. Org. Chem., 1987, 52, 339 CrossRef CAS.
- K. Miller, B. Alvarez, C. Battershill, P. Northcote and H. Parthasarathy, Mar. Biol. (Berlin), 2001, 139, 235 Search PubMed.
- B. Alvarez, P. R. Bergquist and C. N Battershill, N. Z. J. Mar. Freshwater Res., 2002, 36, 151 Search PubMed.
- R. W. M. Van Soest, J. C. Breakman, D. J. Faulkner, E. Hajdu, M. K. Harper and J. Vacelet, Bull. Inst. R. Sci. Nat. Belg., Biol., 1996, 66, 89 Search PubMed.
- T. Samaai, R. Keyzers and M. Davies-Coleman, Zootaxa, 2004, 584, 1 Search PubMed.
- W. H. de Weerdt, Beaufortia, 1985, 36, 81 Search PubMed.
- R. W. M. Van Soest in Systema Porifera. Guide to the supraspecific classification of sponges and spongiomorphs (Porifera), eds. J. N. A. Hooper and R. W. M. Van Soest, Kluwer Academic/Plenum, New York, 2002, p. 521 Search PubMed.
- W. H. de Weerdt in Systema Porifera. Guide to the supraspecific classification of sponges and spongiomorphs (Porifera), eds. J. N. A. Hooper and R. W. M. Van Soest, Kluwer Academic/Plenum, New York, 2002, p. 852 Search PubMed.
- E. M. Antunes, D. R. Beukes, M. Kelly, T. Samaai, L. R. Barrows, K. M. Marshall, C. Sincich and M. T. Davies-Coleman, J. Nat. Prod, 2004, 67, 1268 CrossRef CAS.
- X. Fu, P. L. Ng, F. J. Schmitz, M. B. Hossain, D. van der Helm and M. Kelly-Borges, J. Nat. Prod., 1996, 59, 1104 CrossRef CAS.
- S. P. Gunasekera, P. J. McCarthy, R. E. Longley, S. A. Pomponi and A. E. Wright, J. Nat. Prod., 1999, 62, 1208 CrossRef CAS.
- S. Sakemi, H. H. Sun, C. W. Jefford and G. Bernardinelli, Tetrahedron Lett., 1989, 30, 2517 CrossRef CAS.
- L. C. Chang, S. Otero-Quintero, J. N. A. Hooper and C. A. Bewley, J. Nat. Prod., 2002, 65, 776 CrossRef CAS.
- D. B. Stierle and D. J. Faulkner, J. Nat. Prod., 1991, 54, 1131 CrossRef CAS.
- J. R. Carney, P. J. Scheuer and M. Kelly-Borges, Tetrahedron, 1993, 49, 8483 CrossRef CAS.
- Personal communication from Dr R. W. M. Van Soest.
- N. K. Utkina, A. E. Makarchenko, V. A. Denisenko and P. S. Dmitrenok, Tetrahedron Lett., 2004, 45, 7491 CrossRef CAS.
- E. W. Schmidt, M. K. Harper and D. J. Faulkner, J. Nat. Prod., 1995, 58, 1861 CrossRef CAS.
- N. K. Utkina, A. V. Gerasimenko and D. Yu. Popov, Russ. Chem. Bull., 2003, 52, 258 CrossRef CAS.
- D. C. Radisky, E. S. Radisky, L. R. Barrows, B. R. Copp, R. A. Kramer and C. M. Ireland, J. Am. Chem. Soc., 1993, 115, 1632 CrossRef CAS.
- J. F. Hu, J. A. Schetz, M. Kelly, J. N. Peng, K. K. H. Ang, H. Flotow, C. Y. Leong, S. B. Ng, A. D. Buss, S. P. Wilkins and M. T. Hamann, J. Nat. Prod., 2002, 65, 476 CrossRef CAS.
- R. E. Longley, O. J. McConnell, E. Essich and D. Harmody, J. Nat. Prod., 1993, 56, 915 CrossRef CAS.
- M. Alvarez, M. A. Bros, G. Gras, W. Ajana and J. A. Joule, Eur. J. Org. Chem., 1999, 1173 CrossRef CAS.
- F. Yamada, S. Hamabuchi, A. Shimizu and M. Somei, Heterocycles, 1995, 41, 1905 CrossRef CAS.
- X. L. Tao, J. F. Cheng, S. Nishiyama and S. Yamamura, Tetrahedron, 1994, 50, 2017 CAS.
- X. L. Tao, S. Nishiyama and S. Yamamura, Chem. Lett., 1991, 1785 CAS.
- E. V. Sadanandan and M. P. Cava, Tetrahedron Lett., 1993, 34, 2405 CrossRef CAS.
- D. Roberts, J. A. Joule, M. A. Bros and M. Alvarez, J. Org. Chem., 1997, 62, 568 CrossRef CAS.
- D. Roberts, M. Alvarez and J. A. Joule, Tetrahedron Lett., 1996, 37, 1509 CrossRef CAS.
- A. J. Peat and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 1028 CrossRef CAS.
- D. Roberts, L. Venemalm, M. Alvarez and J. A. Joule, Tetrahedron Lett., 1994, 35, 7857 CAS.
- H. H. Sun, S. Sakemi, N. Burres and P. McCarthy, J. Org. Chem., 1990, 55, 4964 CrossRef CAS.
- M. Ishibashi, T. Iwasaki, S. Imai, S. Sakamoto, K. Yamaguchi and A. Ito, J. Nat. Prod., 2001, 64, 108 CrossRef CAS.
- D. A. Venables, G. P. Concepción, S. S. Matsumoto, L. R. Barrows and C. M. Ireland, J. Nat. Prod., 1997, 60, 408 CrossRef CAS.
- A. Casapullo, A. Cutignano, I. Bruno, G. Bifulco, C. Debitus, L. Gomez-Paloma and R. Riccio, J. Nat. Prod., 2001, 64, 1354 CrossRef CAS.
- L. R. Barrows, D. C. Radisky, B. R. Copp, D. S. Swaffar, R. A. Kramer, R. L. Warters and C. M. Ireland, Anti-Cancer Drug Des., 1993, 8, 333 CAS.
- S. S. Matsumoto, H. M. Haughey, D. M. Schmehl, D. A. Venables and C. M. Ireland, Anti-Cancer Drugs, 1999, 10, 39 CAS.
- T. Izawa, S. Nishiyama and S. Yamamura, Tetrahedron Lett., 1994, 35, 917 CrossRef CAS.
- T. Izawa, S. Nishiyama and S. Yamamura, Tetrahedron, 1994, 50, 13593 CrossRef CAS.
- M. Iwao, O. Motoi, T. Fukuda and F. Ishibashi, Tetrahedron, 1998, 54, 8999 CrossRef CAS.
- G. A. Kraus and N. Selvakumar, J. Org. Chem., 1998, 63, 9846 CrossRef CAS.
- J. D. White, K. M. Yager and T. Yakura, J. Am. Chem. Soc., 1994, 116, 1831 CrossRef CAS.
- E. V. Sadanandan, S. K. Pillai, M. V. Lakshmikantham, A. D. Billimoria, J. S. Culpepper and M. P. Cava, J. Org. Chem., 1995, 60, 1800 CrossRef CAS.
- K. M. Aubart and C. H. Heathcock, J. Org. Chem., 1999, 64, 16 CrossRef CAS.
- M. Makosza, J. Stalewski and O. S. Maslennikova, Synthesis, 1997, 1131 CrossRef CAS.
- V. Bénéteau, A. Pierré, B. Pfeiffer, P. Renard and T. Besson, Bioorg. Med. Chem. Lett., 2000, 10, 2231 CrossRef CAS.
- Y. Kita, M. Egi, T. Takada and H. Tohma, Synthesis, 1999, 885 CrossRef CAS.
- Y. Kita, M. Egi and H. Tohma, Chem. Commun., 1999, 143 RSC.
- H. Wang, N. H. Al-Said and J. W. Lown, Tetrahedron Lett., 1994, 35, 4085 CrossRef CAS.
- R. Zhao, B. Oreski and J. W. Lown, Bioorg. Med. Chem. Lett., 1996, 6, 2169 CrossRef CAS.
- D. A. Venables, L. R. Barrows, P. Lassota and C. M. Ireland, Tetrahedron Lett., 1997, 38, 721 CrossRef.
- Y. Moro-oka, T. Fukuda and M. Iwao, Tetrahedron Lett., 1999, 40, 1713 CrossRef CAS.
- G. J. Hooper, M. T. Davies-Coleman, M. Kelly-Borges and P. S. Coetzee, Tetrahedron Lett., 1996, 37, 7135 CrossRef CAS.
- B. R. Copp, C. M. Ireland and L. R. Barrows, J. Org. Chem., 1991, 56, 4596 CrossRef CAS.
- N. B. Perry, J. W. Blunt, J. D. McCombs and M. H. G. Munro, J. Org. Chem., 1986, 51, 5478 CrossRef CAS.
- N. B. Perry, J. W. Blunt and M. H. G. Munro, Tetrahedron, 1988, 44, 1727 CrossRef CAS.
- J. Kobayashi, J.-F. Cheng, M. Ishibashi, H. Nakamura, Y. Ohizumi, Y. Hirata, T. Sasaki, H. Lu and J. Clardy, Tetrahedron Lett., 1987, 28, 4939 CrossRef CAS.
- N. B. Perry, J. W. Blunt, M. H. G. Munro, T. Higa and R. Sakai, J. Org. Chem., 1988, 53, 4127 CrossRef CAS.
- J.-F. Cheng, Y. Ohizumi, M. R. Wälchli, H. Nakamura, Y. Hirata, T. Sasaki and J. Kobayashi, J. Org. Chem., 1988, 53, 4621 CrossRef CAS.
- J. Kobayashi, J.-F. Cheng, S. Yamamura and M. Ishibashi, Tetrahedron Lett., 1991, 32, 1227 CrossRef CAS.
- J. W. Blunt, M. H. G. Munro, C. N. Battershill, B. R. Copp, J. D. McCombs, N. B. Perry, M. Prinsep and A. M. Thompson, New J. Chem., 1990, 14, 761 Search PubMed.
- B. R. Copp, K. F. Fulton, N. B. Perry, J. W. Blunt and M. H. G. Munro, J. Org. Chem., 1994, 59, 8233 CrossRef CAS.
- A. Yang, B. J. Baker, J. Grimwade, A. Leonard and J. B. McClintock, J. Nat. Prod., 1995, 58, 1596 CrossRef CAS.
- M. D'Ambrosio, A. Guerriero, G. Chiasera, F. Pietra and M. Tatò, Tetrahedron, 1996, 52, 8899 CrossRef CAS.
- F. Reyes, R. Martín, A. Rueda, R. Fernández, D. Montalvo, C. Gómez and J. M. Sánchez-Puelles, J. Nat. Prod., 2004, 67, 463 CrossRef CAS.
- S. P. Gunasekera, P. J. McCarthy, R. E. Longley, S. A. Pomponi, A. E. Wright, E. Lobkovsky and J. Clardy, J. Nat. Prod., 1999, 62, 173 CrossRef CAS.
- M.-G. Dijoux, W. R. Gamble, Y. F. Hallock, J. H. Cardellina, R. van Soest and M. R. Boyd, J. Nat. Prod., 1999, 62, 636 CrossRef.
- J. Ford and R. J. Capon, J. Nat. Prod., 2000, 63, 1527 CrossRef CAS.
- S. P. Gunasekera, I. A. Zuleta, R. E. Longley, A. E. Wright and S. A. Pomponi, J. Nat. Prod., 2003, 66, 1615 CrossRef CAS.
- R. E. Lill, D. A. Major, J. W. Blunt, M. H. G. Munro, C. N. Battershill, M. G. McLean and R. L. Baxter, J. Nat. Prod., 1995, 58, 306 CrossRef.
- F. B. Furrow, C. D. Amsler, J. B. McClintock and B. J. Baker, Mar. Biol. (Berlin), 2003, 143, 443 Search PubMed.
- H. Tohma, Y. Harayama, M. Hashizume, M. Iwata, M. Egi and Y. Kita, Angew. Chem., Int. Ed., 2002, 41, 348 CrossRef CAS.
- H. Tohma, Y. Harayama, M. Hashizume, M. Iwata, Y. Kiyono, M. Egi and Y. Kita, J. Am. Chem. Soc., 2003, 125, 11235 CrossRef CAS.
- Y. Kita, H. Tohma, M. Inagaki, K. Hatanaka and T. Yakura, J. Am. Chem. Soc., 1992, 114, 2175 CrossRef CAS.
- S. Nishiyama, J.-F. Cheng, X. L. Tao and S. Yamamura, Tetrahedron Lett., 1991, 32, 4151 CrossRef CAS.
- K. Marshall Aubart and C. H. Heathcock, J. Org. Chem., 1999, 64, 16 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |