Simple indole alkaloids and those with a non-rearranged monoterpenoid unit
Masanori Somei and Fumio Yamada
Division of Pharmaceutical Sciences, Graduate School of Natural Science and Technology, Kanazawa University, Japan
First published on 10th January 2005
Abstract
Covering: January 2003 to December 2003. Previous review: Nat. Prod. Rep., 2004, 21, 211
This review covers newly isolated simple indole alkaloids, structure determinations, total syntheses and biological activities reported in the literature in 2003.
 Masanori Somei Masanori Somei | Masanori Somei was born in 1941 in Chiba, Japan. He attended Tokyo University (1961–1970) and obtained his Ph.D. degree (1970) under the supervision of Professor Toshihiko Okamoto. He then moved to ITSUU Laboratory (1970–1976). After postdoctoral work (1975–1976) with Professor William G. Dauben (University of California, Berkeley), he was promoted to associate professor in 1976 and full professor in 1984, his current position. He received the Pharmaceutical Science of Japan Award for Young Scientists (1971) and the Kametani award (2001). His main research interests are verification of his 1-hydroxyindole hypotheses, their synthetic applications and creation of suitable new reactions for realising syntheses of indole alkaloids as simply as possible. He proposed the originality rate, the intellectual property factor, and the application potential factor for evaluating the effectiveness of organic synthesis. |
 Fumio Yamada Fumio Yamada | Fumio Yamada is an associate professor of Kanazawa University. He was born in 1955 in Toyama, Japan and received his Batchelor of Pharmaceutical Science (1978) from Kanazawa University and his Ph.D. degree (1988) from Tokyo University. He joined the Faculty of Pharmaceutical Sciences, Kanazawa University in 1980 and was promoted to his current position in 1997. He worked as a postdoctoral fellow with Professor Alan. P. Kozikowski at University of Pittsburgh and Mayo Clinic Jacksonville from 1990 to 1991. He received the award of encouragement in 1999 from the Hokuriku branch of Japan Pharmaceutical Society. His current research interests are the synthesis of biologically active indole alkaloids as simply as possible and creation of new reactions suitable for attaining simple syntheses of the target molecules. |
1 Introduction
This review covers the literature on simple indole alkaloids and those with a non-rearranged monoterpenoid unit from the beginning of 2003 up to the end of 2003. In this series, marine natural products and peptide alkaloids have also been surveyed. As a result, there will be some overlap with the marine alkaloids and the peptide alkaloids containing the indole moiety. Reviews on the synthesis of melatonin1 and betalains2 have appeared.2 Simple indole alkaloids
2.1 Non-tryptamines
Extensive fractionation of the crude organic extract from a Puerto Rican collection of Lyngbya majuscula has led to the discovery of a new indole derivative (1).3 The structure was deduced by spectroscopic methods (1H and 13C NMR, HSQC, TOCSY, and HMBC experiments).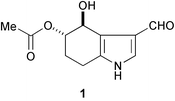
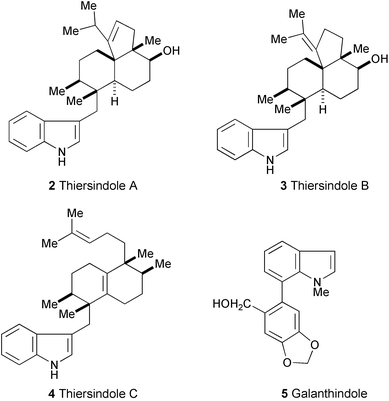
Thiersindoles A–C (2–4), new 3-substituted indole diterpenoids, have been isolated from organic extracts of a new Penicillium species (P. thiersii).4 Their structures, including absolute stereochemistry, were determined by analysis of NMR data (1H and 13C NMR, NOESY, DEPT, HMQC, and HMBC experiments) and application of Mosher's method. The [6,6,5] tricyclic diterpenoid skeleton in compounds 2 and 3 is unprecedented in any of the known members of this class.
A new indole alkaloid, galanthindole (5), has been isolated from Galanthus plicatus ssp. byzantinus
(Amaryllidaceae), a plant native to northwestern Turkey.5 The structure was determined by spectral analysis (1H and 13C NMR, COSY, HMQC, NOESY, and HMBC spectra).
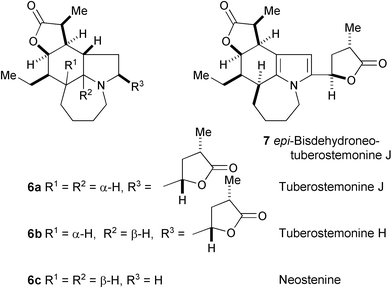
Bioactivity-directed fractionation of the crude extract of Stemona tuberosa has led to the isolation and characterisation of four new alkaloids, tuberostemonine J (6a), tuberostemonine H (6b), neostenine (6c), and epi-bisdehydroneotuberostemonine J (7).6 Their structures were determined by spectroscopic analysis (1H and 13C NMR, and DEPT spectra), and their absolute structures elucidated by X-ray crystallographic analysis. These alkaloids were examined for antitussive activity in the guinea pig after cough induction by citric acid aerosol stimulation; 6c showed significant antitussive activity. Studies of the structure–activity relationships for these alkaloids and synthetic analogues revealed that the saturated tricyclic pyrrolo[3,2,1-jk]benzazepine nucleus is the primary key structure contributing to the antitussive activity, and that the all-cis configurations at the three ring junctions are the optimal structure for the antitussive activity of stenine-type Stemona alkaloids.
The aquatic peptide aeruginosin EI461 was isolated from the cyanobacterium Microcyctis aeruginosa and structure 8a was proposed (Scheme 1).7 Valls' group had prepared a putative structure 8a for aeruginosin EI461 from O-methyl-D-tyrosine,8 but the synthetic material was not identical with the natural product. A corrected structure (8b) for aeruginosin EI461 was proposed and the structure proven by synthesis. O-Methyl-L-tyrosine was converted to the azabicyclic ketone 9. Successive couplings of the dipeptide 10 with L-Hpla and NH4OH, and then a deprotection step gave aeruginosin EI461 (8b).
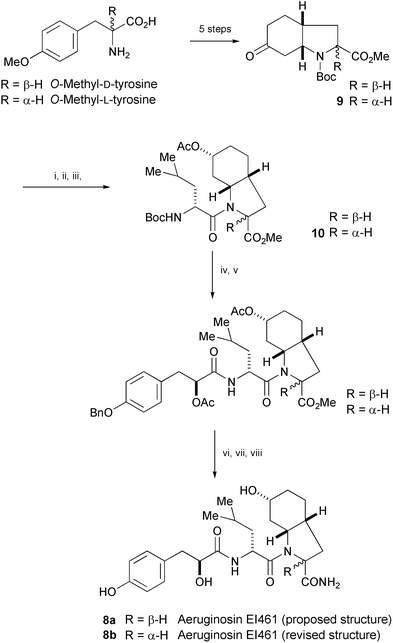 | ||
| Scheme 1 Reagents and conditions: i, NaBH4, MeOH; ii, Ac2O, DMAP, PyBOP; iii, 1. TFA, CH2Cl2, 0 °C, 2. Boc-D-Leu, CH2Cl2, PyBOP, NMM, rt; iv, TFA, CH2Cl2, 0 °C; v, AcO-L-Hpla(Bn), CH2Cl2, PyBOP, NMM, rt; vi, 0.1 N LiOH, THF, rt; vii, HOBt·H2O, EDC, NMM, THF, 0 °C then NH4OH, rt; viii, H2, 10% Pd/C, rt. | ||
The first stereoselective total synthesis of (+)-benzastatin E (11), isolated from Streptomyces nitrosporeus 30643,9 has been accomplished in 11 steps from commercially available (S)-2-indolinecarboxylic acid (12) (Scheme 2).10 The key reaction is a diastereoselective Grignard addition to 2-acylindole 13 leading to 14, which was further converted into (+)-benzastatin E (11). The previously unknown absolute stereochemistry of (+)-11 was determined as (9S,10R).
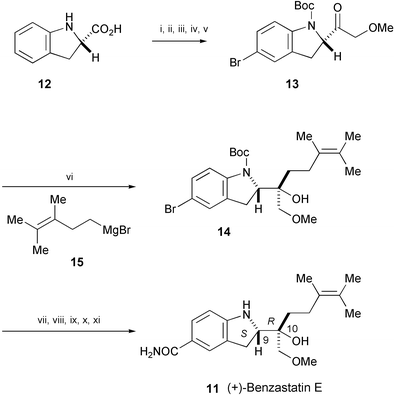 | ||
| Scheme 2 Reagents and conditions: i, MeOH, H2SO4, 80 °C; ii, Boc2O, CH2Cl2, rt; iii, NBS, DMF, 0 °C; iv, i-PrMgBr, Me(MeO)NH·HCl, THF, −20 to −10 °C; v, MeOCH2Sn(n-Bu)3, n-BuLi, THF; vi, 15, THF, −78 °C; vii, HCO2H, CH2Cl2, 40 °C; viii, Me2C(OMe)2, PPTS, CH2Cl2, rt; ix, t-BuLi, CO2, Et2O, −78 to 0 °C; x, CDI, 28% aq. NH3, THF, rt; xi, PPTS, MeOH, rt. | ||
Stereoselective synthesis of (−)-monatin (16), isolated from the bark of the roots of Schlerochiton ilicifolius,11 has been achieved by chelation-controlled cycloaddition of the nitrone 17 with the allyl alcohol 18 in the presence of MgBr2·OEt2 to produce 19 (Scheme 3).12 Cycloadduct 19 was converted into (−)-monatin (16) through the lactone 20.
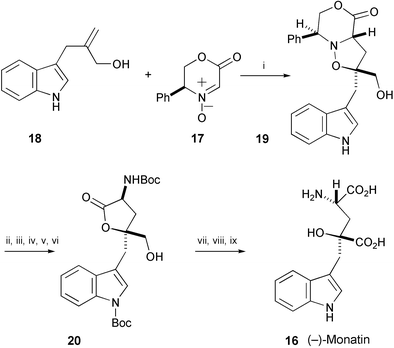 | ||
| Scheme 3 Reagents and conditions: i, MgBr2·OEt2, CH2Cl2, rt; ii, TBSCl, imidazole, DMF; iii, Boc2O, DMAP, MeCN; iv, HF·pyridine, THF; v, H2, Pd(OH)2/C, MeOH; vi, Boc2O, MeCN; vii, PDC, DMF; viii, HCl, HCO2H; ix, NaOH, MeOH then Amberlite® IR-120-H+ form, aq. NH3. | ||
The total synthesis of (±)-alantrypinone (21), isolated from the extracts of the fungus Penicillium thymicola,13 has been accomplished employing a novel aza-Diels–Alder reaction of 3-methyleneoxindole (22) and the azadiene 23 as the key step (Scheme 4).14 The reaction sequence comprises 8 steps starting from anthranilic acid. Mild acid hydrolysis of the adduct 24 afforded (±)-alantrypinone (21).
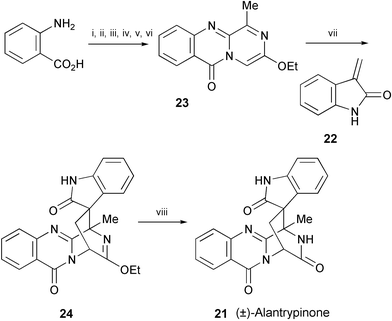 | ||
| Scheme 4 Reagents and conditions: i, glycine ethyl ester, EDCI, CH2Cl2, rt; ii, Fmoc-L-Ala–OH, EDCI, MeCN, rt; iii, PPh3, Br2, Et3N, CH2Cl2, rt; iv, piperidine, CH2Cl2, rt; v, Na2CO3, Et3O+BF4−, CH2Cl2, rt; vi, DDQ, PhH, reflux; vii, 22, CHCl3, rt; viii, 1 N HCl, AcOEt, rt. | ||
A total synthesis of (±)-epiuleine (25), isolated from Aspidosperma subincanum,15 has been achieved starting from 4-methoxypyridine and indolyl magnesium bromide (Scheme 5).16 The key finding is the isolation of the thermodynamic enolate as the vinyl triflate 26 in the 1,4-reduction of dihydropyridone (27). Heck vinylation of 26 followed by reductive methylation afforded 28, which was then converted into (±)-epiuleine (25) according to the reported procedure.17,18
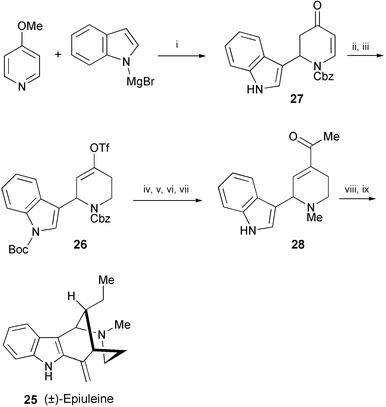 | ||
| Scheme 5 Reagents and conditions: i, CbzCl, THF; ii, Boc2O, DMAP; iii, L-Selectride, N-(5-chloro-2-pyridyl)triflimide, 0 °C; iv, n-butyl vinyl ether, Pd(OAc)2, dppf, THF, 40 °C; v, 1,4-cyclohexadiene, Pd/C; vi, CH2O, NaBH3CN; vii, TFA; viii, EtMgBr, CuCl, 0 °C; ix, p-TsOH, 40 °C. | ||
The first total synthesis of the inhibitor of 3α-hydroxysteroid dehydrogenase, 0231B (29), isolated from a fermentation broth of Streptomyces sp. HKI0231,19 has been achieved starting from 6-methylindole (30) (Scheme 6).20 Introduction of an acyl group into the 3-position of 30, cyclisation of the side chain at the 3-position of 31 to the 4-position and subsequent elimination of the phenyl group, and conjugate addition of the salicylamide derivative afforded 32. Compound 32 was then converted into 0231B (29).
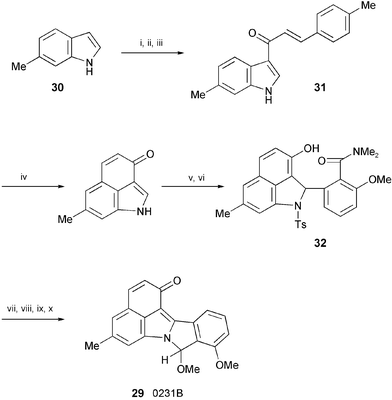 | ||
| Scheme 6 Reagents and conditions: i, trimethylacetyl chloride, DMAP, Et3N, CH2Cl2; ii, trans-p-methylcinnamoyl chloride, AlCl3, CH2Cl2; iii, 1 N NaOH, MeOH; iv, AlCl3, CHCl2CHCl2, 80 °C; v, TsCl, pyridine; vi, N,N-dimethyl-O-methylsalicylamide, s-BuLi, TMEDA, THF; vii, O2, KOH, CuCl, MeOH; viii, PhMe, reflux; ix, LiAlH4, THF, −40 °C; x, CSA, MeOH, reflux. | ||
The total synthesis of anhydrolycorinone (33) and hippadine (34), isolated from Amaryllidaceae plants,21 has been accomplished by an iron-mediated construction of the indole nucleus (Scheme 7).22 Nucleophilic addition of the lithium ester enolate 35 to the iron complex 36, followed by an iron-mediated oxidative alkylamine cyclisation, afforded the iron complex 37. This complex was converted into anhydrolycorinone (33) by an intramolecular palladium-mediated biaryl coupling. The oxidation of 33 with DDQ provided hippadine (34).
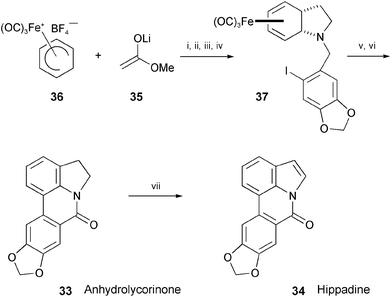 | ||
| Scheme 7 Reagents and conditions: i, THF, −78 °C; ii, DIBAL-H, PhMe, −78 °C; iii, iodopiperonylamine·HCl, MeOH, NaBH3CN, 4 Å MS; iv, Cp2FePF6, Na2CO3, CH2Cl2, 25 °C; v, Me3NO, Me2CO, reflux; vi, Pd(PPh3)4, DMF, 100 °C, air; vii, DDQ, CH2Cl2, reflux. | ||
The first total synthesis of (−)-21-isopentenylpaxilline (38), isolated from the sclerotioid ascostromata of Eupenicillium shearii (NRRL3324),23 has been achieved starting from 39 and 40 (Scheme 8).24,25 Key elements of the synthesis include the stereocontrolled construction of the eastern fragment (41), a highly efficient union of the eastern and western fragments (41 and 42, respectively), and the construction of 43, entailing mesylation followed by treatment with a Grignard reagent. Compound 43 was further converted into (−)-21-isopentenylpaxilline (38).
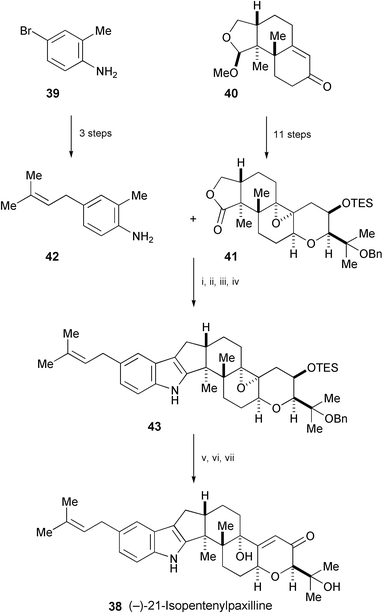 | ||
| Scheme 8 Reagents and conditions: i, 42, MeLi, Et2O, TMSCl, 0 °C, then s-BuLi, 0 °C to rt, then 0 °C quench with 41, 0 °C to rt; ii, PhMe, 110 °C; iii, MsCl, DMAP, CH2Cl2; iv, t-BuMgCl, PhMe, 110 °C; v, TBAF, THF; vi, Ph3BiCO3, PhH; vii, 10% Pd/C, 1-methyl-1,4-cyclohexadiene, MeOH, 65 °C. | ||
The first total synthesis of (−)-penitrem D (44), isolated from Penicillium crustosum,26 has been accomplished in a similar convergent and stereocontrolled manner (Scheme 9).27 Union of the western hemisphere (45) and the eastern hemisphere (46) was achieved employing the 2-substituted indole synthesis. After oxidation of 47 and subsequent selective removal of the TMS and TES groups, a Sc(OTf)3-promoted cationic cascade reaction completed construction of the nonacyclic system 48, which was then converted into (−)-penitrem D (44).
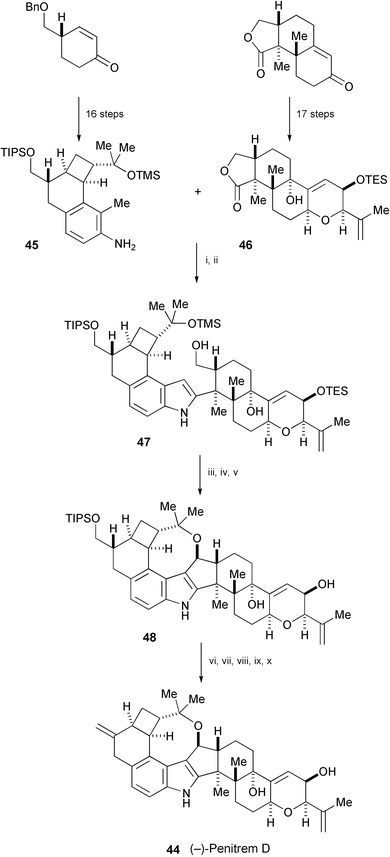 | ||
| Scheme 9 Reagents and conditions: i, n-BuLi, THF, −78 °C to rt, then TMSCl, 0 °C, then s-BuLi, 0 °C, then 46, THF–Et2O (1 : 1), 0 °C; ii, SiO2, CHCl3; iii, SO3·pyridine, DMSO–Et3N (4 : 1); iv, 1 N HCl, THF–H2O (5 : 1); v, Sc(OTf)3, PhH; vi, Ac2O, DMAP, Et3N, CH2Cl2; vii, TBAF, THF, 0 °C to rt; viii, o-nitrophenylselenocyanide, P(n-Bu)3, THF; ix, m-CPBA, NaHCO3, CH2Cl2, 0 °C to rt; x, K2CO3, MeOH, 0 °C to rt. | ||
2.1.1 Indole auxins and phytoalexins
Three new cytotoxic 3,6-disubstituted indoles (49a–c) have been isolated from the mycelium of a strain identified as Streptomyces sp. (BL-49-58-005), which was separated from a Mexican marine invertebrate.28 Their structures were established by analysis of spectral data (1H and 13C NMR, COSY, HMQC, and HMBC experiments). Cytotoxic assays for these compounds were performed against a panel of 14 different tumor cell lines. Compound 49a showed the best activity against K-562 (leukemia), with a GI50 value of 8.46 µM.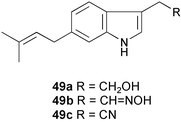
Hinckdentine A (50a) is an alkaloid isolated from the bryozoan Hincksinoflustra denticulate.29 The first synthesis of unnatural 8-desbromohinckdentine A (50b) was achieved using a 20-step reaction sequence from readily available starting materials 51 and 52 (Scheme 10).30 The quaternary center of hinckdentine A was formed using an uncommon pinacol-like rearrangement of 53 to produce key intermediate 54. The lactam 56 was synthesised by employing a seven-membered ring-forming intramolecular aldol condensation of 55 and subsequent reduction with complete stereocontrol. The dihydropyrimidine ring of 57 was prepared by means of a reaction sequence in which the Pd-catalysed amination of 56 is the key step. Bromination of 57 in HCO2H afforded 8-desbromohinckdentine A (50b).
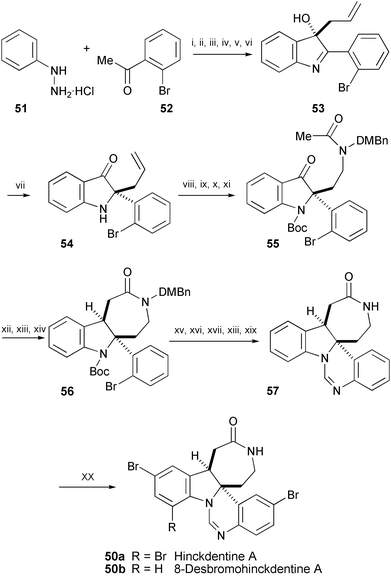 | ||
Scheme 10 Reagents and conditions: i, polyphosphoric acid, 120 °C; ii, NaNO2, AcOH, rt; iii, Na2S2O4, NaOH, H2O, EtOH, reflux; iv, PbO2, PhH, reflux; v, 1 N HCl, PhMe, rt, then reduced pressure, 105 °C; vi, allylmagnesium bromide, THF, rt; vii, 88% HCO2H, PhMe, reflux; viii, Boc2O, DMAP, MeCN, rt; ix, O3, MeOH, −78 °C to rt; x, 2,4-dimethoxybenzylamine, AcOH, NaBH3CN, MeOH, THF, rt; xi, Ac2O, pyridine, 0 °C to rt; xii, LDA, THF, −78 °C, then AcOH, THF, −78 °C to rt; xiii, MsCl, pyridine, 0 °C to rt; xiv, Mg turnings, MeOH, rt; xv, Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH, NaOt-Bu, Pd(DPPF)2Cl2, DPPF, PhMe, 140 °C; xvi, 2 N HCl, THF, rt; xvii, 20% TFA, CH2Cl2, 0 °C to rt; xviii, 96% HCO2H–2-propanol (1 : 1), 75 °C; xix, 96% HCO2H, 100 °C; xx, Br2, 96% HCO2H, rt. NH, NaOt-Bu, Pd(DPPF)2Cl2, DPPF, PhMe, 140 °C; xvi, 2 N HCl, THF, rt; xvii, 20% TFA, CH2Cl2, 0 °C to rt; xviii, 96% HCO2H–2-propanol (1 : 1), 75 °C; xix, 96% HCO2H, 100 °C; xx, Br2, 96% HCO2H, rt. | ||
Wasalexin A (58) and desmethyl analogue 59 have been prepared starting from phenylacetyl chloride (Scheme 11).31 Following formylation of the key intermediate3260, the resulting enol was converted to the enamine 61. Treatment of 61 with carbon disulfide, followed by methylation, afforded 58 or 59 depending on the conditions. The Z configuration of the double bond of 59 was determined on the basis of NOE experiments. A synthesis of arvelexin33 (62) from 4-methoxyindole (63) employing a Grignard reagent has also been developed.31
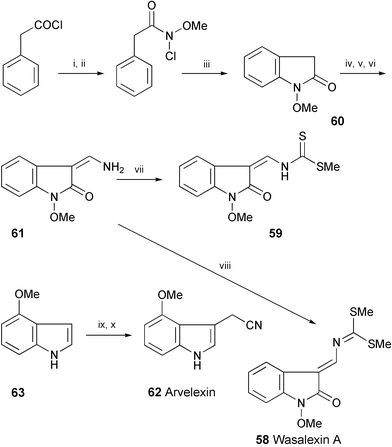 | ||
| Scheme 11 Reagents and conditions: i, MeONH2·HCl, Na2CO3; ii, t-BuOCl; iii, Ag2CO3, TFA; iv, HCO2Et, NaH; v, SOCl2; vi, NH4OH, MeOH; vii, NaH (1.1 eq.), CS2, then MeI (1.1 eq.); viii, NaH (2.1 eq.), CS2, then MeI (2.1 eq.); ix, MeI, Mg, Et2O; x. BrCH2CN. | ||
2.1.2 Carbazoles and indolocarbazoles
Two new carbazole alkaloids, murrayanine (64) and 8,8″-biskoenigine (65) have been isolated from Murraya koenigii.34 Their structures were elucidated by spectral data (1H and 13C NMR, ROESY, and HMBC experiments). Compound 65 showed antiosteoporotic activity in the CAT-B model with IC50 1.3 µg ml−1. The synthesis of 65 from koenigine (66) was carried out through oxidative coupling using a solid-state reaction (Scheme 12).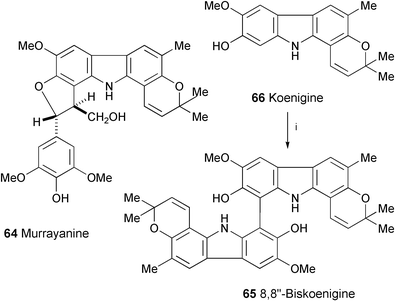 | ||
| Scheme 12 Reagents and conditions: i, FeCl3·6H2O in the solid state. | ||
Two novel quasi-racemic bisindole alkaloids, cycloaplysinopsin A (67a) and B (67b) have been isolated from tropical Indo-Pacific scleractinian corals of the family Dendrophylliidae.35 Their structures were elucidated by spectroscopic analysis (ESIMS, 1H and 13C NMR, COSY, and NOE experiments).
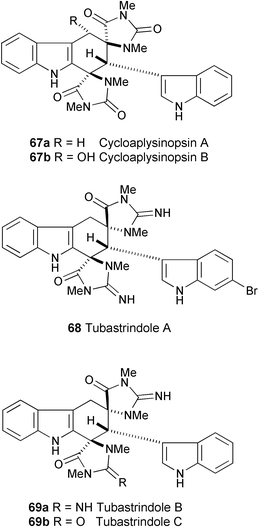
Three novel indole alkaloids named tubastrindoles A (68), B (69a), and C (69b) have been isolated from a stony coral, Tubastraea sp.36 Their structures were elucidated on the basis of spectral data (1H and 13C NMR, COSY, and HMBC experiments). Neither compound exhibited antibacterial, antifungal or cytotoxic activity.
A direct total synthesis of 9-methoxycarbazole-3-carbaldehyde (70), isolated from Murraya euchrestifolia Hayata,37 has been achieved based on a novel methodology for the preparation of 1-methoxyindoles (Scheme 13).38,39 Compound 71 was converted into the aldehyde 72 in 4 steps. Allylation of 72 followed by ring closing metathesis and DDQ oxidation afforded 9-methoxycarbazole-3-carbaldehyde (70).39
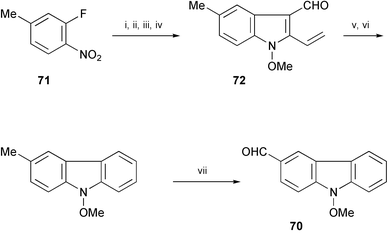 | ||
| Scheme 13 Reagents and conditions: i, methyl cyanoacetate, NaH, THF, 0 to 60 °C; ii, NaH, DMF, CH2CHCH2Br, 0 °C to rt; iii, NaCl, DMSO, 155 °C; iv, DIBAL-H, dry PhMe, −78 °C; v, allyltributyltin, n-BuLi, THF, −78 °C; vi, Grubbs' catalyst, CH2Cl2, rt to reflux; vii, DDQ, AcOH, rt. | ||
Mukonine (73a) has been isolated from the Indian curry-leaf tree, Murraya koenigii.40 Mukonidine (73b) has been independently isolated from the stem bark of Murraya koenigii41 and from the root bark of Clausena excavata.42 The iron-mediated total synthesis of these alkaloids has been achieved by oxidative cyclisation with air as the oxidising agent (Scheme 14).43 Electrophilic substitution of the methyl esters 74a and 74b with the iron-complex salt 36 led to the iron complexes 75a and 75b. The oxidative cyclisation of 75a and 75b by air in protic medium followed by demetalation provided mukonine (73a) and mukonidine (73b), respectively.
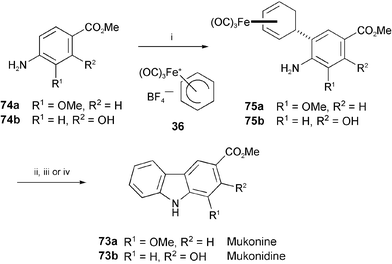 | ||
| Scheme 14 Reagents and conditions: i, 36, MeCN, heat; ii, air, PhH, TFA, 25 °C; iii, Cp2FePF6, Na2CO3, ClCH2CH2Cl, 83 °C; iv, PhH, p-chloranil, 110 °C. | ||
The neuronal cell protecting carbazole alkaloid, carbazomadurin A (76), isolated from the microorganism Actinomadura madurae 2808-SV1,44 has been synthesised in 9 steps and 11% overall yield from isovanillic acid (77) (Scheme 15).45 The key steps in the synthesis are the following palladium-catalysed reactions: Buchwald–Hartwig amination of 78 with 79, oxidative cyclisation of 80, and Stille coupling of 81 with 82. The resulting compound 83 was converted into carbazomadurin A (76).
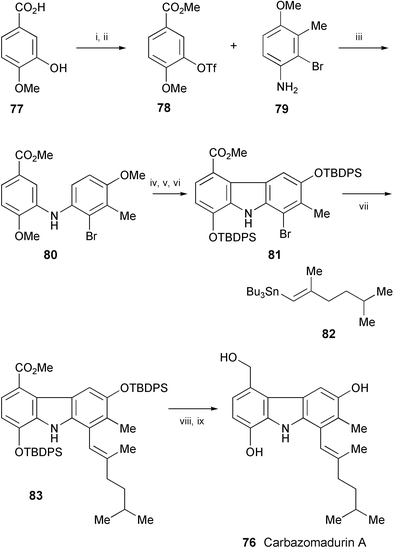 | ||
| Scheme 15 Reagents and conditions: i, MeOH, H2SO4, 65 °C; ii, 2,6-lutidine, Tf2O, CH2Cl2, −10 °C to rt; iii, Pd(OAc)2, BINAP, Cs2CO3, PhMe, 100 °C; iv, Pd(OAc)2, dioxane, AcOH, 100 °C; v, BBr3, CH2Cl2, −78 °C to rt; vi, t-BuPh2SiCl, imidazole, DMF, 70 °C; vii, 82, Pd(PPh3)4, PhMe, 110 °C; viii, DIBAL-H, PhMe, 0 °C; ix, TBAF, THF, rt. | ||
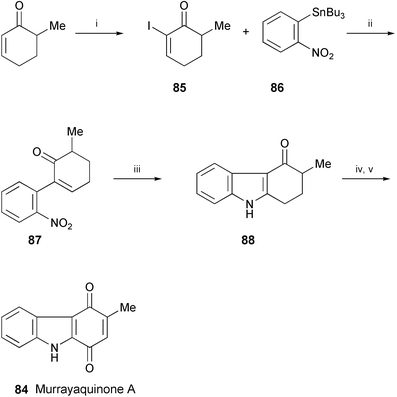
A formal total synthesis of murrayaquinone A (84), isolated from the root bark of Murraya eucrestifolia,46 has been achieved using two sequential palladium-catalysed reactions as the key steps in a novel synthetic route to 1,2-dihydro-4(3H)-carbazolones (Scheme 16).47 An intermolecular Stille cross-coupling of 85 with arylstannane 86 followed by a reductive N-heteroannulation of the resulting compound 87 afforded the carbazolone 88, which was converted into murrayaquinone A (84) according to the literature procedures.48,49,50
 | ||
| Scheme 16 Reagents and conditions: i, I2, CCl4, pyridine; ii, PdCl2(PhCN)2, AsPh3, CuI, NMP, 80 °C; iii, Pd(dba)2, dppp, 1,10-phenanthroline, CO, DMF, 80 °C; iv, Pd/C. Ph2O, 230 °C, 1,2,4-trimethylbenzene; v, Fremy's salt. | ||
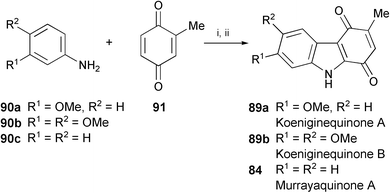
Koeniginequinones A (89a) and B (89b) have been isolated from the stem bark of Murraya koenigii.51 The total syntheses of these two compounds, and of murrayaquinone A (84) have been achieved employing regioselective addition of the arylamines 90a–c to 2-methyl-1,4-benzoquinone (91) followed by a palladium(II)-catalysed oxidative cyclisation (Scheme 17).52
 | ||
| Scheme 17 Reagents and conditions: i, AcOH, MeOH, H2O; ii, Pd(OAc)2, Cu(OAc)2, AcOH, heat. | ||
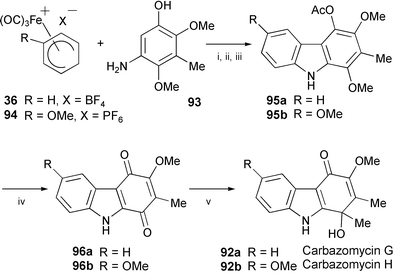
The total synthesis of the carbazole-1,4-quinol alkaloids carbazomycins G (92a) and H (92b), isolated from Streptoverticillium ehimense,53 has been achieved by a convergent iron-mediated construction of the carbazole framework (Scheme 18).54 Electrophilic substitution of the arylamine 93 using the iron-complex salts 36 and 94, followed by oxidative iron-mediated arylamine cyclisation, afforded the carbazole derivatives 95a and 95b, respectively. These carbazoles were transformed to the corresponding carbazole-1,4-quinones 96a and 96b by oxidation with cerium(IV) ammonium nitrate. Finally, regioselective addition of methyllithium at C-1 provided carbazomycins G (92a) and H (92b).
 | ||
| Scheme 18 Reagents and conditions: i, MeCN; ii, Ac2O, Et3N, DMAP, CH2Cl2; iii, activated MnO2, CH2Cl2; iv, (NH4)2Ce(NO3)6, MeCN, H2O; v, MeLi, THF, −78 °C. | ||
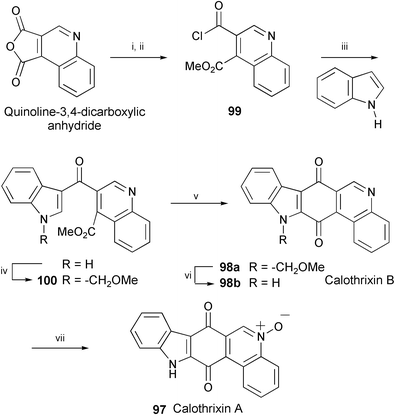
The total syntheses of calothrixins A (97) and B (98b), isolated from Calothrix cyanobacteria,55 have been accomplished starting from quinoline-3,4-dicarboxylic anhydride (Scheme 19).56 The key steps employed in the construction of the pentacyclic ring system are Friedel–Crafts acylation of indole with 99 and metalation of the N–MOM derivative 100. Deprotection of 98a with BBr3 afforded calothrixin B (98b), which was further converted into calothrixin A (97) by Kelly's procedure.57
 | ||
| Scheme 19 Reagents and conditions: i, dry MeOH, reflux; ii, SOCl2, reflux; iii, indole, MeMgCl, ZnCl2, CH2Cl2, rt; iv, NaH, MOMCl, THF, rt; v, LHMDS, TMEDA, THF, −78 °C to rt; vi, BBr3, CH2Cl2, 0 °C to rt; vii, m-CPBA. | ||
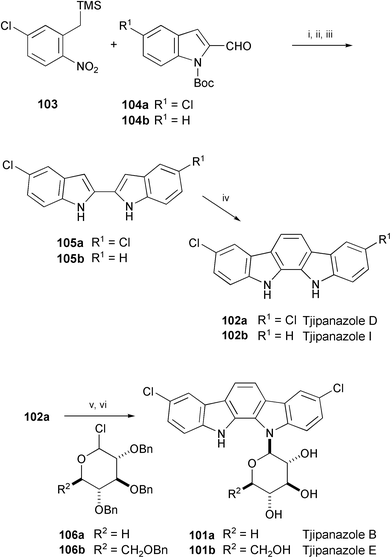
The total syntheses of tjipanazoles B (101a), D (102a), E (101b), and I (102b), isolated from Tolypothrix tjipanasensis,58 have been achieved starting from TMS-nitro compound 103 and indole-2-carbaldehyde derivatives 104a and 104b (Scheme 20).59 The key step is a condensation of 2,2′-biindoles 105a and 105b with (dimethylamino)acetaldehyde diethyl acetal to afford tjipanazoles D (102a) and I (102b). Tjipanazole D (102a) was further converted into tjipanazoles B (101a) and E (101b) by glycosidation with 106a and 106b, respectively.
 | ||
| Scheme 20 Reagents and conditions: i, cat. TBAF, 103, 104a or 104b; ii, TFAA, then DBU; iii, P(OEt)3 or Pd(OAc)2, PPh3; iv, (dimethylamino)acetaldehyde diethyl acetal, AcOH; v, 45% KOH, Aliquat 336, 106a or 106b; vi, Pd(OH)2, H2. | ||
2.2 Non-isoprenoid tryptamines
Bacillamide (107) has been isolated as a new algicide from the marine bacterium Bacillus sp SY-1.60 The structure was elucidated by spectroscopic analysis (1H, 13C, and 15N NMR, HMBC, and HSQC experiments). Compound 107 showed algicidal activity against the harmful dinoflagellate, Cochlodinium polykrikoides, with an LC50 of 3.2 µg ml−1.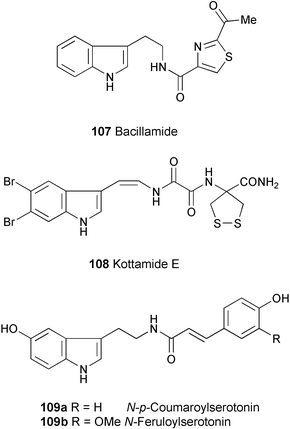
Kottamide E (108), a novel indole alkaloid containing dibrominated indole enamide, oxalic acid diamide, and 4-amino-1,2-dithiolane-4-carboxamide moieties, has been isolated from the New Zealand ascidian Pycnoclavella kottae.61 The structure was established by spectroscopic analysis (1H, 13C, and 15N NMR, HMBC, and HSQC experiments).
Aciculosporium take (Ascomycota; Clavicipitaceae), causes the witches' broom disease in bamboo, particularly Phyllostachys bambusoides. N-p-Coumaroylserotonin (109a) and N-feruloylserotonin (109b) have been found to accumulate in the diseased twigs.62 Compound 109a possesses antifungal activity, and A. take was unable to grow at concentrations of 109a higher than 100 µg ml−1. However, A. take grew at 100 µg ml−1 of 109b.
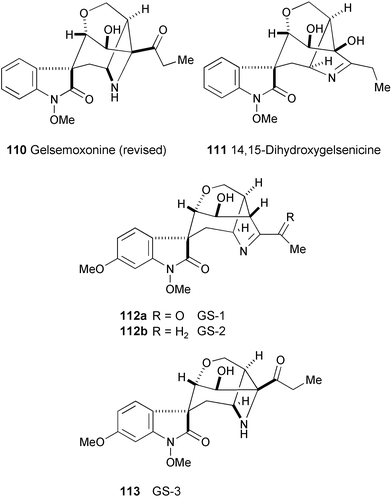
The structure of gelsemoxonine,63 isolated from Gelsemium elegans Benth., has been revised to a novel oxindole alkaloid (110) having an azetidine unit, based on the reinvestigation of its spectral data and the result of X-ray single crystallographic analysis.64 A new alkaloid, 14,15-dihydroxygelsenicine (111), which was presumed to be a biosynthetic precursor of 110, has also been isolated.
Three new gelsedine-type oxindole alkaloids, GS-1 (112a), GS-2 (112b), and GS-3 (113), have been isolated from the stems and leaves of cultivated Carolina jasmine (Gelsemium sempervirens Ait. f.).65 Their structures were determined by spectroscopic analysis (1H and 13C NMR, and HMBC experiments). The absolute configuration of these alkaloids was determined by comparison of their CD spectra with that of gelsemicine.
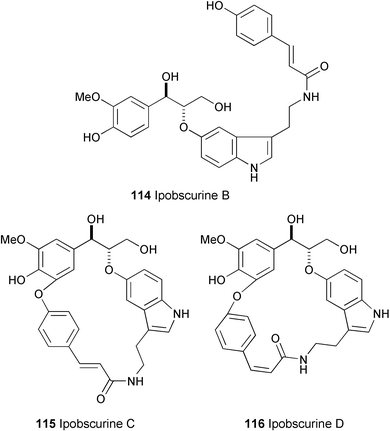
Ipobscurines B–D (114–116) have been isolated from the methanolic seed extract of Ipomoea obscura.66 Their structures were established on the basis of spectral data (HR-EIMS, 1H and 13C NMR, and NOESY spectra). Moreover, total synthesis of the racemic erythro- and threo-ipobscurine B 4′,4″-dimethyl ethers and comparison with the corresponding derivative of natural (−)-ipobscurine B proved the erythro configuration of the latter.
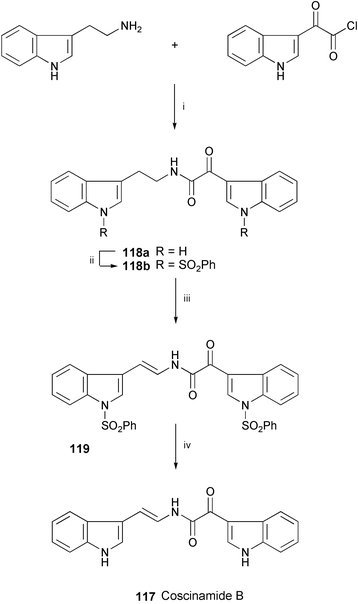
The synthesis of coscinamide B (117), isolated from Coscinoderma sp.,67 has been achieved starting from tryptamine, which was condensed with 3-indolylglyoxyl chloride to give 118a (Scheme 21).68 After sulfonylation of 118a, one-pot benzylic bromination of the resulting 118b and subsequent in situ dehydrobromination in the absence of an added base produced 119. Alkaline hydrolysis of 119 furnished coscinamide B (117).
 | ||
| Scheme 21 Reagents and conditions: i, MeCN, rt; ii, PhSO2Cl, BnEt3NCl, NaOH, CH2Cl2, rt; iii, NBS, (PhCO2)2O, CCl4, reflux; iv, K2CO3, MeOH, H2O, reflux. | ||
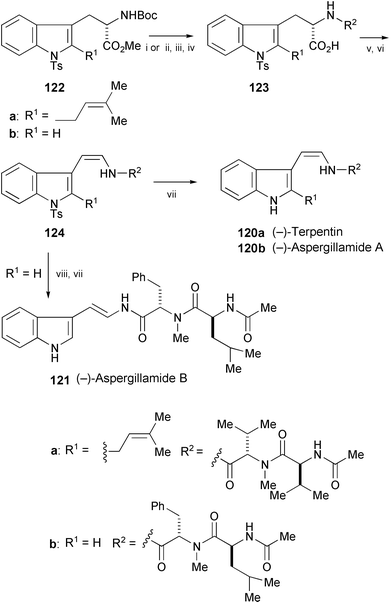
Terpentin (120a) is a novel peptide recently isolated from Aspergillus terreus which inhibits the cell cycle at G2/M phase (Scheme 22).69 The related aspergillamide A (120b) and the corresponding E-enamide stereoisomer, aspergillamide B (121), have been isolated from the genus Aspergillus and they showed modest in vitro cyctotoxicity toward the human carcinoma cell line HCT-116.70 The total syntheses of these natural products have been achieved starting from tryptophan derivatives 122a and 122b.71 As a key reaction, an oxidative decarboxylation–elimination of the carboxylic acids 123a and 123b was employed to construct the indolic enamide moiety of 124a and 124b. Compound 124a was converted into (−)-terpentin (120a). Deprotection of 124b gave (−)-aspergillamide A (120b). Isomerisation of 124b to the E-isomer followed by deprotection afforded (−)-aspergillamide B (121).
 | ||
| Scheme 22 Reagents and conditions: i, 1. 10% HF, MeCN, 2. HOBT, EDC, DIEA, DMF, 0–25 °C, Boc–NMe-L-Val–OH, 3. 10% HF, MeCN, 4. HATU, DIEA, DMF, 0–25 °C, Boc-L-Val–OH, 5. TMSOTf, 2,6-lutidine, 6. MeOH; ii, 1. TFA, CH2Cl2, 2. HOBT, EDC, DIEA, DMF, 0–25 °C, Boc-L-NMePhe–OH, 3. TFA, CH2Cl2, 4. HATU, DIEA, DMF, 0–25 °C, Boc-L-Leu–OH, 5. TFA, CH2Cl2; iii, Et3N, Ac2O, CH2Cl2; iv, LiOH, THF, H2O; v, Pb(OAc)4, Cu(OAc)2, pyridine, THF, 0 °C to rt; vi, PS-TBD, CH2Cl2, 0 °C; vii, Mg, MeOH, rt, 0 °C to rt; viii, KI, AcOH. | ||
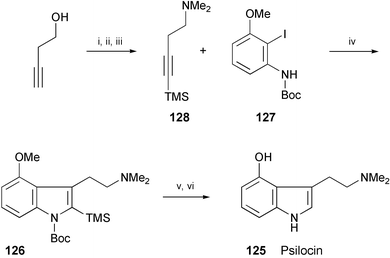
A synthesis of psilocin (125), reported to enter the central nervous system and cause powerful psychotomimetic effects,72 has been achieved (Scheme 23).73 The key step is the preparation of 126 by the palladium-catalysed cyclisation of the protected aniline derivative 127 and the silyl acetylene 128. Removal of the protecting groups of 126 afforded psilocin (125).74
 | ||
| Scheme 23 Reagents and conditions: i, TsCl, Et3N, CH2Cl2; ii, Me2NH, rt; iii, n-BuLi, Et2O, −10 °C, then TMSCl; iv, Pd(OAc)2, PPh3, NEt4Cl, i-Pr2EtN, DMF, 80 °C; v, TFA, 25 °C; vi, BBr3, CH2Cl2, −78 to 25 °C. | ||
The large-scale syntheses of psilocin (125) and psilocybin (129), the principal hallucinogenic constituents of magic mushroom, have been achieved without chromatographic purification (Scheme 24).75 The key step in the synthesis of 129 is the isolation of the dibenzyl-protected zwitterionic derivative 130.
![Reagents and conditions: i, Ac2O, pyridine; ii, (COCl)2; iii, Me2NH; iv, LiAlH4; v, [(BnO)2PO]2O, n-BuLi, THF, −78 °C to 0 °C; vi, Pd/C, H2.](/image/article/2005/NP/b316241a/b316241a-s24.gif) | ||
| Scheme 24 Reagents and conditions: i, Ac2O, pyridine; ii, (COCl)2; iii, Me2NH; iv, LiAlH4; v, [(BnO)2PO]2O, n-BuLi, THF, −78 °C to 0 °C; vi, Pd/C, H2. | ||
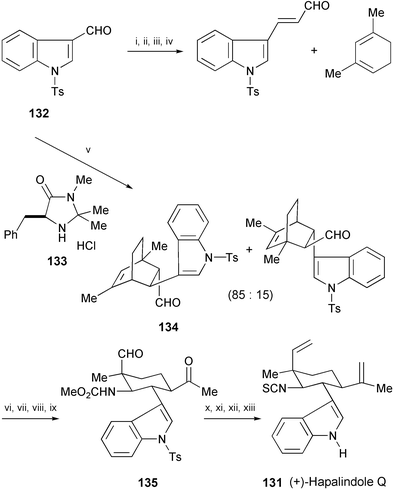
The total synthesis of (+)-hapalindole Q (131), isolated from the terrestrial blue-green alga Hapalosiphon fontinalis,76 has been achieved in 12 steps starting from the indole-3-carbaldehyde derivative 132 (Scheme 25).77 The key step is a Diels–Alder reaction mediated by the organocatalyst 133 to provide the critical intermediate 134 with high enantioselectivity. The intermediate then underwent a series of functional group transformation to converge with 135, which was further converted into (+)-hapalindole Q (131).
 | ||
| Scheme 25 Reagents and conditions: i, malonic acid, pyridine, pyrrolidine, reflux; ii, EtOH, cat. H2SO4, reflux, Dean–Stark; iii, DIBAL-H, CH2Cl2, −10 to 0 °C; iv, Dess–Martin periodinane, CH2Cl2, 0 °C or DMSO, (COCl)2, CH2Cl2, −78 °C, Et3N; v, DMF–MeOH (1 : 1) (5% H2O); vi, NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH; vii, 1. DPPA, Et3N, PhMe, reflux, 2. MeOH, sealed tube, 150 °C. viii, K2OsO2(OH)4, DABCO, MeSO2NH2, K2CO3, K3Fe(CN)6, THF, H2O; ix, NaIO4/SiO2, CH2Cl2; x, KOt-Bu, Ph3PCH3I, THF; xi, KOt-Bu, Ph3PCH3I, PhMe, 60 °C; xii, TBAF, THF, reflux; xiii, TCDI, CH2Cl2, 0 °C. | ||
A formal synthesis of (−)-peduncularine (136), isolated from the Tasmanian shrub Aristotelia peduncularis,78 has been completed by preparing the key intermediate (−)-137 in 5 steps from imide 138 (Scheme 26).79 The key reaction is a ring-closing metathesis of 139 to form 137 using Grubbs′ catalyst (140). Compound 137 has been previously converted into (−)-peduncularine (136).80
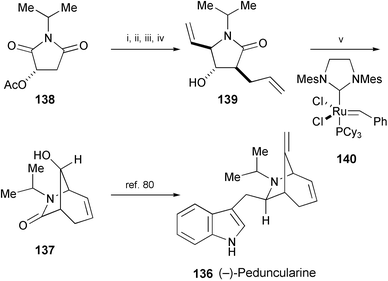 | ||
| Scheme 26 Reagents and conditions: i, NaBH4, HCl, EtOH; ii, PhSO2H, CaCl2, CH2Cl2, rt; iii, CH2CHMgBr, ZnCl2, THF, rt, then, H2SO4; iv, LDA, THF, 0 °C, then CH2CHCH2Br, −78 °C to −20 °C; v, 140, 40 °C. | ||
A brief synthesis of the spiropyrrolidinyloxindoles (±)-horsfiline (141a) and (±)-coerulescine (141b) has been achieved, starting from itaconic acid and (respectively) p-anisidine or o-iodoaniline (Scheme 27).81 Tandem radical cyclisation of iodoaryl alkenyl azides 142 is the key step in both syntheses.
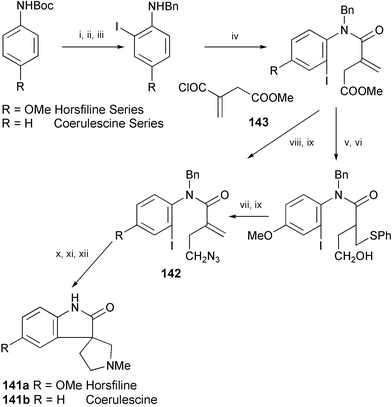 | ||
| Scheme 27 Reagents and conditions: i, t-BuLi, Et2O then ICH2CH2I; ii, TFA, CH2Cl2; iii, ZnCl2, PhCHO, MeOH, NaBH3CN; iv, Et3N, 143, PhH; v, PhSH, DBU, THF; vi, DIBAL-H, PhMe; vii, m-CPBA, CH2Cl2, then PhMe, heat; viii, LiCl, NaBH4, EtOH, THF; ix, PPh3, (PhO)2P(O)N3, DEAD, THF; x, (Me3Si)3SiH, AIBN, PhH; xi, CH2O, NaBH3CN, MeCN; xii, Na, NH3. | ||
The structures of D-seco Corynanthe-type oxindoles Us-7 and Us-8, isolated from a Rubiaceous plant, Uncaria attenuata Korth,82 were proposed as 144 and 145, respectively (Scheme 28). Aimi's group has succeeded in the syntheses of 145 and 146 starting from (S)-glycidol through 147 and 148.83 The synthetic 145 and 146 are completely identical in all respects (chromatographic behavior, mass, IR, UV, CD, 1H and 13C NMR, [α]D) with natural Us-8 and Us-7, respectively. Therefore, the structure of Us-7 was revised from 144 (with the (3R,7S) configuration), to 146 (with the (3S,7R) configuration), and the absolute stereochemistry at C-15 of both alkaloids was established.
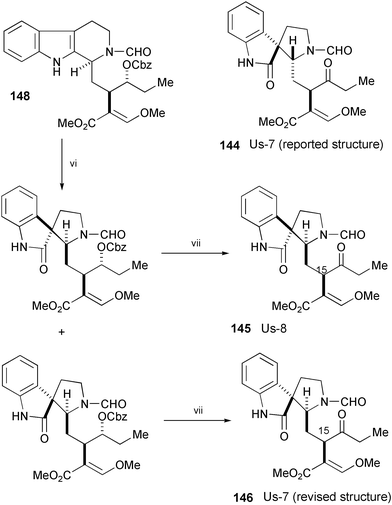 | ||
| Scheme 28 Reagents and conditions: i, tryptamine, AlMe3 in hexane, CH2Cl2, rt; ii, Cbz2O, CeCl3, THF, 60 °C; iii, POCl3, PhH, reflux; iv, NaBH4, MeOH, 0 °C; v, C6H5OCHO, CH2Cl2, rt; vi, t-BuOCl, Et3N, CH2Cl2, −30 °C, then 1 drop AcOH, aq. MeOH, reflux; vii, H2, 10% Pd/C, EtOH, rt, then Dess–Martin periodinane, rt. | ||
2.2.1 Piperazinediones
Marine fungi producing antifungal compounds were screened against Pyricularia oryzae and a novel diketopiperazine (149) has been isolated from the fungus M-3 isolated from the marine alga Porphyra yezoensis.84 The structure was elucidated by spectroscopic methods (1H and 13C NMR, COSY, HMQC, and HMBC experiments). The MIC of 149 against Pyricularia oryzae was 0.36 µM.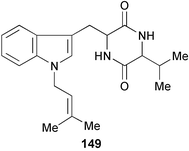
A brief total synthesis of the cell cycle inhibitor tryprostatin B (150), isolated from a marine strain (BM939) of Aspergillus fumigatus,85 has been achieved in 32% overall yield from the readily available cyclo-(L-Trp-L-Pro) (151) (Scheme 29).86 Tandem C-3 prenylation/cyclisation of 151 gave the corresponding pentacyclic pyrroloindoles 152 and 153. These compounds were then submitted to acid-catalysed opening of the newly formed ring, with concomitant migration of the prenyl group to the indole C-2 position, giving trifluoroacetate ester 154 and tryprostatin B (150), respectively. Exposure of compound 154 to Et3N gave tryprostatin B (150).
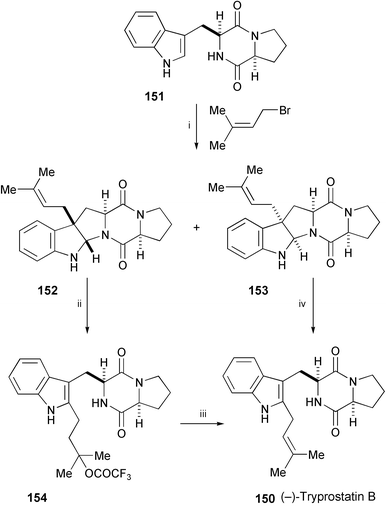 | ||
| Scheme 29 Reagents and conditions: i, prenyl bromide, Mg(NO3)2·6H2O, AcOH–NaOAc buffer, rt; ii, CF3CO2H–CH2Cl2 (1 : 10), rt; iii, Et3N, MeOH, rt; iv, Yb(OTf)3, MeNO2, reflux. | ||
An asymmetric total synthesis of spirotryprostatin A (155), isolated from Aspergillus fumigatus,87 has been achieved utilising the asymmetric [1,3]-dipolar cycloaddition reaction of methylene indolinone 156 as the key step (Scheme 30).88 Compound 157 was further converted into spirotryprostatin A (155) through 158.
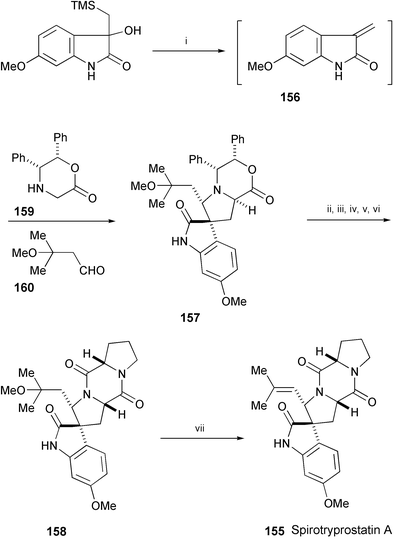 | ||
| Scheme 30 Reagents and conditions: i, TFA, PhMe, 0 °C, then 159, 160, 3 Å MS, PhMe, −15 ° to 0 °C; ii, H2, Pd(OH)2, THF, MeOH; iii, n-PrCHO, AcOH, 65 °C; iv, L-Pro–OBn·HCl, BOP, Et3N, MeCN; v, H2, Pd/C, EtOH, MeOH; vi, WSC, Et3N, MeCN; vii, p-TsOH·H2O, 3 Å MS, PhMe, 110 °C. | ||
The total synthesis of spirotryprostatin B (161), isolated from the fermentation broth of Aspergillus fumigatus BM939,89 has been accomplished by employing the MgI2-catalysed ring-expansion reaction of spiro[oxindole-3,1′-vinylcyclopropane] (162) in the presence of alkynyl imine 163 as a key reaction (Scheme 31).90 The resulting 164 was converted into 165 through the coupling of the L-proline fragment by a Kociensky–Julia reaction. From 165, the synthesis of spirotryprostatin B (161) was completed in 4 steps following the Danishefsky route.91
![Reagents and conditions: i, piperylene, [Rh(OAc)2]2, PhH, CH2Cl2, reflux; ii, 163, MgI2, THF, sealed tube, 75 °C; iii, Pd(PPh3)4, 1,3-dimethylbarbituric acid, CH2Cl2, 30 °C; iv, Et3N, Boc-l-ProCl, CH2Cl2, rt; v, NMO·H2O, OsO4, THF, t-BuOH, H2O, rt; vi, Pb(OAc)4, AcOEt, rt; vii, NaClO2, 2-methyl-2-butene, t-BuOH, pH 3.6 buffer, rt; viii, CH2N2, Et2O, rt; ix, TBAF, THF, rt; x, H2, Pd/BaSO4, quinoline, EtOH, rt; xi, 166, LHMDS, THF, −78 °C; xii, 1. PhSeCl, LHMDS, THF, 0 °C, 2. DMSO, THF, 0 °C; xiii, 1. TFA, CH2Cl2, rt, 2. Et3N, CH2Cl2, rt.](/image/article/2005/NP/b316241a/b316241a-s31.gif) | ||
| Scheme 31 Reagents and conditions: i, piperylene, [Rh(OAc)2]2, PhH, CH2Cl2, reflux; ii, 163, MgI2, THF, sealed tube, 75 °C; iii, Pd(PPh3)4, 1,3-dimethylbarbituric acid, CH2Cl2, 30 °C; iv, Et3N, Boc-L-ProCl, CH2Cl2, rt; v, NMO·H2O, OsO4, THF, t-BuOH, H2O, rt; vi, Pb(OAc)4, AcOEt, rt; vii, NaClO2, 2-methyl-2-butene, t-BuOH, pH 3.6 buffer, rt; viii, CH2N2, Et2O, rt; ix, TBAF, THF, rt; x, H2, Pd/BaSO4, quinoline, EtOH, rt; xi, 166, LHMDS, THF, −78 °C; xii, 1. PhSeCl, LHMDS, THF, 0 °C, 2. DMSO, THF, 0 °C; xiii, 1. TFA, CH2Cl2, rt, 2. Et3N, CH2Cl2, rt. | ||
The first total synthesis of paraherquamide A (167), isolated from cultures of Penicillium paraherquei,92 has been achieved (Scheme 32).93 The key steps in this asymmetric, stereocontrolled synthesis include enantioselective synthesis of an α-alkylated-β-hydroxyproline derivative, coupling of the gramine derivative94168 with the diketopiperazine 169, an intramolecular SN2′ cyclisation of 170, and a palladium-mediated ring closure to produce 171. Following a sequence of reactions, 171 was converted to (−)-paraherquamide A (167).
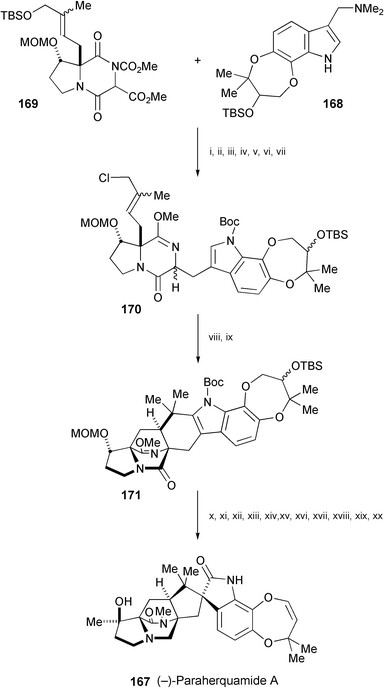 | ||
| Scheme 32 Reagents and conditions: i, P(n-Bu)3, MeCN; ii, HMPA, H2O, LiCl, 105 °C; iii, Me3OBF4, Cs2CO3, CH2Cl2; iv, DMAP, Et3N, (Boc)2O; v, TBAF, THF; vi, MsCl, collidine, CH2Cl2, 7 °C, then HMPA, n-Bu3BnNCl; vii, TBSOTf, 2,6-lutidine, CH2Cl2; viii, NaH, THF, heat; ix, PdCl2, AgBF4, MeCN, then propylene oxide, then NaBH4, EtOH; x, aq. HCl, THF; xi, 2-hydroxypyridine, PhMe, reflux; xii, DIBAL-H, CH2Cl2; xiii, NaH, MeI, DMF; xiv, bromocatecholborane, CH2Cl2; xv, Dess–Martin periodinane, CH2Cl2; xvi, TFA, CH2Cl2; xvii, t-BuOCl, pyridine; xviii, TsOH, THF, H2O; xix, (PhO)3PMeI, DMPU; xx, MeMgBr, THF. | ||
A short enantioselective total synthesis of okaramine N (172), isolated from the fungus Penicillium simplicissum (ATCC 90288),95,96 has been accomplished. Starting from (S)-tryptophan methyl ester, it was converted into 173 by reductive N-alkylation with 3-methyl-2-butenal, followed by acylation with 174 (Scheme 33).97 The key reaction in the synthesis is a Pd-promoted conversion of 173 to the dihydroindoloazocine tricycle 175. Cyclisation of 175, followed by an ene reaction with N-methyltriazolinedione and photooxidation, produced okaramine N (172).
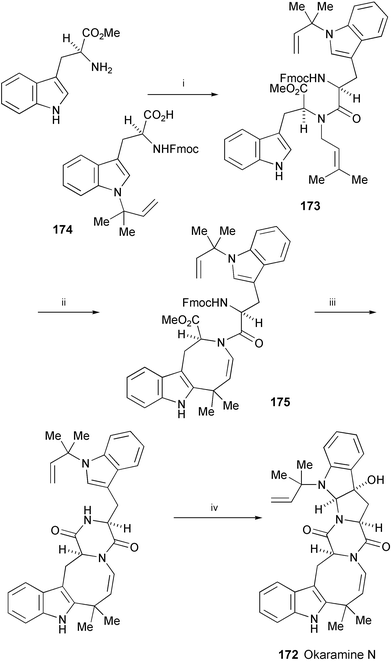 | ||
| Scheme 33 Reagents and conditions: i, 3-methyl-2-butenal, CH2Cl2, then removal of CH2Cl2, then NaBH4, MeOH, then BOPCl, 174; ii, Pd(OAc)2, AcOH, dioxane, H2O, O2, 25 °C; iii, Et2NH, THF; iv, N-methyltriazolinedione, CH2Cl2, −5 °C, then removal of CH2Cl2, then O2, sunlamp, MeOH, methylene blue, −28 °C, then Me2S, then 110 °C. | ||
2.2.2 Physostigmine and related compounds
A new 16N-ethoxycarbonyl derivative (176) of 3,12-dihydroroquefortin has been isolated from the fermentation broth of Penicillium aureovirens VKM FW-766.98 The structure was assigned on the basis of spectral analysis (HR-EIMS, 1H and 13C NMR, DEPT, NOESY, HMQC, and HMBC experiments).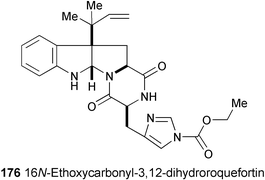
A formal total synthesis of (±)-physostigmine (177), isolated from calabar beans,99 has been achieved (Scheme 34).100 The key step in the synthesis is a new vicarious nucleophilic substitution reaction between p-nitroanisole and the C-silylated derivative of N-methylpyrrolidinone (178). Subsequent conversion of 179 to the tricyclic framework of physostigmine followed a well-established protocol and provided the key intermediate 180. The final 3 steps to obtain (±)-physostigmine (177) are efficient and well-established.101
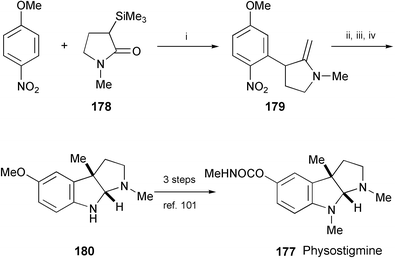 | ||
| Scheme 34 Reagents and conditions: i, TASF, THF, −78 °C, then DDQ; ii, MeI, CsOH·H2O, PhMe, n-Bu4NBr, rt; iii, H2, 10% Pd/C, AcOEt, 50 psi; iv, LiAlH4, THF. | ||
A total synthesis of (−)-pseudophrynaminol (181), isolated from the skin of the Australian frog Pseudophryne coriacea, 102 has been achieved by a sequence involving 3-allylindol-2-one (182) as a key intermediate (Scheme 35).103 The enantioselective construction of the quaternary carbon in 182 was performed through a tandem cascade reaction of 2-allyloxyindolin-3-one (183), olefination, isomerisation, and asymmetric Claisen rearrangement. Compound 182 was converted into (−)-pseudophrynaminol (181) through the reductive cyclisation of 184.
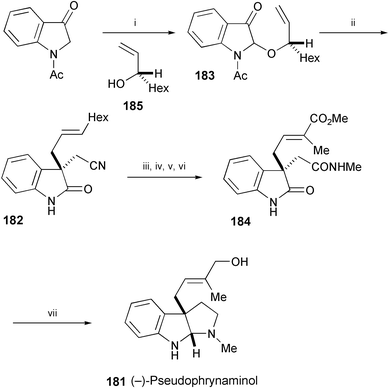 | ||
| Scheme 35 Reagents and conditions: i, Br2, CH2Cl2, 0 °C, then 185, MeCN, DMF, 4 Å MS, rt; ii, (EtO)2POCH2CN, KOt-Bu, DMF, −78 °C to rt; iii, 35% NaOH aq., MeOH, reflux; iv, HOC6F5, Et3N, EDC, THF, then MeNH2, rt; v, OsO4, NMO, MeCN, rt, then NaIO4, dioxane, H2O; vi, Ph3PC(Me)CO2Me, pyridine, MeCN, reflux; vii, LiAlH4, THF, reflux. | ||
A three-step total synthesis of debromoflustramine B (186), isolated from the marine byrozoan Flustra foliacea,104 has been completed starting from tryptamine (Scheme 36).105 The key step is the zinc triflate mediated alkylation of 187 to afford 188. Reduction of 188 with Red-Al gave debromoflustramine B (186).106
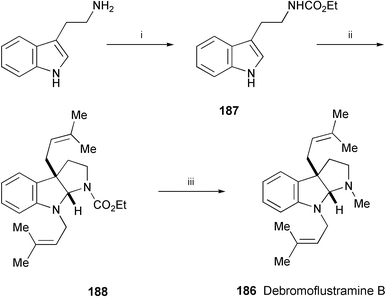 | ||
| Scheme 36 Reagents and conditions: i, ClCO2Et, CH2Cl2, aq. NaHCO3, 0 °C; ii, prenyl bromide, Zn(OTf)2, n-Bu4NI, i-Pr2NEt, PhMe, rt; iii, Red-Al, PhMe, reflux. | ||
The convergent total synthesis of (+)-okaramine J (189), isolated from fermentation extracts of Penicillium simplicissimum and Aspergillus aculeatus cultured on okara,95 has been achieved in a linear sequence of 12 steps from L-tryptophan t-butyl ester (Scheme 37).107 A key reaction is the acid-catalysed room-temperature aza-Claisen rearrangement of the N-reverse-prenylated hexahydro[2,3-b]pyrroloindole 190 to the C-prenylated derivative 191. Removal of the t-butyl ester of 191 followed by coupling with 192 afforded 193, which was converted into (+)-okaramine J (189) by removal of protecting group and subsequent cyclisation.
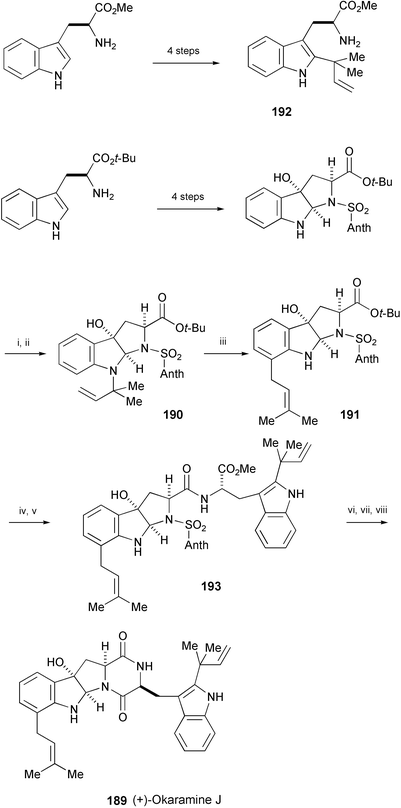 | ||
Scheme 37 Reagents and conditions: i, HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC(Me)2Br, CuCl, i-Pr2NEt, THF; ii, H2, Pd/Al2O3, AcOEt; iii, TFA, CH2Cl2; iv, TMSOTf, 2,6-lutidine, CH2Cl2; v, PyBop, Et3N, 192, THF; vi, Al/Hg, THF, H2O; vii, 10% KOH, MeOH, 1,4-dioxane; viii, HBTU, i-Pr2NEt, CH2Cl2. CC(Me)2Br, CuCl, i-Pr2NEt, THF; ii, H2, Pd/Al2O3, AcOEt; iii, TFA, CH2Cl2; iv, TMSOTf, 2,6-lutidine, CH2Cl2; v, PyBop, Et3N, 192, THF; vi, Al/Hg, THF, H2O; vii, 10% KOH, MeOH, 1,4-dioxane; viii, HBTU, i-Pr2NEt, CH2Cl2. | ||
The enantioselective total synthesis of (−)-idiospermuline (194), isolated from the seed of Idiospermum australiense,108 has been achieved starting from isatine in 6% overall yield (Scheme 38).109,110 The first key step in the synthesis is stereocontrolled formation of the vicinal quaternary carbon centers of 195 by dialkylation of the lithium dienolate of dihydroisoindigo (196) with enantiopure ditriflate 197. The second key step is a catalyst-controlled diastereoselective Heck reaction of 198 to afford an inseparable 6 : 1 mixture of 199 and 200. Catalytic hydrogenation of this mixture, followed by reduction of the carbonyl group and treatment with Na in NH3 provided (−)-idiospermuline (194) and isomer 201, which could be separated readily by preparative HPLC.
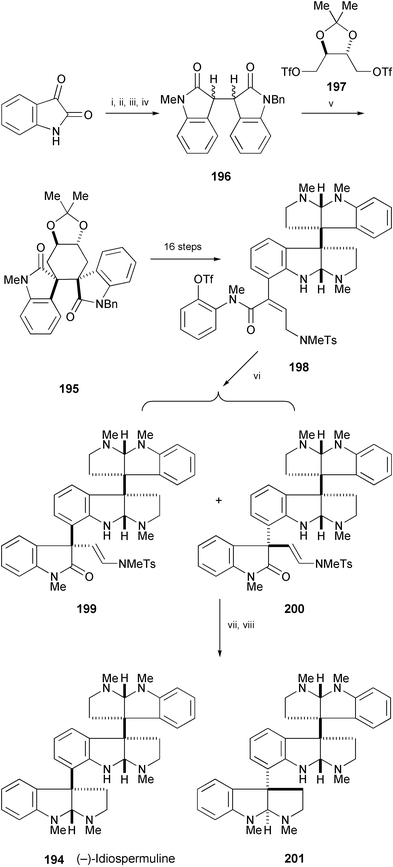 | ||
| Scheme 38 Reagents and conditions: i, NaH, BnBr, DMF, rt; ii, oxindole, AcOH, HCl, 110 °C; iii, Cs2CO3, MeI, DMF, rt; iv, PtO2, H2, AcOEt, rt; v, LHMDS, THF, HMPA, 197, −40 °C; vi, Pd(OAc)2, (S)-Tol-BINAP, PMP, MeCN, 80 °C; vii, Pd(OH)2, 1500 psi H2, 80 °C, EtOH; viii, 1. Red-Al, PhMe, rt, 2. Na, NH3, THF, −78 °C. | ||
Hodgkinsine (202) has been isolated from the leaves of Hodgkinsonia frutescens111 and hodgkinsine B (203) from Amazon Psychotria species (Scheme 39).112 The enantioselective total syntheses of these alkaloids from meso-chimonanthine (204) have been accomplished113,114 by highly concise sequences.115 The key reaction in the synthesis is an asymmetric intramolecular Heck reaction of 205, prepared from 204 in 4 steps, to afford enantioenriched 206 and 207. Saturation of the alkene double bond of 206 and 207, followed by reductive deprotection–cyclisation, furnished hodgkinsine (202) and hodgkinsine B (203). The stereorational total synthesis of 203 established the relative and absolute configuration of this Psychotria natural product.
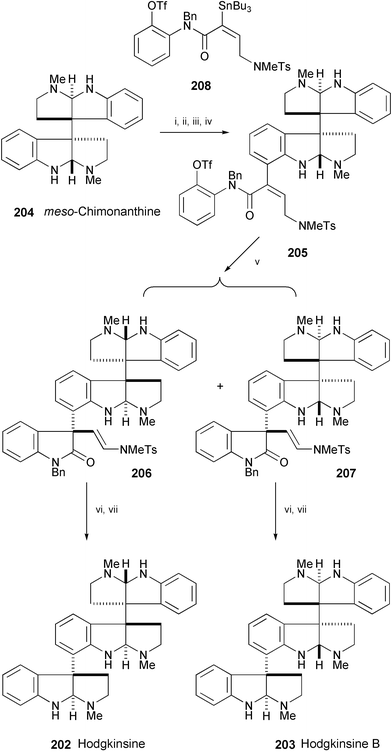 | ||
| Scheme 39 Reagents and conditions: i, Boc2O, NaHMDS, THF, rt; ii, 1. s-BuLi, TMEDA, THF, −78 °C, 2. diiodoethane, −78 °C to 0 °C; iii, TMSOTf, CH2Cl2; iv, 208, Pd2(dba3)·CHCl3, P(2-furyl)3, CuI, DMA, rt; v, Pd(OAc)2, (R)-Tol-BINAP, PMP, MeCN, 80 °C; vi, Pd(OH)2, 80 psi H2, 80 °C, EtOH; vii, Na, NH3, THF, −78 °C. | ||
2.2.3 Pyrrolo[3,2-e]indoles and pyrrolo[2,3-f]indoles
A novel chiral dipyrrolobenzoquinone derivative, terreusinone (209a), has been isolated as a potent UV-A protectant from the marine algicolous fungus Aspergillus terreus.116 The structure was established by spectroscopic analysis (1H and 13C NMR, COSY, TOCSY, DEPT, HMBC, and HSQC experiments) and the absolute stereochemistry was determined by Horeau's method117 and quantum calculations. 209a exhibited a UV-A absorbing activity with an ED50 value of 70 µg ml−1.Preparative-scale fermentation of terreusinone (209a) with Streptomyces sp. has resulted in the isolation of a new oxidised metabolite, terreusinol (209b).118 The structure was determined by spectral analysis (1H and 13C NMR, DEPT, HMQC, and HMBC experiments). Terreusinol (209b) showed UV-A protecting activity with an ED50 value of 150 µM, making it more active than oxybenzone (ED50
= 350 µM) currently being used as sunscreen.
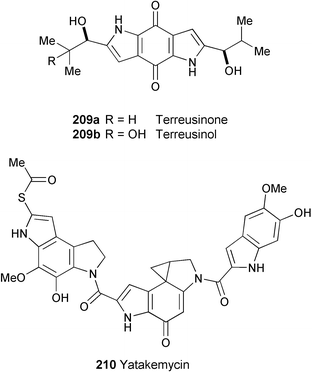
Yatakemycin (210), a novel antibiotic belonging to the family of CC-1065 and the duocarmycins, known DNA alkylating reagents, has been found in the culture broth of Streptomyces sp. TP-A0356.119 The structure was elucidated by spectroscopic methods (CID-MS/MS, 1H and 13C NMR, DOF-COSY, HMQC, and HMBC experiments). 210 inhibited the growth of pathogenic fungi such as Aspergillus fumigatus and Candida albicans with MIC values of 0.01–0.03 µg ml−1, lower than amphotericin B (MIC = 0.1–0.5 µg ml−1) and itraconazole (MIC = 0.03–0.2 µg ml−1). 210 also showed potent cytotoxicity against cancer cell lines with an IC50 of 0.01–0.03 µg ml−1.
Five new alkaloids, dictyodendrins A–E (211, 212, 213a, 213b and 214, respectively), have been isolated from the Japanese marine sponge Dictyodendrilla verongiformis as telomerase inhibitors.120 Their structures were elucidated by spectroscopic (HR-FABMS, 1H and 13C NMR, ROESY, and HMBC experiments) and chemical methods. Dictyodendrins are tyramine-based pyrrolocarbazole derivatives containing three or four p-hydroxyphenyl groups. They inhibited telomerase completely at a concentration of 50 µg ml−1.
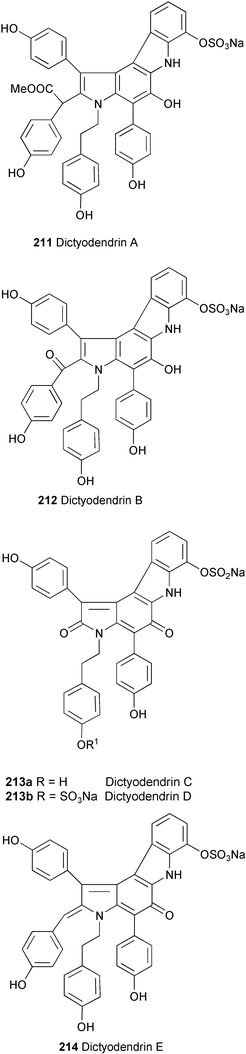
A total synthesis of seco-duocarmycin SA (215), a highly potent cytostatic agent, has been achieved (Scheme 40).121 Starting from 2-methoxy-4-nitroaniline (216) the synthetic protocol contains a Fischer indole synthesis using phenylhydrazone 217 to introduce the heterocyclic scaffold, and a radical 5-exo-trig-cyclisation of 218 to furnish the chloromethylindoline derivative 219 as key reactions.
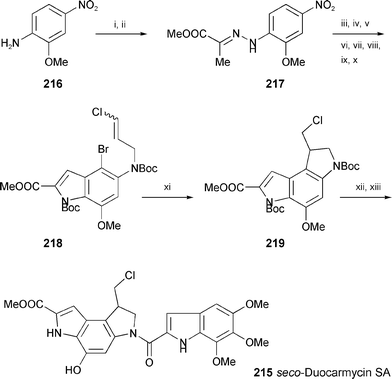 | ||
| Scheme 40 Reagents and conditions: i, NaNO2, HCl, then SnCl2; ii, NaOAc, methyl pyruvate, MeOH; iii, PPA, xylene; iv, AlCl3, CH2Cl2; v, BnBr, K2CO3; vi, Boc2O, DMAP; vii, Lindlar catalyst, quinoline, AcOEt, H2; viii, Boc2O, THF; ix, NBS, THF; x, NaH, DMF, then (E/Z)-1,3-dichloropropene; xi, (Me3Si)3SiH, AIBN, PhH; xii, 4 M HCl–AcOEt, then TMI-CO2H, EDC, DMF; xiii, NH4HCO2, Pd/C, THF. | ||
(+)-Duocarmycins A (220) and SA (221) are prominent members of the potent antitumor antibiotics isolated from Streptomyces species (Scheme 41).122 A convergent synthesis of (+)-duocarmycin A (220) has been achieved starting from 222 through 223, whose flexible strategy also enabled a straightforward synthesis of (+)-duocarmycin SA (221).123 The key reaction is the copper-mediated aryl amination reaction which has been used for forming all of the aryl–nitrogen bonds present in the duocarmycins. The intermediates 224 and 225 obtained by the aryl amination were converted into 220 and 221, respectively.
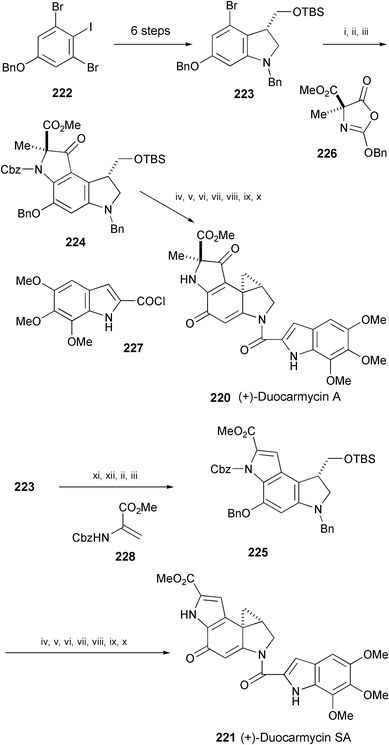 | ||
| Scheme 41 Reagents and conditions: i, n-BuLi, THF, −78 °C; then 226 in PhMe, −78 °C; ii, NBS, DMF, 23 °C; iii, CuI, CsOAc, DMSO, 23 °C; iv, TrocCl, NaHCO3, MeCN, 70 °C; v, Zn, KH2PO4, THF–H2O (5 : 1), 23 °C; vi, 227, pyridine, CH2Cl2, 0 °C; vii, TBAF, THF, 23 °C; viii, MsCl, pyridine, 0 °C; ix, H2, Pd/C, AcOEt, EtOH, 23 °C; x, CsCO3, MeCN, 23 °C; xi, n-BuLi, THF, −78 °C, then I2; xii, 228, Pd(OAc)2, P(O-tolyl)3, Et3N, MeCN, 90 °C. | ||
2.2.4 Indoloiminoquinones
Discorhabdins S (229), T (230), and U (231), three new discorhabdin analogues, have been isolated from a deep-water marine sponge of the genus Batzella.124 Their structures were elucidated based on spectroscopic methods (HR-ESIMS, 1H and 13C NMR, DEPT, HMQC, and HMBC experiments). 229, 230, and 231 showed in vitro cytotoxicity against murine P-388 tumor cells, with IC50 values of 3.08, >5, and 0.17 µM, respectively. Cytotoxicity was also observed for A-549 human lung adenocarcinoma cells (IC50 values of >5, >5, and 0.17 µM, respectively), and for PANC-1 human pancreatic cells (IC50 values of 2.6, 0.7, and 0.069 µM, respectively).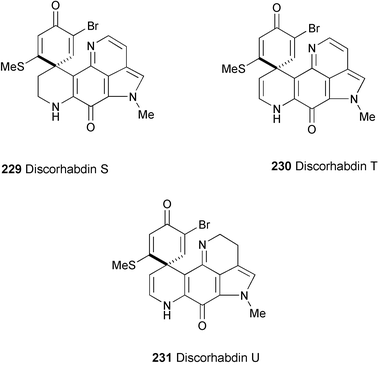
The synthesis of the marine alkaloid sebastianine A (232), isolated from the ascidian Cystodytes dellechiajei,125 has been accomplished via hetero-Diels–Alder reaction of indole-4,7-dione (233), prepared from 4,7-dimethoxyindole (234) in 3 steps, with trifluoroacetamidocinnamaldehyde dimethylhydrazone (235) (Scheme 42).126 Subsequent cyclisation of 236 under alkaline conditions gave sebastianine A (232).
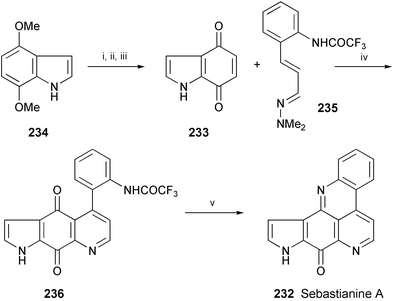 | ||
| Scheme 42 Reagents and conditions: i, NaH, Boc2O, THF, rt; ii, CAN, MeCN–H2O (4 : 1), rt; iii, TFA, CH2Cl2, rt; iv, PhMe, reflux then MnO2; v, 1 N NaOH, CH2Cl2. | ||
The first total synthesis of (+)-discorhabdin A (237), isolated from marine sponges such as New Zealand sponges of the genus Latrunculia127 and the Okinawa sponge Prianos melanos,128 has been achieved starting from L-tyrosine methyl ester hydrochloride (Scheme 43).129 The key step of the synthesis involves both a diastereoselective oxidative spirocyclisation of 238 using a hypervalent iodine(III) reagent and an efficient construction of the labile and highly strained N,S-acetal core (239). Removal of the tosyl group of 239 gave discorhabdin A (237) in an optically pure form. Discorhabdin A shows potent antitumor activity.
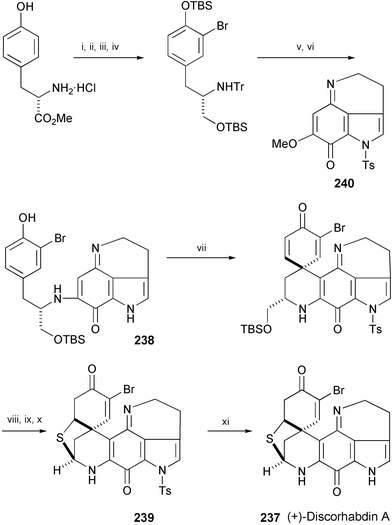 | ||
| Scheme 43 Reagents and conditions: i, TrCl, Et3N, DMF; ii, NBS, DMF; iii, DIBAL-H, CH2Cl2, −78 °C to rt; iv, TBSCl, DBU, CH2Cl2, 0 °C; v, TBAF, THF, 0 °C; vi, 0.1 N HCl, MeOH, then 240, MeOH; vii, PIFA-MK10, CF3CH2OH; viii, BF3·Et2O, CH2Cl2, 0 °C to rt; ix, Pb(OAc)4, CH2Cl2–MeOH (2 : 1), 0 °C; x, p-MeOC6H4CH2SH, 30% HBr–AcOH, −78 to 4 °C then aq. MeNH2; xi, NaOMe, THF, MeOH, 0 °C. | ||
2.2.5 β- and γ-Carbolines
Five new manzamine type alkaloids, 32,33-dihydro-31-hydroxymanzamine A (241), 32,33-dihydro-6-hydroxymanzamine A-35-one (242), 32,33-dihydro-6,31-dihydroxymanzamine A (243), des-N-methylxestomanzamine A (244), and 1,2,3,4-tetrahydronorharman-1-one (245), have been isolated from an Indonesian sponge.130 Their structures have been assigned on the basis of spectral data (HR-ESIMS, 1H and 13C NMR, COSY, HMQC, and HMBC experiments). The structure of 241 was confirmed by X-ray crystallographic analysis. The bioactivity and SAR of the manzamines against malaria, TB, and leishmania are also presented.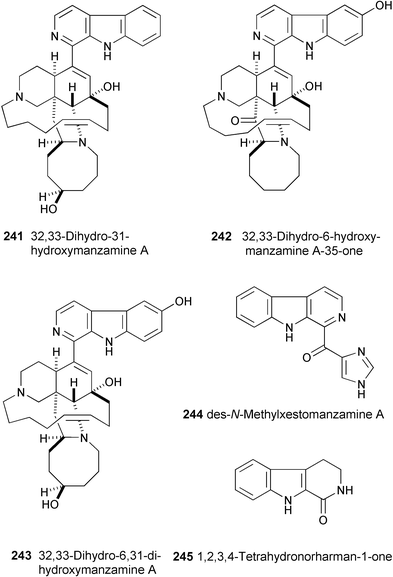
Two new compounds, taraxacines A (246a) and B (246b), have been isolated and characterised from the fresh aerial parts of Taraxacum formosanum.131 Structures of 246a and 246b were determined by spectral analysis (1H and 13C NMR, HMQC, and HMBC experiments).
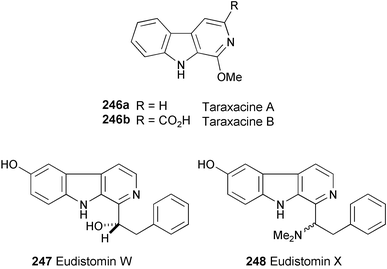
Chemical investigation of the Micronesian ascidian Eudistoma sp. has afforded two new eudistomins, W (247) and X (248).132 Their structures were established on the basis of spectroscopic analysis (1H and 13C NMR, COSY, and HMBC experiments). 248 exhibited antibiotic activity toward Bacillus subtilis, Staphyloccocus aureus, and Escherichia coli, and was also found to be fungicidal against Candida albicans in an agar diffusion assay. 247 was selectively active against C. albicans but showed no antibacterial activity.
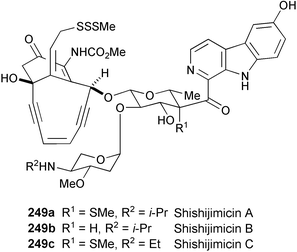
Three novel enediyne antitumor antibiotics, shishijimicins A (249a), B (249b), and C (249c), have been isolated from a lipophilic extract of the thin encrusting orange ascidian Didemnum proliferum collected in southern Japan.133 Their structures were determined by spectral analysis (1H and 13C NMR, COSY, HOHAHA, HMBC, NOESY, and HMQC experiments). Shishijimicins show extremely potent cytotoxicity. The IC50 values of shishijimicins A, B, and C against P388 cells are 0.47, 2.0, and 1.7 pg ml−1, respectively.
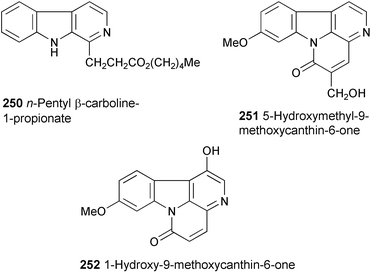
Three new β-carboline alkaloids, n-pentyl β-carboline-1-propionate (250), 5-hydroxymethyl-9-methoxycanthin-6-one (251), and 1-hydroxy-9-methoxycanthin-6-one (252), have been isolated from the roots of Eurycoma longifolia.134 Their structures were determined by spectral data (HR-FABMS, 1H and 13C NMR, COSY, NOESY, HMQC, and HMBC experiments) and by chemical transformation.
Investigation of the extract of the sponge Cymbastela
(order Halichondrida, family Axinellidae), screened against Caco-2 and A-431 cell lines, has led to the isolation of cytotoxic milnamide D (253).135 The structure was assigned on the basis of spectral analysis (1H and 13C NMR, COSY, HMQC, and HMBC experiments). In an assay for inhibition of tubulin polymerisation, the IC50 value was 16.90 µM. In an HCT-116 cytotoxicity assay, the IC50 value was 66.8 nM.
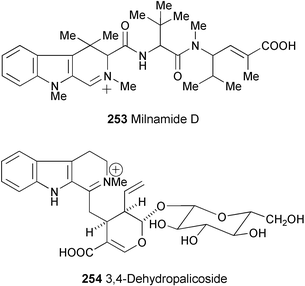
From the stem of Strychnos vanprukii, a new vincosan-type gluco-indole alkaloid, 3,4-dehydropalicoside (254) has been isolated.136 The structure was elucidated based on spectroscopic evidence (ES-MS/MS, 1H and 13C NMR, and HMBC spectra).
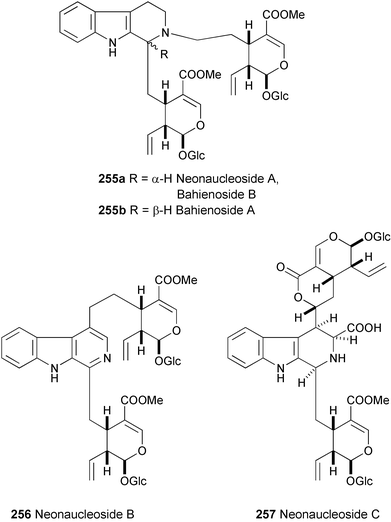
Indole alkaloid glycosides linked with a second secoiridoid glucoside unit have been isolated from the dried roots of Neonauclea sessilifolia and named as neonaucleosides A (255a), B (256), and C (257).137 Their structures were determined by spectroscopic analysis (HR-ESIMS, 1H and 13C NMR, ROESY, and HMBC experiments). The stereochemistry at C-3 of 255a was confirmed by the synthesis of 255a and its C-3 epimer (255b). Independently, the same alkaloids, 255b
(major) and 255a
(minor), have been isolated from the aerial parts of Psychotria bahiensis collected in Trinidad, West Indies138 and named as bahienosides B and A, respectively.
Extraction of the leaves of Chimarrhis turbinata has led to the isolation of a new Corynanthean-type indole alkaloid, turbinatine (258).139 The structure was elucidated based on spectroscopic data (1H and 13C NMR, NOESY, HMQC, TOCSY, and HMBC experiments). An evaluation of the DNA-damaging activities was performed by means of a bioassay using mutant strains of Saccharomyces cerevisiae; 258 was weakly active.
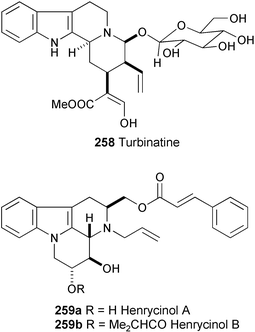
Henrycinols A (259a) and B (259b) have been isolated from the roots of Melodinus henryi Craib.140 Their structures were established on the basis of spectral analysis (HR-FABMS, 1H and 13C NMR, NOESY, and HMBC experiments). The relative configuration of 259a and 259b was determined by NOESY analysis.
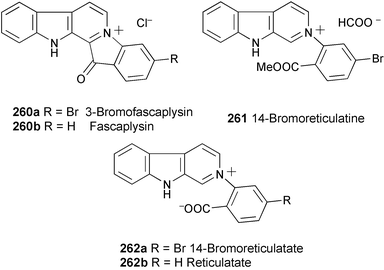
Three new alkaloids, 3-bromofascaplysin (260a), 14-bromoreticulatine (261), and 14-bromoreticulatate (262a), have been isolated along with the known reticulatate 262b from four organisms: two collections of the sponge Fascaplysinopsis reticulata and two collections of the tunicate Didemnum sp.141 Their structures were determined by spectral analysis (1H and 13C NMR, HMBC, and NOESY spectra). These compounds were less cytotoxic than fascaplysin (260b).
Three new indoloquinazolidine-type alkaloids, 263a, 263b, and 264, have been isolated from Araliopsis tabouensis.142 Their structures were determined by spectral analysis (1H and 13C NMR, COSY, HOHAHA, HMQC, and HMBC experiments). The antimalarial activities of compounds 263a, 263b, and 264 were evaluated against Plasmodium falciparum D6 and W2 clones. The IC50 values in an antimalarial bioassay for 263b and 264 against D6 are 1.8 and 3.3 µg ml−1. The corresponding IC50 values against W2 are both >4.7 µg ml−1. 263a is not active.
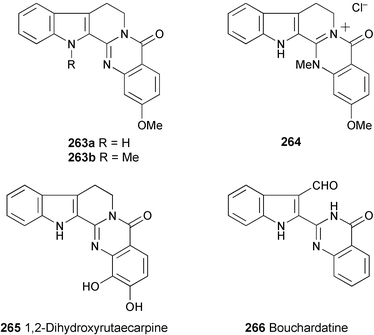
Two new alkaloids, 1,2-dihydroxyrutaecarpine (265) and 2-(2-[3-formylindolyl])-(3H)-quinazolin-4-one (bouchardatine, 266), have been isolated from the aerial parts of Bouchardatia neurococca (Rutaceae).143 Their structures were determined by spectroscopic evidence (API-ESMS, 1H and 13C NMR, COSY, and HMBC spectra).
A novel indole alkaloid, macrodasine A (267), incorporating fused spirocyclic tetrahydrofuran rings onto a macroline-like moiety, has been isolated from a Malayan Alstonia spcies.144 The structure is also notable for the presence of a spiroacetal moiety in the alkaloid. The structure was established by spectroscopic analysis (1H and 13C NMR, COSY, and HMBC experiments).
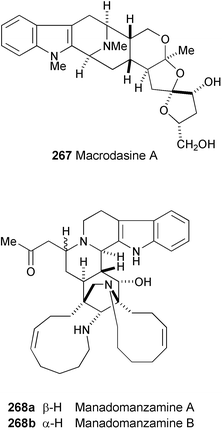
Two novel alkaloids, manadomanzamines A (268a) and B (268b), have been isolated from an Indonesian sponge Acanthostrongylophora sp. (Haplosclerida: Petrosiidae).145 The structures were elucidated based on spectroscopic data (1H and 13C NMR, COSY, HMQC, and HMBC experiments). Their absolute configuration and conformation were determined by CD, NOESY, and molecular modeling analysis. Manadomanzamines exhibited strong activity against Mycobacterium tuberculosis with MIC values of 1.9 and 1.5 µg ml−1, respectively. They also exhibit activities against human immunodeficiency virus and AIDS opportunistic fungal infections.
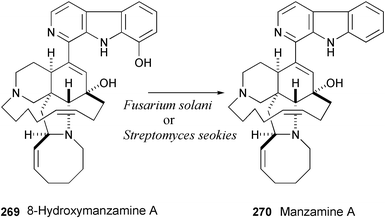
8-Hydroxymanzamine A (269) has been isolated from sponges of the genera Amphimedon, Xestospongia, and Pachypellina.146 The stereoselective dehydroxylation of 269 to form manzamine A (270), isolated from sponges of the genera Haliclona and Pellina,147 has been completed by fermentation with Fusarium solani or Streptomyces seokies.148 This unique biocatalytic conversion is important due to the fact that manzamine A (270) has more desirable biological activity when assayed in a murine model against malaria.
6-Methoxy-2-methyl-1,2,3,4-tetrahydro-β-carboline (271a) has been isolated from a variety of plant sources, such as Phalaris tuberosa149 and Horsfieldia superba (Scheme 44).150 Bauerine A (272) has been isolated from the blue–green alga Dichotrix baueriana.151 These β-carbolines have been prepared152 using a Stille cross-coupling of 273a and 273b with arylstannanes 274a and 274b, prepared from nitroanilines 275a and 275b, to give 276a and 276b, respectively. Subsequent palladium-catalysed N-heteroannulation of 276a and 276b afforded 6-methoxy-2-methyl-1,2,3,4-tetrahydro-β-carboline (271a) and 271b, respectively. N-Methylation of 271b followed by deprotection and oxidation using MnO2 provided bauerine A (272).
 | ||
| Scheme 44 Reagents and conditions: i, NaNO2, KI, H2SO4; ii, (n-Bu3Sn)2, PhMe, (PPh3)2PdCl2, PPh3, 80 °C; iii, 273a or 273b Pd(dba)2, CuI, AsPh3, NMP; iv, Pd(dba)2, dppp, DMF, 1,10-phenanthroline, CO, 80 °C; v, NaH, MeI, THF, 0 °C; vi, 3 N HCl, AcOEt; vii, MnO2, PhMe. | ||
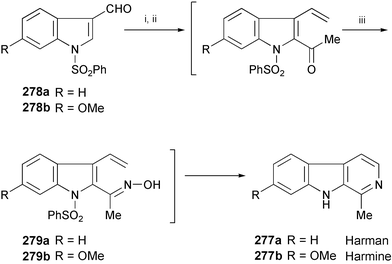
Harman (277a) and harmine (277b) were found to inhibit HIV replication in H9 lymphocyte cells.153 These compounds also showed significant antitumor activity (Scheme 45).154 Kusurkar's group has synthesised 277a and 277b starting from the indole-3-carbaldehyde derivatives 278a and 278b.155 The key step is electrocyclic cyclisation of 279a and 279b leading to harman (277a) and harmine (277b), respectively.
 | ||
| Scheme 45 Reagents and conditions: i, Ph3PMeI, KOt-Bu, THF; ii, LDA, THF, N,N-dimethylacetamide; iii, NH2OH·HCl, NaOAc, o-dichlorobenzene, heat. | ||
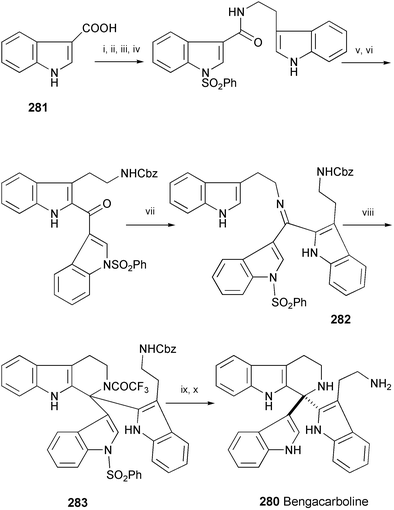
The first synthesis of marine alkaloid (±)-bengacarboline (280) has been achieved in 9 steps from indole-3-carboxylic acid (281) and tryptamine (Scheme 46).156 The main features of this synthesis are the use of a microwave irradiation procedure for the formation of ketimine 282 and of trifluoroacetic anhydride to produce the tetrahydro-β-carboline derivative 283.
 | ||
| Scheme 46 Reagents and conditions: i, n-BuLi, THF; ii, PhSO2Cl; iii, SOCl2, neat; iv, tryptamine, K2CO3, AcOEt, H2O; v, POCl3, neat; vi, CbzCl, Na2CO3; vii, tryptamine, ZnCl2, 1,4-Me2C6H4, microwaves; viii, TFAA, CH2Cl2, reflux; ix, H2, Pd/C, MeOH; x, K2CO3, MeOH, H2O. | ||
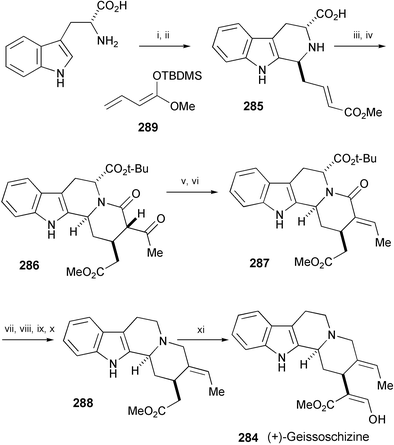
A synthesis of (+)-geissoschizine (284), a biosynthetic precursor of a variety of monoterpenoid indole alkaloids, has been performed starting from D-tryptophan (Scheme 47).157 The key steps in the synthesis involve a vinylogous Mannich reaction to prepare the carboline 285, and an intramolecular Michael addition that leads to the tetracyclic Corynantheane derivative 286. Compound 286 was then transformed into 287 by a sequence that allowed the stereospecific introduction of the E-ethylidene moiety. Selective reduction of the lactam in 287 followed by removal of the carboxy group by radical decarbonylation gave 288. Formylation of 288 afforded (+)-geissoschizine (284).
 | ||
| Scheme 47 Reagents and conditions: i, HCO2H, Ac2O, HCl; ii, 289; iii, isobutylene, H2SO4; iv, diketene, DMAP, PhMe, then KOt-Bu; v, NaBH4, MeOH; vi, NaOMe, MeOH, 50 °C, then AcCl; vii, Me3OBF4, 2,6-t-Bu2Py, then NaBH4; viii, TFA, PhSMe; ix, i-BuOCOCl, NaSePh; x, n-Bu3SnH, AIBN, heat; xi, LDA, HCO2Me. | ||
The first total synthesis of racemic tangutorine (290), isolated from the leaves of Nitraria tangutorum,158 has been performed in 7 steps starting from 291 (Scheme 48).159 The key reactions in the synthesis are dithionite reduction of 292 to 293 and acidic cyclisation of 293 to 294. This was then converted into tangutorine (290) by reduction, dehydration of the resulting alcohol, and reduction of the ester group. The separation of the enantiomers of tangutorine was successfully performed by chiral HPLC.
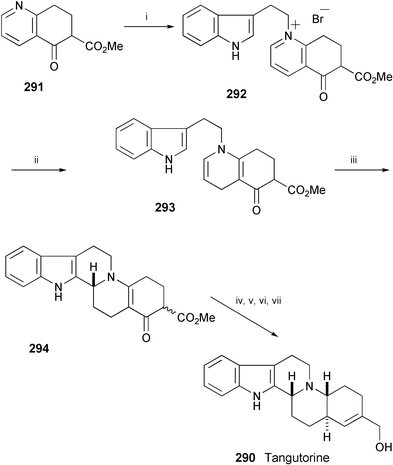 | ||
| Scheme 48 Reagents and conditions: i, tryptophyl bromide, 100 °C; ii, Na2S2O4, NaHCO3, MeOH, H2O, rt; iii, HCl, MeOH, rt; iv, NaBH4, AcOH, rt; v, MsCl, Et3N, CH2Cl2, 0 °C; vi, DBU, 100 °C; vii, LiAlH4, THF. | ||
A stereoselective total synthesis of (±)-tangutorine (290) has been achieved in 19 steps starting from tryptamine (Scheme 49).160 The key reactions are Heck coupling of 295 with methyl acrylate to give 296 and a formal intramolecular aza-[3+3] cycloaddition of 297 to afford 298. Compound 298 was further converted into (±)-tangutorine (290) in 9 steps.
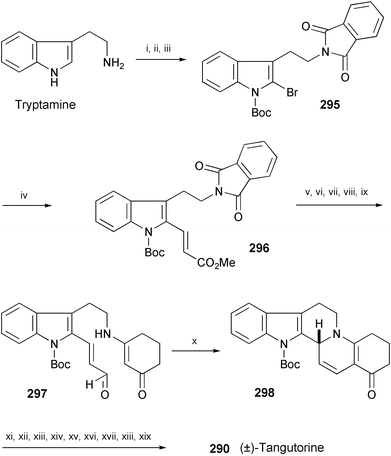 | ||
| Scheme 49 Reagents and conditions: i, phthalic anhydride, Et3N, PhMe; ii, pyridine·HBr·Br2, CHCl3, THF, −10 °C; iii, Boc2O, DMAP, CH2Cl2, rt; iv, methyl acrylate, Pd(PPh3)4, Cy2NMe, PhMe, 95 °C; v, DIBAL-H, CH2Cl2, −78 °C; vi, NaBH4, i-PrOH, H2O, rt; vii, AcOH, i-PrOH, H2O, 85 °C; viii, 1,3-cyclohexanedione, PhMe, reflux; ix, MnO2, CH2Cl2, rt; x, cyclohexylamine, AcOH, EtOH–PhMe (2 : 3), Na2SO4, 95 °C; xi, 20% Pd(OH)2, H2, AcOEt; xii, Na, i-PrOH, THF, rt; xiii, pyridine–SO3, DMSO, Et3N; xiv, Boc2O, DMAP, CH2Cl2; xv, NaH, (EtO)2CO, THF, reflux; xvi, NaBH4, MeOH, rt; xvii, MsCl, Et3N, CH2Cl2, −78 °C; xviii, DBU, THF, reflux; xix, LiAlH4, THF, 0 °C. | ||
The total synthesis of (−)-raumacline (299), isolated from plant cell cultures of Rauwolfia serpentina Benth.,161 has been achieved. Starting from L-tryptophan, it was converted into the aminonitrile (300) (Scheme 50).162 The key steps are a cis-specific Pictet–Spengler reaction of 300 to yield 301 and a stereoselective Dieckmann cyclisation of 302 to give 303. Reduction of the ester group followed by the formation of lactone and reduction afforded (−)-raumacline (299).
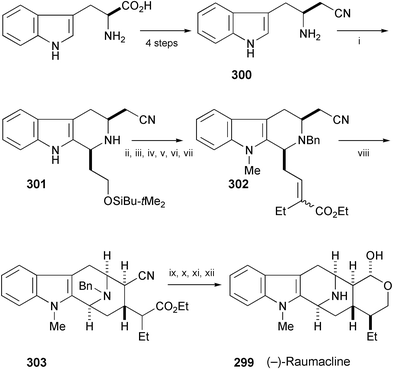 | ||
| Scheme 50 Reagents and conditions: i, TBSOCH2CH2CHO, 3 Å MS; ii, CH2Cl2, TFA, −78 °C to rt; iii, BnBr, 70 °C; iv, MeI, NaH, 0 °C; v, TBAF; vi, Swern oxidation; vii, (EtO)2POCHEtCO2Et, NaH; viii, LiNEt2, THF, −78 °C; ix, LiBH4; x, TsOH·H2O, THF, reflux; xi, DIBAL-H; xii, H2, Pd/C. | ||
Isocryptolepine (304), isolated from the root of the West African plant Cryptolepis sanguinolenta,163 has been synthesised in 3 steps (Scheme 51).164 A selective Buchwald–Hartwig amination of 4-chloroquinoline (305) with 2-chloroaniline (306) afforded 307. Subsequent intramolecular arylation of 307, followed by methylation of 308, produced 304.
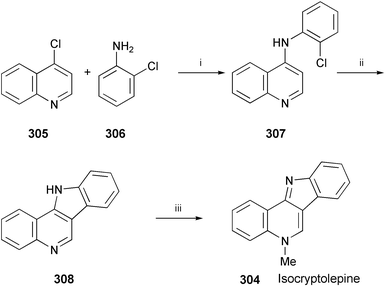 | ||
| Scheme 51 Reagents and conditions: i, Pd2(dba)3, XANTPHOS, Cs2CO3, dioxane, reflux; ii, Pd2(dba)3, P(t-Bu)3, K3PO4, dioxane, pressure tube, 120 °C; iii, MeI, DMF, 80 °C, then Na2CO3. | ||
3 Isoprenoid tryptamines
Efforts directed towards the total synthesis of communesins A (309a) and B (309b) have been reported,165 starting from the ergot alkaloid aurantioclavine166 (310) (Scheme 52). The key step is the preparation of the intermediate 311 by a [4+2] cycloaddition reaction. Based on the 1H and 13C NMR chemical shift data of 311 and 312, the authors suggest that the structure of the recently isolated microfilament-disrupting alkaloid nomofungin167 (313), should be revised to 309b.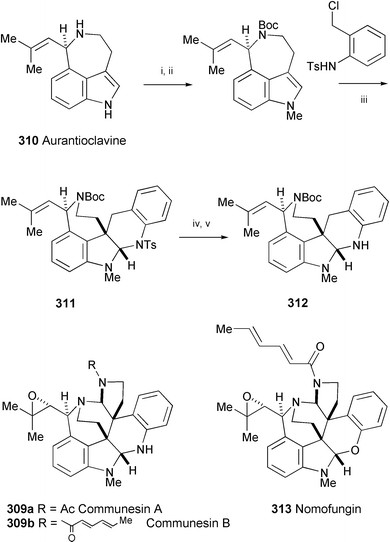 | ||
| Scheme 52 Reagents and conditions: i, (Boc)2O; ii, NaH, MeI; iii, Cs2CO3, CH2Cl2; iv, Mg, NH4Cl, MeOH; v, TLC separation. | ||
3.1 Ergot alkaloids
A new pentacyclic oxindole alkaloid, speradine A (314), has been isolated from the culture broth of the fungus Aspergillus tamarii.168 The structure and relative stereochemistry were determined on the basis of spectral data (1H, 13C, and 15N NMR, HMBC, and NOESY experiments) and a single crystal X-ray diffraction analysis.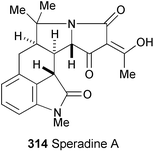
4 Bisindole alkaloids
1,1,3-Tris(3-indolyl)butane (315) has been isolated from a North Sea bacterium that is closely related to Vibrio parahaemolyticus (98% homology).169 The structure was established on the basis of spectral data (1H and 13C NMR, COSY, and HMQC experiments). 315 was inactive against a range of bacteria and fungi.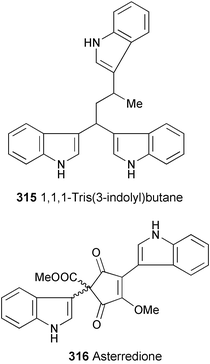
A novel cyclopentenedione, asterredione (316) has been isolated from Aspergillus terreus occurring in the rhizosphere of Opuntia versicolor, using bioassay-guided fractionation.170 The structure of 316 was elucidated by spectroscopic analysis (HR-FABMS, 1H and 13C NMR, and HMBC experiments). 316 was evaluated for cytotoxicity in a panel of three sentinel cancer cell lines, NCI–H460 (non-small cell lung cancer), MCF-7 (breast cancer), and SF-268 (CNS glioma), and was found to be moderately active. A pathway for the biosynthetic origin of 316 from asterriquinone D is proposed.
The biomimetically divergent synthesis of hamacanthins A (317b) and B (318b), isolated from a deep-water marine sponge Hamacantha sp.,171 has been demonstrated through intramolecular transamidation–cyclisation (Scheme 53).172 Removal of the Boc group in 2-oxoethanamide (319), obtained in 7 steps from 320, followed by heating in ClCH2CH2Cl provided 317a and 318a. When 319 was reacted in ethanol, the cyclisation proceeded regioselectively to yield only 318a. Deacetylation of 317a and 318a afforded hamacanthins A (317b) and B (318b), respectively.
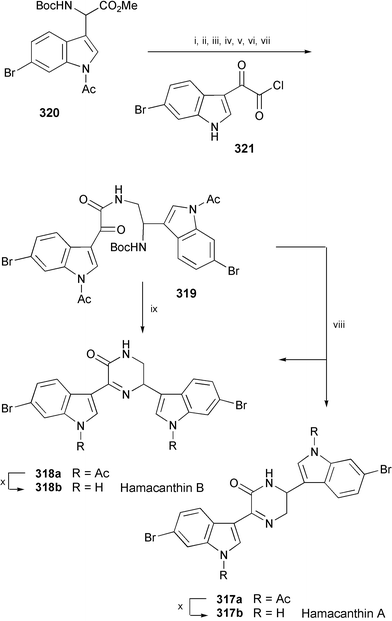 | ||
| Scheme 53 Reagents and conditions: i, 10% LiOH, THF–MeOH (1 : 1), rt; ii, i-BuOCOCl, N-methylmorpholine, DME, −15 °C, then NaBH4, rt; iii, TsCl, DMAP, Et3N, CH2Cl2, −20 °C; iv, NaN3, DMF, 80 °C; v, PPh3, H2O, THF, reflux, then 321, Et3N, THF, 0 °C to rt; vi, 10% KOH, EtOH, reflux; vii, Ac2O, DMAP, Na2CO3, THF, rt; viii, HCO2H, CH2Cl2, rt, then ClCH2CH2Cl, pH 4, reflux; ix, HCO2H, CH2Cl2, rt, then EtOH, pH 4, reflux; x, NH4OH, THF, MeOH. | ||
5 Peptide alkaloids containing a tryptophan residue
The cytotoxic hemiasterlins (322a–d) have been isolated from the marine sponges Hemiasterella minor,173Cymbastella sp.,174Auletta sp.,175 and Siphonochalina sp.176322a and a number of structural analogues have been synthesised and evaluated in cell-based assays for both cytotoxic and antimitotic activity in order to explore the SAR for this promising anticancer drug lead.177 The synthesis of 322a, the analogues, and a discussion of how their biological activities vary with their structures are also presented.177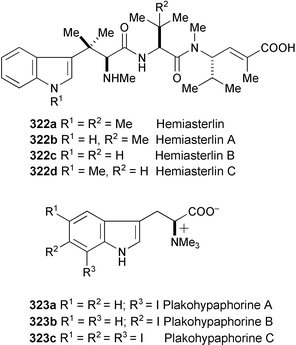
Three novel iodine-containing tryprophan betaines, plakohypaphorine A (323a), B (323b), and C (323c), have been isolated from the organic extract of the Caribbean marine sponge Plakortis simplex.178 Their structures were determined on the basis of spectral data (HR-ESIMS, 1H and 13C NMR, ROESY, HMQC, and HMBC experiments). This is the first report of iodoindole compounds from either marine or terrestrial sources.
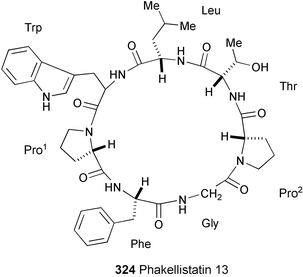
A new cyclic heptapeptide, phakellistatin 13 (324) has been isolated from the sponge Phakellia fusca Thiele, collected from Yongxing Island (China).179 The structure was elucidated on the basis of spectral data (1H and 13C NMR, TOCSY, DQFCOSY, HMQC, NOESY, and HMBC experiments). 324 showed strong cytotoxicity against the human hepatoma BEL-7404 cell line with an ED50 of <10−2
µg ml−1, but it was not active against the HL-60 cell line.
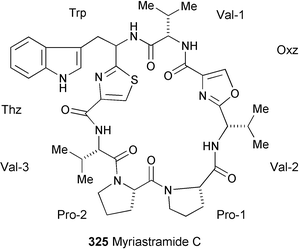
Chemical investigation of the Philippines marine sponge Myriastra clavosa has resulted in the isolation of a new cyclic octapeptide, myriastamide C (325).180 The structure was determined on the basis of spectral data (1H and 13C NMR, COSY, and NOESY experiments) and a series of degradation and derivatisation studies.
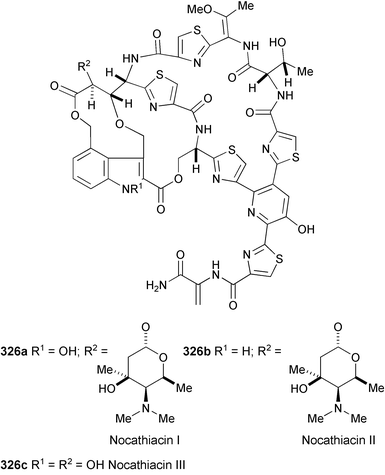
Thiazolyl peptide antibiotics, nocathiacin I (326a), II (326b), and III (326c), have been identified in a culture of Nocardia sp. WW-12651 (ATCC 202099).181 Their structures were elucidated by spectroscopic methods (HR-ESIMS, 1H, 13C, and 15N NMR, COSY, HMQC, and HMBC experiments). They exhibit potent in vitro activity (ng ml−1) against a wide spectrum of Gram-positive bacteria, including multiple-drug resistant pathogens such as methicillin-resistant Staphylococcus aureus
(MRSA), multi-drug resistant Enterococcus faecium
(MREF) and fully penicillin-resistant Streptococcus pneumoniae
(PRSP), and demonstrate excellent in vivo efficacy in a systemic Staphylococcus aureus mouse infection model.182
Six new bicyclic peptides, celogentins D–H (327, 328a,b, 329a,b) and J (329c) have been isolated from the seeds of Celosia argentea.183 Their structures, including absolute stereochemistry, were determined on the basis of spectral data (1H and 13C NMR, COSY, HOHAHA, HMQC, and HMBC experiments) and chemical means. 328a,b and 329a–c showed potent inhibition of tubulin polymerisation, while the inhibitory activity of 327 was modest. The structure–activity relationship indicates that the ring size of the bicyclic ring system including the unusual crosslinked Leu, Trp, and His residues is be important for their biological activity. IC50 values for celogentins D–H (327, 328a,b, 329a,b) and J (329c) are 20, 2.5, 3.0, 4.0, 2.0, and 3.0.µg ml−1, respectively.
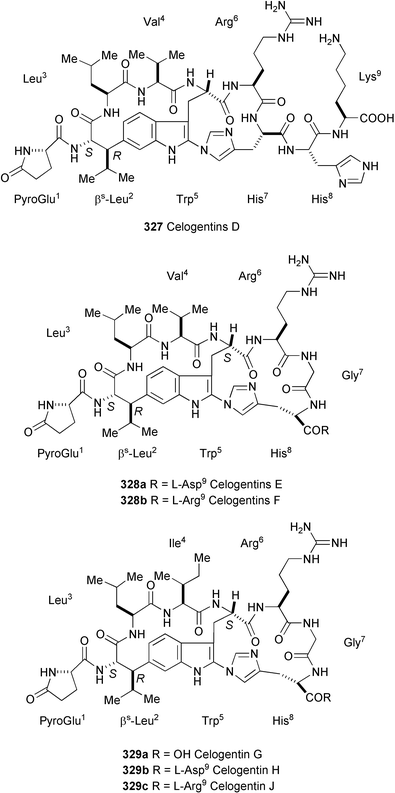
Three new kapakahines, E–G (330–332) have been isolated from the marine sponge Cribrochalina olemda.184 Their structures were elucidated by spectroscopic methods (FAB-MS/MS, 1H and 13C NMR, HMQC, NOE, and HMBC experiments). 330 showed cytotoxicity against P388 murine leukemia cells with an IC50 of 5.0 µg ml−1; 331 and 332 showed only weak cytotoxicity at this concentration.
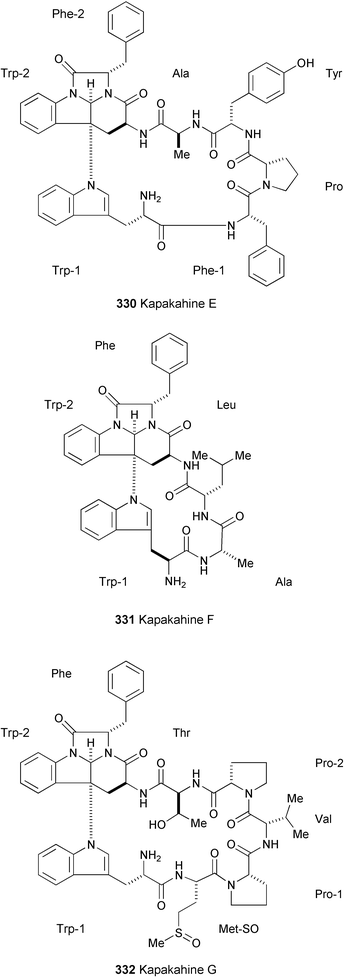
The synthesis of the octacyclopeptide phakellistatin 10 (333), isolated from marine sponges of genus Phakellia,185 has been accomplished using a combination of solid and solution-phase techniques (Scheme 54).186 Starting from 334, obtained by anchoring Fmoc-protected valine to 2-chlorotrityl chloride resin as a solid support, stepwise elongation of the linear sequence was carried out in seven cycles up to the desired length using Fmoc-amino acids, resulting in the formation of 335. Removal of the Fmoc group from the N-terminal residue followed by cleavage from the resin afforded the linear peptide 336. After the cyclisation reaction of 336, removal of the side-chain protecting groups gave phakellistatin 10 (333).
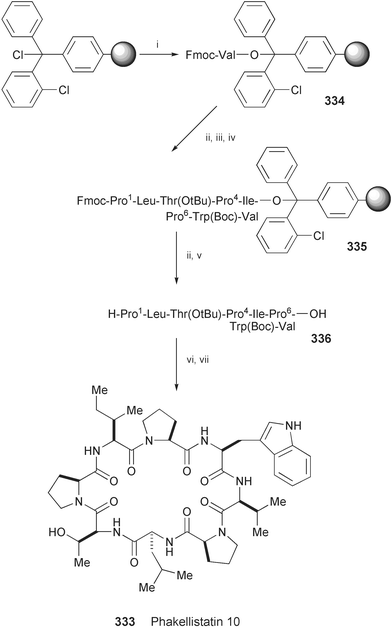 | ||
| Scheme 54 Reagents and conditions: i, Fmoc–Val–OH, DIEA, CH2Cl2; ii, 20% piperidine, DMF; iii, HOBt, HBTU, Fmoc-amino acid, NMM, DMF; iv, seven cycles of ii and iii; v, AcOH, CF3CH2OH, CH2Cl2; vi, HATU, DIEA, CH2Cl2; vii, TFA, TIS, H2O. | ||
The total synthesis of microsclerodermin E (337), isolated from the sponge Microscleroderma sp.,187 has been achieved using the methods for assembling four unusual amino acid units, which include 3-amino-10-(p-ethoxyphenyl)-2,4,5-trihydroxydeca-7,9-dienoic acid (AETD), 4-amino-3-hydroxybutanoic acid (GABOB), (R)-Trp-2-carboxylic acid, and a pyrrolidinone fragment (Scheme 55).188 Starting from the AETD unit 338 obtained from δ-gluconolactone in 17 steps, dipeptide fragment 339 was prepared by coupling with the GABOB unit 340. (R)-Trp-2-carboxylic acid derivative 341, prepared from the D-tryptophan derivative 342, was coupled with the pyrrolidinone fragment 343 and then with the dipeptide fragment 339 to produce 344. This was converted into microsclerodermin E (337) in 4 steps.
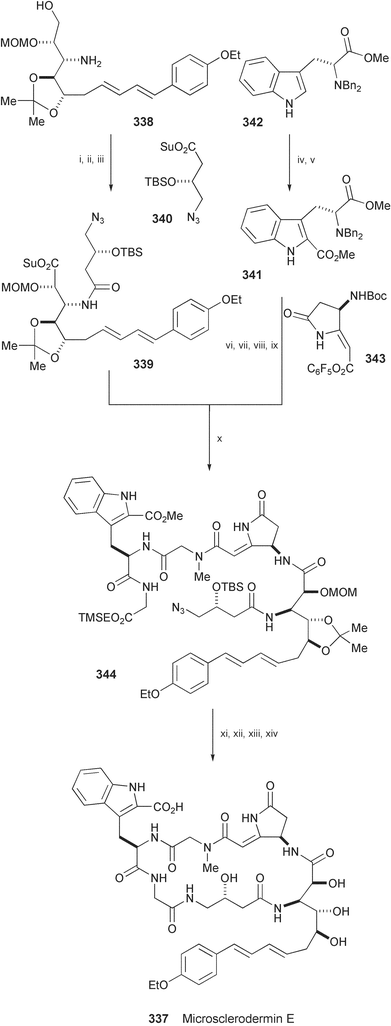 | ||
| Scheme 55 Reagents and conditions: i, 340, AcOEt, reflux; ii, Dess–Martin periodinane, then NaClO2, resorcinol, NaH2PO4; iii, HOSu, EDCI; iv, t-BuOCl, Et3N, −78 °C, then TMSCN, BF3·Et2O; v, aq. KOH, MeOH, then MeI, KHCO3, DMF; vi, Pd(OH)2, H2, MeOH; vii, BocN(Me)CH2CO2H, EDCl, HOBt, DIEA; viii, aq. LiOH, MeOH, then NH2CH2CO2TMSE, EDCI, HOBt, DIEA; ix, TFA, then 343, NaHCO3, DMF, 45 °C; x, TFA, then 339, NaHCO3, MeCN; xi, TBAF, THF, xii, PMe3, THF; xiii, DPPA, DMF; xiv, LiOH, MeOH, THF. | ||
The first stereoselective total synthesis of anti-HIV agent chloropeptin I (345), isolated from Streptomyces sp. WK-3419,189 has been completed from boronic ester 346,190 which was prepared from the corresponding Boc-protected amino acid (Scheme 56).191 The key steps in the synthesis are the Cu-mediated formation of biaryl ether 347 and the Pd-mediated intramolecular cross coupling of 348 leading to chloropeptin I (345). Chloropeptin I exhibits notable activity against HIV-1-induced cytopathicity and syncyctium formation in CD-4+ lymphocytes.
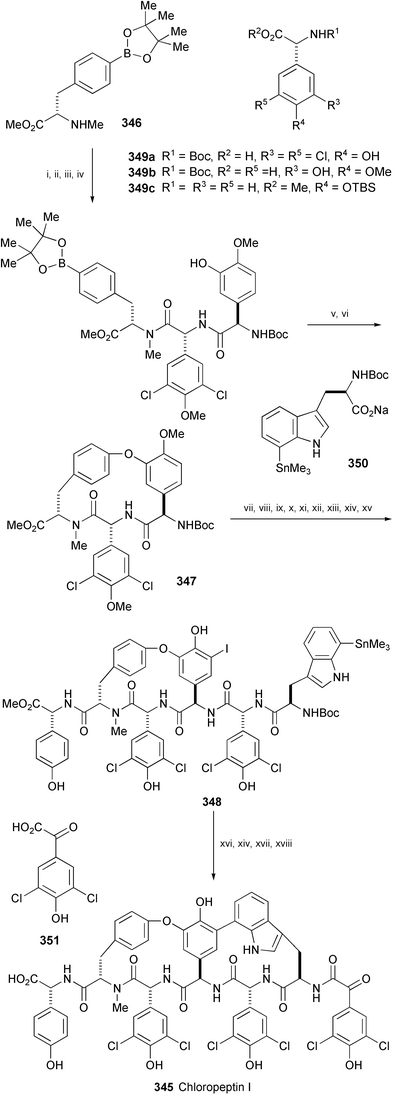 | ||
| Scheme 56 Reagents and conditions: i, 349a, DEPBT, NaHCO3, THF, 0 to 22 °C; ii, TMSCHN2, CH2Cl2, MeOH, 22 °C; iii, HCl(g), MeOH, −78 to 22 °C; iv, 1-hydroxy-7-azabenzotriazole, EDC, NaHCO3, 349b, THF, DMF, 0 to 22 °C; v, NaIO4, NH4OAc, acetone, H2O, 22 °C; vi, Cu(OAc)2, Et3N, 4 Å MS, MeOH, CH2Cl2, 22 °C; vii, HCl(g), MeOH, then NaHCO3; viii, TFAA, 2,6-lutidine, CH2Cl2, 0 to 22 °C; ix, AlBr3, EtSH, 0 °C; x, 349c, 1-hydroxy-7-azabenzotriazole, EDC, NaHCO3, THF, 0 to 22 °C; xi, NIS, MeCN, 22 °C; xii, HCl, MeOH, 45 °C, then NaHCO3; xiii, HATU, collidine, 349a, CH2Cl2, THF; xiv, HCl, MeOH, 22 °C; xv, 350, HATU, collidine, CH2Cl2, THF, 0 to 22 °C; xvi, Pd(Pt-Bu3)2, CsF, dioxane, 50 °C; xvii, 351, 1-hydroxy-7-azabenzotriazole, EDC, CH2Cl2, DMF, 0 to 22 °C; xviii, LiOH, H2O, THF, 0 °C. | ||
Diazonamide A is a potent antimitotic natural product, isolated from the colonial ascidian Diazona angulata.192 Harran's group revised its structure to 352 (Scheme 57).193 Two novel syntheses of 352 have been reported. Nicolaou's group achieved the synthesis of (−)-352 using tyrosine methyl ester and 4-bromoindole as starting materials, and converting them into 353 and 354, respectively.194 The key reactions in the synthesis are Suzuki coupling between aryl halide 353 and boronate 354 and an SmI2-induced heteropinacol coupling of 355 to produce the 12-membered heterocyclic core. This macrocycle was converted, via356, into (−)-diazonamide A (352) by the following sequence of reactions: oxazole construction, macrolactamisation, aminal formation, chlorination, and peptide coupling.
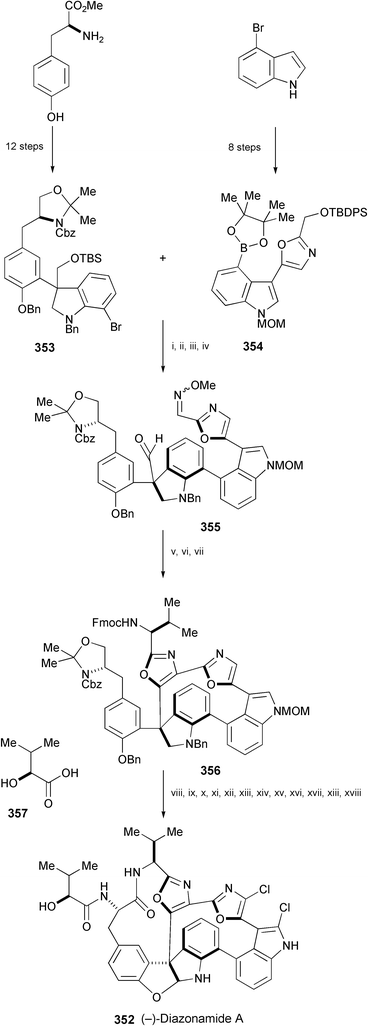 | ||
| Scheme 57 Reagents and conditions: i, Pd(dppf)Cl2·CH2Cl2, K2CO3, DME, 85 °C; ii, TBAF, THF, 45 °C; iii, Et3N, pyridine·SO3, DMSO, CH2Cl2, 0 °C; iv, MeONH2·HCl, DMSO; v, SmI2, DMA, THF, then sat. aq. NH4Cl, solvent removal, then Fmoc–Val–OH, EDC, HOBt, DMF; vi, TPAP, NMO, 4 Å MS, CH2Cl2; vii, POCl3, pyridine, 70 °C; viii, aq. HF, MeCN, 0 °C; ix, IBX, DMSO; x, NaClO2, NaH2PO4, resorcinol, DMSO, H2O; xi, Et2NH, THF; xii, HATU, collidine, DMF, CH2Cl2; xiii, H2, Pd(OH)2/C, EtOH; xiv, CbzCl, NaHCO3, dioxane; xv, NCS, CCl4, THF, 60 °C; xvi, BCl3, CH2Cl2, −78 °C, then aq. NaOH, THF, −78 to 25 °C; xvii, DIBAL-H, THF, −78 to 25 °C; xviii, 357, EDC, HOBt, NaHCO3, DMF. | ||
Another total synthesis of (−)-diazonamide A (352) has been accomplished starting from 7-bromotryptophan methyl ester (Scheme 58).195 The key reactions in the synthesis are oxidative cycloaddition of 358 to produce the aminal 359 and photoinduced macrocyclisation of 360 to generate 361. Compound 361 was converted into (−)-diazonamide A (352) by the following sequence of reactions: reductive removal of phenol, acylation, chlorination, deprotection, and condensation with (S)-α-hydroxy isovaleric acid (357).
![Reagents and conditions: i, PhI(OAc)2, LiOAc, 2,2,2-trifluoroethanol, inverse addition, −20 °C; ii, PhSH, Na2CO3, DMF, rt; iii, TeocCl, CH2Cl2, aq. K2CO3; iv, LiOH, aq. MeOH; v, 7-hydroxytryptamine, TBTU, i-Pr2NEt, DMF; vi, Ac2O, pyridine, CH2Cl2, THF; vii, DDQ, THF, H2O; viii, PPh3, (CCl3)2, Et3N, CH2Cl2, 15 min; ix, hν
(300 nm), LiOH, MeCN, H2O; x, 4-nitrophenyltriflate, K2CO3, DMF; xi, 20% Pd(OH)2/C, H2, AcOEt, MeOH, rt; xii, diallyldicarbonate, Et3N, THF, rt, then TeocCl, Et3N, rt, then [Pd(PPh3)4], morpholine, 0 °C; xiii, 2,3,4,5,6,6-hexachloro-2,4-cyclohexadien-1-one, DMF, rt; xiv, (Me2N)3SSiMe3F2, DMF, rt; xv, 357, (EtO)2P(O)CN, N-methylmorpholine, THF, rt.](/image/article/2005/NP/b316241a/b316241a-s58.gif) | ||
| Scheme 58 Reagents and conditions: i, PhI(OAc)2, LiOAc, 2,2,2-trifluoroethanol, inverse addition, −20 °C; ii, PhSH, Na2CO3, DMF, rt; iii, TeocCl, CH2Cl2, aq. K2CO3; iv, LiOH, aq. MeOH; v, 7-hydroxytryptamine, TBTU, i-Pr2NEt, DMF; vi, Ac2O, pyridine, CH2Cl2, THF; vii, DDQ, THF, H2O; viii, PPh3, (CCl3)2, Et3N, CH2Cl2, 15 min; ix, hν (300 nm), LiOH, MeCN, H2O; x, 4-nitrophenyltriflate, K2CO3, DMF; xi, 20% Pd(OH)2/C, H2, AcOEt, MeOH, rt; xii, diallyldicarbonate, Et3N, THF, rt, then TeocCl, Et3N, rt, then [Pd(PPh3)4], morpholine, 0 °C; xiii, 2,3,4,5,6,6-hexachloro-2,4-cyclohexadien-1-one, DMF, rt; xiv, (Me2N)3SSiMe3F2, DMF, rt; xv, 357, (EtO)2P(O)CN, N-methylmorpholine, THF, rt. | ||
A formal total synthesis of TMC-95 A (362a) and B (362b), isolated from the fermentation broth of Apiospora montagnei Sacc. TC1093,196 has been accomplished starting from N-Cbz-serine methyl ester (Scheme 59).197 The key steps in the synthesis include a stereoselective modified Julia olefination reaction between sulfone 363 and 7-iodoisatin (364), a diastereoselective dihydroxylation to construct 365, and macrocyclisation leading to 366 with limited use of protecting group chemistry. Compound 366 has been converted into TMC-95 A (362a) and B (362b) by Danishefsky's group.198
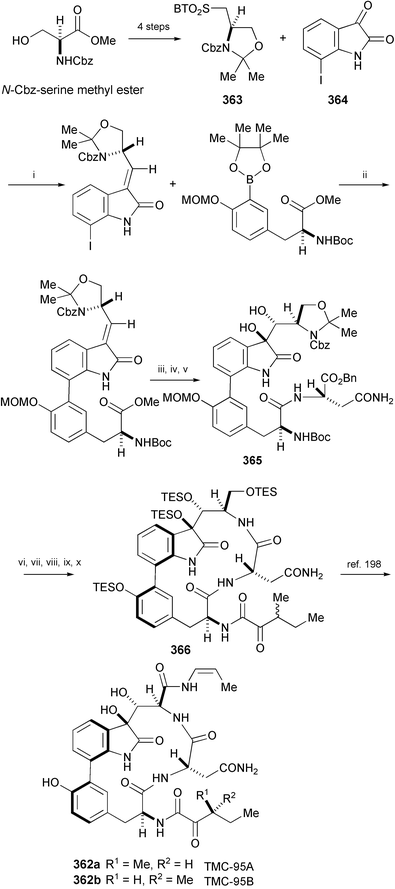 | ||
| Scheme 59 Reagents and conditions: i, LiHMDS, DMF, DMPU, 0 °C; ii, K2CO3, Pd(dppf)Cl2, aq. DME; iii, LiOH, THF, H2O, 0 °C; iv, H2N–Asn–OBn, 1-hydroxy-7-azabenzotriazole, EDCI, NMM, CH2Cl2, 0 °C; v, OsO4, pyridine, H2O, 0 °C, then NaHSO3, THF, MeOH; vi, TFA–H2O (1 : 1); vii, 3-methyl-2-oxopentanoic acid, 1-hydroxy-7-azabenzotriazole, EDCI, THF; viii, Pd black, H2, EtOH; ix, EDCI, 1-hydroxy-7-azabenzotriazole, CH2Cl2–DMF (1 : 1), 1 µM; x, TESOTf, 2,6-lutidine, CH2Cl2, DMF, 0 to rt. | ||
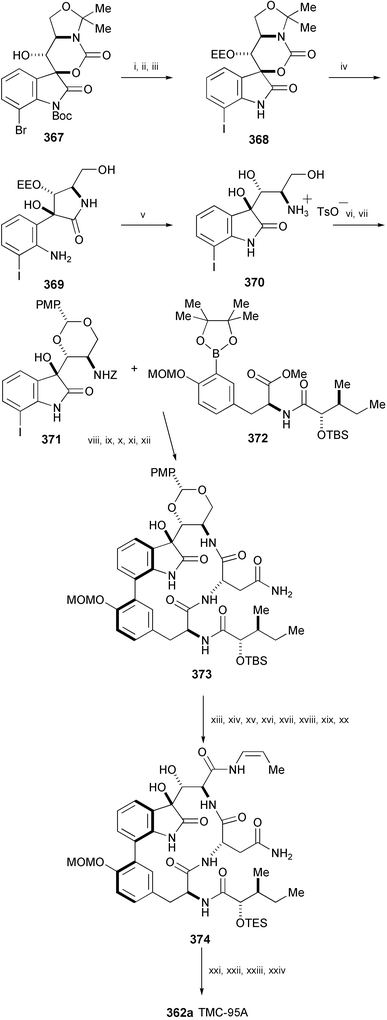
An efficient, convergent total synthesis of TMC-95A (362a) has been achieved starting from 367 (Scheme 60).199 The key transformations in the synthesis are a new deprotection protocol for the removal of the hindered carbamate and acetonide of 368, conversion of the aniline 369 into the oxindole 370 by lowering the pH, a Suzuki–Miyaura coupling reaction of 371 with 372, and macrolactamisation to creat the macrocyclic framework (373), and a mild decarboxylative anti-elimination to selectively produce the (Z)-1-propenylamide, part of structure 374. Compound 374 was then converted into TMC-95A (362a) by the following sequence of reactions: removal of the TES group, selective oxidation of the C-35 alcohol with Dess–Martin periodinane, protection of the C-7 alcohol with chloroacetyl anhydride, and exposure to acid and then base in one pot.
 | ||
| Scheme 60 Reagents and conditions: i, Mg(ClO4)2, MeCN, 50 °C; ii, ethyl vinyl ether, PPTS, THF, 35 °C; iii, n-BuLi, ICH2CH2I, THF, −60 °C to rt; iv, KOt-Bu, H2O, Et2O, rt; v, H2O, TsOH, MeOH, rt; vi, ZCl, Et3N, DMF, 0 °C to rt; vii, p-MeOC6H4CH(OMe)2, TsOH, THF; viii, Pd(PPh3)4, Na2CO3, DMF, H2O, 95 °C; ix, LiOH, THF, H2O; x, H–Asn–OBn·TFA, EDC·HCl, HOBt, DMF, 0 °C; xi, H2, Pd(OH)2/C, THF–H2O (1 : 1); xii, EDC·HCl, 1-hydroxy-7-azabenzotriazole, DMF, 0 °C; xiii, TBAF, 4 Å MS, THF; xiv, TESCl, imidazole, DMF; xv, Zn(OTf)2, EtSH, NaHCO3, CH2Cl2; xvi, pyridine·SO3, Et3N, CH2Cl2, DMSO, rt; xvii, NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH, H2O, rt; xviii, L-allo-Thr–OBn·TFA, EDC·HCl, HOBt, DMF, 0 °C; xix, H2, Pd(OH)2/C, THF–H2O (1 : 2); xx, DEAD, PPh3, 4 Å MS, 0 °C to rt; xxi, HF·pyridine, THF; xxii, Dess–Martin periodinane, CH2Cl2; xxiii, (ClCH2CO)2O, pyridine, CH2Cl2, 0 °C; xxiv, aq. HCl (1 M)–THF (3 : 1), rt, then sat. aq. NaHCO3. | ||
6 References
- D. Strack, T. Vogt and W. Schliemann, Phytochemistry, 2003, 62, 247 CrossRef CAS.
- H. M. Hügel and F. Nurlawis, Heterocycles, 2003, 60, 2349.
- L. M. Nogle and W. H. Gerwick, J. Nat. Prod., 2003, 66, 217 CrossRef CAS.
- C. Li, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2003, 66, 1232 CAS.
- N. Unver, G. I. Caya, C. Werner, R. Verpoorte and B. Gözler, Planta Med., 2003, 69, 869 CrossRef CAS.
- H.-S. Chung, P.-M. Hon, G. Lin, P. P.-H. But and H. Dong, Planta Med., 2003, 69, 914 CrossRef CAS.
- A. Ploutno, M. Shoshan and S. Carmeli, J. Nat. Prod., 2002, 65, 973 CrossRef CAS.
- N. Valls, M. Vallribera, S. Carmeli and J. Bonjoch, Org. Lett., 2003, 5, 447 CrossRef CAS.
- W. G. Kim, J.-P. Kim, H. Koshino, K. Shin-Ya, H. Seto and I.-D. Yoo, Tetrahedron, 1997, 53, 4309 CrossRef CAS.
- N. Toda, M. Ori, K. Takami, K. Tago and H. Kogen, Org. Lett., 2003, 5, 269 CrossRef CAS.
- R. Vleggaar, L. G. J. Ackerman and P. S. Steyn, J. Chem. Soc., Perkin Trans. 1, 1992, 3095 RSC.
- O. Tamura, T. Shiro, A. Toyao and H. Ishibashi, Chem. Commun., 2003, 2678 RSC.
- T. O. Larsen, K. Frydenvang, J. C. Frisvad and C. Christophersen, J. Nat. Prod., 1998, 61, 1154 CrossRef CAS.
- A. S. Kende, J. Fan and Z. Chen, Org. Lett., 2003, 5, 3205 CrossRef CAS.
- A. J. Gaskell and J. A. Joule, Chem. Ind. (London), 1967, 1089 CAS.
- E. S. Tasber and R. M. Garbaccio, Tetrahedron Lett., 2003, 44, 9185 CrossRef CAS.
- M. Natsume and Y. Kitagawa, Tetrahedron Lett., 1980, 21, 839 CrossRef CAS.
- D. S. Grierson, M. Harris and H. P. Husson, Tetrahedron, 1983, 39, 3683 CrossRef CAS.
- P. Klenwächter, B. Schlegel, I. Groth, A. Härtl and U. Gräfe, J. Antibiot., 2001, 54, 510 CAS.
- T. Komoda, Y. Shinoda and S. Nakatsuka, Biosci. Biotechnol. Biochem., 2003, 67, 659 CrossRef CAS.
- S. Ghosal, H. P. Rao, D. K. Jaiswal, Y. Kumar and W. A. Frahm, Phytochemistry, 1981, 20, 2003 CrossRef CAS.
- H.-J. Knölker and S. Filali, Synlett, 2003, 1752 CrossRef.
- G. N. Belofsky, J. B. Gloer, D. T. Wicklow and P. E. Dowd, Tetrahedron, 1995, 51, 3959 CrossRef CAS.
- A. B. Smith, III and H. Cui, Org. Lett., 2003, 5, 587 CrossRef.
- A. B. Smith, III and H. Cui, Helv. Chim. Acta, 2003, 86, 3908 CrossRef.
- A. E. de Jesus, P. S. Steyn, F. R. van Heerden, R. Vleggaar, P. L. Wessels and W. E. Hull, J. Chem. Soc., Perkin Trans. 1, 1983, 1847 RSC.
- A. B. Smith, III, N. Kanoh, H. Ishiyama, N. Minakawa, J. D. Rainier, R. A. Hartz, Y. S. Cho, H. Cui and W. H. Moser, J. Am. Chem. Soc., 2003, 125, 8228 CrossRef.
- J. M. S. López, M. M. Insua, Ju. P. Baz, J. L. F. Puentes and L. M. C. Hernández, J. Nat. Prod., 2003, 66, 863.
- A. J. Blackman, T. W. Hambly, K. Picker, W. C. Taylor and N. Thirasasana, Tetrahedron Lett., 1987, 28, 5561 CrossRef CAS.
- Y. Liu and W. W. McWhorter, Jr., J. Am. Chem. Soc., 2003, 125, 4240 CrossRef CAS.
- M. S. C. Pedras, P. B. Chumala and M. Suchy, Phytochemistry, 2003, 64, 949 CrossRef CAS.
- Y. Kikugawa and M. Kawase, J. Am. Chem. Soc., 1984, 106, 5728 CrossRef CAS.
- F. Yamada, T. Hashizume and M. Somei, Heterocycles, 1998, 47, 509 CAS.
- Y.-S. Wang, H.-P. He, Y.-M. Shen, X. Hong and X.-J. Hao, J. Nat. Prod., 2003, 66, 416 CrossRef CAS.
- I. Mancini, G. Guella, H. Zibrowius and F. Pietra, Tetrahedron, 2003, 59, 8757 CrossRef CAS.
- T. Iwagawa, M. Miyazaki, H. Okamura, M. Nakatani, M. Doe and K. Takemura, Tetrahedron Lett., 2003, 44, 2533 CrossRef CAS.
- C. Ito, T.-S. Wu and H. Furukawa, Chem. Pharm. Bull., 1988, 36, 2377 CAS.
- N. Selvakumar, M. K. Khera, B. Y. Reddy, D. Srinivas, A. M. Azhagan and J. Iqbal, Tetrahedron Lett., 2003, 44, 7071 CrossRef CAS.
- T. Kawasaki and M. Somei, Heterocycles, 1990, 31, 1605 CAS.
- D. P. Chakraborty, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Grisebach, G. W. Kirby, Springer, Wien, 1977, vol. 34, p. 299. Search PubMed.
- D. P. Chakraborty, S. Roy and R. Guha, J. Indian Chem. Soc., 1978, 55, 1114 CAS.
- T.-S. Wu, S.-C. Huang, J.-S. Lai, C.-M. Teng, F.-N. Ko and C.-S. Kuoh, Phytochemistry, 1993, 32, 449 CrossRef CAS.
- H.-J. Knölker and M. Wolpert, Tetrahedron, 2003, 59, 5317 CrossRef CAS.
- N. Kotoda, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, J. Antibiot., 1997, 50, 770 CAS.
- H.-J. Knölker and J. Knöll, Chem. Commun., 2003, 1170 RSC.
- H. Furukawa, T.-S. Wu, T. Ohta and C.-S. Kuoh, Chem. Pharm. Bull., 1985, 33, 4132 CAS.
- T. L. Scott and B. C. G. Söderberg, Tetrahedron, 2003, 59, 6323 CrossRef CAS.
- G. Bringmann, A. Ledermann and G. Francois, Heterocycles, 1995, 40, 293 CAS.
- Y. Miki and H. Hachiken, Synlett, 1993, 333 CrossRef CAS.
- K. Matsuo and S. Ishida, Chem. Pharm. Bull., 1994, 42, 1325 CAS.
- S. Saha and B. K. Chowdhury, Phytochemistry, 1998, 48, 363 CrossRef CAS.
- H.-J. Knölker and K. R. Reddy, Heterocycles, 2003, 60, 1049.
- M. Kaneda, T. Naid, T. Kitahara, S. Nakamura, T. Hirata and T. Suga, J. Antibiot., 1988, 41, 602 CAS.
- H.-J. Knölker, W. Fröhner and K. R. Reddy, Eur. J. Org. Chem., 2003, 740 CrossRef CAS.
- R. W. Richards, J. M. Rothschild, A. C. Willis, N. M. de Chazal, J. Kirk, K. Kirk, J. K. Saliba and G. D. Smith, Tetrahedron, 1999, 55, 13513 CrossRef CAS.
- P. H. Bernardo and C. L. L. Chai, J. Org. Chem., 2003, 68, 8906 CrossRef CAS.
- T. R. Kelly, Y. Zhao, M. Cavero and M. Torneiro, Org. Lett., 2000, 2, 3735 CrossRef CAS.
- R. Bonjouklian, T. A. Smitka, L. E. Doolin, R. M. Molloy, M. Debono, S. A. Shaffer, R. E. Moore, J. B. Stewart and G. M. L. Patterson, Tetrahedron, 1991, 47, 7739 CrossRef CAS.
- J. T. Kuethe, A. Wong and I. W. Davies, Org. Lett., 2003, 5, 3721 CrossRef CAS.
- S.-Y. Jeong, K. Ishida, Y. Ito, S. Okada and M. Murakami, Tetrahedron Lett., 2003, 44, 8005 CrossRef CAS.
- D. R. Appleton and B. R. Copp, Tetrahedron Lett., 2003, 44, 8963 CrossRef CAS.
- E. Tanaka, C. Tanaka, N. Mori, Y. Kuwahara and M. Tsuda, Phytochemistry, 2003, 64, 965 CrossRef CAS.
- L.-Z. Lin, G. A. Cordell, C.-Z. Ni and J. Clardy, J. Phytochem., 1991, 30, 1311 Search PubMed.
- M. Kitajima, N. Kogure, K. Yamaguchi, H. Takayama and N. Aimi, Org. Lett., 2003, 5, 2075 CrossRef CAS.
- M. Kitajima, A. Urano, N. Kogure, H. Takayama and N. Aimi, Chem. Pharm. Bull., 2003, 51, 1211 CrossRef CAS.
- K.-J. Siems, R. Weigl, M. Kaloga, J. Schulz and E. Eich, Phytochemistry, 2003, 62, 247 CrossRef CAS.
- H. R. Bokesch, L. K. Pannell, T. C. McKee and M. R. Boyd, Tetrahedron Lett., 2000, 41, 6305 CrossRef CAS.
- M. Chakrabarty, R. Basak and Y. Harigaya, Synthesis, 2003, 2011 CrossRef CAS.
- T. Kagamizono, N. Sakai, K. Arai, K. Kobinata and H. Osada, Tetrahedron Lett., 1997, 38, 1223 CrossRef CAS.
- S. G. Toske, P. R. Jensen, C. A. Kauffman and W. Fenical, Tetrahedron, 1998, 54, 13459 CrossRef CAS.
- S. Su, H. Kakeya, H. Osada and J. A. Porco, Jr., Tetrahedron, 2003, 59, 8931 CrossRef CAS.
- A. Hofmann, Bull. Narc., 1971, 23, 3 Search PubMed.
- N. Gathergood and P. J. Scammells, Org. Lett., 2003, 5, 921 CrossRef CAS.
- F. Yamada, M. Tamura and M. Somei, Heterocycles, 1998, 49, 451 CAS.
- O. Shirota, W. Hakamata and Y. Goda, J. Nat. Prod., 2003, 66, 885 CrossRef CAS.
- R. E. Moor, C. Cheuk, X.-Q. Yang, G. M. L. Patterson, R. Bonjouklian, T. A. Smita, J. S. Mynderse, R. S. Foster, N. D. Jones, J. K. Swartzendruber and J. B. Deeter, J. Org. Chem., 1987, 52, 1036 CrossRef CAS.
- A. C. Kinsman and M. A. Kerr, J. Am. Chem. Soc., 2003, 125, 14120 CrossRef CAS.
- H.-P. Ros, R. Kyburz, N. W. Preston, R. T. Gallagher, I. R. C. Bick and M. Hesse, Helv. Chim. Acta, 1979, 62, 481 CrossRef CAS.
- D. G. Washburn, R. W. Heidebrecht, Jr. and S. F. Martin, Org. Lett., 2003, 5, 3523 CrossRef CAS.
- W. J. Klaver, H. Hiemstra and W. N. Speckamp, J. Am. Chem. Soc., 1989, 111, 2588 CrossRef CAS.
- D. E. Lizos and J. A. Murphy, Org. Biomol. Chem., 2003, 1, 117 RSC.
- N. Aimi, T. Shimizu, H. Sada, H. Takayama, S. Sakai, S. Wongseripipatana and D. Ponglux, J. Chem. Soc., Perkin Trans. 1, 1997, 187 RSC.
- H. Takayama, R. Fujiwara, Y. Kasai, M. Kitajima and N. Aimi, Org. Lett., 2003, 5, 2967 CrossRef CAS.
- H.-G. Byun, H. Zhang, M. Mochizuki, K. Adachi, Y. Shizuri, W.-J. Lee and S.-K. Kim, J. Antibiot., 2003, 56, 102 CAS.
- C.-B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 534 CAS.
- E. Caballero, C. Avendano and J. C. Menéndez, J. Org. Chem., 2003, 68, 6944 CrossRef CAS.
- C.-B. Cui, H. Kakeya and H. Osada, Tetrahedron, 1997, 53, 59 CrossRef CAS.
- T. Onishi, P. R. Sebahar and R. M. Williams, Org. Lett., 2003, 5, 3135 CrossRef CAS.
- C.-B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 832 CAS.
- C. Meyers and E. M. Carreira, Angew. Chem., Int. Ed., 2003, 42, 694 CrossRef CAS.
- F. von Nussbaum and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 2175 CrossRef CAS.
- M. Yamazaki, E. Okuyama, M. Kobayashi and H. Inoue, Tetrahedron Lett., 1981, 22, 135 CrossRef CAS.
- R. M. Williams, J. Cao, H. Tsujishima and R. J. Cox, J. Am. Chem. Soc., 2003, 125, 12172 CrossRef CAS.
- M. Somei, Y. Karasawa and C. Kaneko, Heterocycles, 1981, 16, 941 CAS.
- Y. Shiono, K. Akiyama and H. Hayashi, Biosci. Biotechnol. Biochem., 2000, 64, 103 CAS.
- Y. Shiono, K. Akiyama and H. Hayashi, Biosci. Biotechnol. Biochem., 2000, 64, 1519 CAS.
- P. S. Baran, C. A. Guerrero and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 5628 CrossRef CAS.
- A. Z. Kozlovsky, V. P. Zhelifonova, T. V. Antipova, V. M. Adanin, S. M. Ozerskaya, N. E. Ivanushkina, F. A. Gollmick and U. Gräfe, Heterocycles, 2003, 60, 1639 CAS.
- S. Takano and K. Ogasawara, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1989, vol. 36, p. 225 Search PubMed and references cited therein.
- P. D. Rege and F. Johnson, J. Org. Chem., 2003, 68, 6133 CrossRef CAS.
- S. Takano, E. Goto, M. Hirama and K. Ogasawara, Chem. Pharm. Bull., 1982, 30, 2641 CAS.
- T. F. Spande, M. W. Edwards, L. K. Pannell and J. W. Daly, J. Org. Chem., 1988, 53, 1222 CrossRef CAS.
- T. Kawasaki, A. Ogawa, Y. Takashima and M. Sakamoto, Tetrahedron Lett., 2003, 44, 1591 CrossRef CAS.
- P. B. Holst, U. Anthoni, C. Christophersen and P. H. Nielsen, J. Nat. Prod., 1994, 57, 997 CrossRef CAS.
- G. H. Tan, X. Zhu and A. Ganesan, Org. Lett., 2003, 5, 1801 CrossRef CAS.
- M. Somei, F. Yamada, T. Izumi and M. Nakajou, Heterocycles, 1997, 45, 2327 CAS.
- J. M. Roe, R. A. B. Webster and A. Ganesan, Org. Lett., 2003, 5, 2825 CrossRef CAS.
- R. K. Duke, R. D. Allan, G. A. R. Johnston, K. N. Mewett, A. D. Mitrovic, C. C. Duke and T. W. Hambley, J. Nat. Prod., 1995, 58, 1200 CrossRef CAS.
- L. E. Overman and E. A. Peterson, Angew. Chem., Int. Ed., 2003, 42, 2521 CrossRef CAS.
- L. E. Overman and E. A. Peterson, Tetrahedron, 2003, 59, 6905 CrossRef CAS.
- E. F. L. J. Anet, G. K. Hughes and E. Ritchie, Aust. J. Chem., 1961, 14, 173 CAS.
- L. Verotta, F. Peterlongo, E. Elisabetsky, T. A. Amador and D. S. Nunes, J. Chromatogr., A, 1999, 841, 165 CrossRef CAS.
- L. E. Overman, J. F. Larrow, B. A. Stearns and J. M. Vance, Angew. Chem., Int. Ed., 2000, 39, 213 CrossRef CAS.
- H. Ishikawa, H. Takayama and N. Aimi, Tetrahedron Lett., 2002, 43, 5637 CrossRef CAS.
- J. F. Kodanko and L. E. Overman, Angew. Chem., Int. Ed., 2003, 42, 2528 CrossRef CAS.
- S. M. Lee, X. F. Li, H. Jiang, J. G. Cheng, S. Seong, H. D. Choi and B. W. Son, Tetrahedron Lett., 2003, 44, 7707 CrossRef CAS.
- A. Horeau, Tetrahedron Lett., 1961, 2, 506 CrossRef.
- X. F. Li, S. M. Lee, H. D. Choi, J. S. Kang and B. W. Son, Chem. Pharm. Bull., 2003, 51, 1458 CrossRef CAS.
- Y. Igarashi, K. Futamata, T. Fujita, A. Sekine, H. Senda, H. Naoki and T. Furumai, J. Antibiot., 2003, 56, 107 CAS.
- K. Warabi, S. Matsunaga, R. W. M. van Soest and N. Fusetani, J. Org. Chem., 2003, 68, 2765 CrossRef CAS.
- L. F. Tietze, F. Haunert, T. Feuerstein and T. Herzig, Eur. J. Org. Chem., 2003, 562 CrossRef CAS.
- T. Yasuzawa, Y. Saitoh, M. Ichimura, I. Takahashi and H. Sano, J. Antibiot., 1991, 44, 445 CAS.
- K. Yamada, T. Kurokawa, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2003, 125, 6630 CrossRef CAS.
- S. P. Gunasekera, I. A. Zuleta, R. E. Longley, A. E. Wright and S. A. Pomponi, J. Nat. Prod., 2003, 66, 1615 CrossRef CAS.
- Y. R. Torres, T. S. Bungni, R. G. S. Berlinck, C. M. Ireland, A. Magalhaes, A. G. Ferreira and R. M. da Rocha, J. Org. Chem., 2002, 67, 5429 CrossRef CAS.
- L. Legentil, J. Bastide and E. Delfourne, Tetrahedron Lett., 2003, 44, 2473 CrossRef CAS.
- N. B. Perry, J. W. Blunt and M. H. G. Munro, Tetrahedron, 1988, 44, 1727 CrossRef CAS.
- J. Kobayashi, J.-F. Cheng, M. Ishibashi, H. Nakamura, Y. Ohizumi, Y. Hirata, T. Sasaki, H. Lu and J. Clardy, Tetrahedron Lett., 1987, 28, 4939 CrossRef CAS.
- H. Tohma, Y. Harayama, M. Hashizume, M. Iwata, Y. Kiyono, M. Egi and Y. Kita, J. Am. Chem. Soc., 2003, 125, 11235 CrossRef CAS.
- K. V. Rao, B. D. Santarsiero, A. D. Mesecar, R. F. Schinazi, B. L. Tekwani and M. T. Hamann, J. Nat. Prod., 2003, 66, 823 CrossRef CAS.
- Y.-L. Leu, L.-S. Shi and A. G. Damu, Chem. Pharm. Bull., 2003, 51, 599 CrossRef CAS.
- P. Schupp, T. Poehner, R. A. Edrada, R. Ebel, A. Berg, V. Wray and P. Proksch, J. Nat. Prod., 2003, 66, 272 CrossRef CAS.
- N. Oku, S. Matsunaga and N. Fusetani, J. Am. Chem. Soc., 2003, 125, 2044 CrossRef CAS.
- P.-C. Kuo, L.-S. Shi, A. G. Damu, C.-R. Su, C.-H. Huang, C.-H. Ke, J.-B. Wu, A.-J. Lin, K. F. Bastow, K.-H. Lee and T.-S. Wu, J. Nat. Prod., 2003, 66, 1324 CrossRef CAS.
- C. Chevallier, A. D. Richardson, M. C. Edler, E. Hamel, M. K. Harper and C. M. Ireland, Org. Lett., 2003, 5, 3737 CrossRef CAS.
- P. Thongphasuk, R. Suttisri, R. Bavovada and R. Verpoorte, Phytochemistry, 2003, 62, 897 CrossRef.
- A. Itoh, T. Tanahashi, N. Nagakura and T. Nishi, Phytochemistry, 2003, 62, 359 CrossRef CAS.
- J. H. A. Paul, A. R. Maxwell and W. F. Reynolds, J. Nat. Prod., 2003, 66, 752 CAS.
- C. L. Cardoso, D. H. S. Silva, D. M. Tomazela, H. Verli, M. C. M. Young, M. Furlan, M. N. Eberlin, V. da and S. Bolzani, J. Nat. Prod., 2003, 66, 1017 CrossRef CAS.
- Y. W. Zhang, R. Yang, Q. Cheng and K. Ofuji, Helv. Chim. Acta, 2003, 86, 415 CrossRef CAS.
- N. L. Segraves, S. Lopez, T. A. Johnson, S. A. Said, X. Fu, F. J. Schmitz, H. Pietraszkiewicz, F. A. Valeriote and P. Crews, Tetrahedron Lett., 2003, 44, 3471 CrossRef CAS.
- E. Christopher, E. Bedir, C. Dunbar, I. A. Khan, C. O. Okunji, B. M. Schuster and M. M. Iwu, Helv. Chim. Acta, 2003, 86, 2914 CrossRef CAS.
- C. Wattanapiromsakul, P. I. Forsterc and P. G. Waterman, Phytochemistry, 2003, 62, 609 CrossRef CAS.
- T.-S. Kam and Y.-M. Choo, Tetrahedron Lett., 2003, 44, 8787 CrossRef CAS.
- J. Peng, J.-F. Hu, A. B. Kazi, Z. Li, M. Avery, O. Peraud, R. T. Hill, S. G. Franzblau, F. Zhang, R. F. Schinazi, S. S. Wirtz, P. Tharnish, M. Kelly, S. Wahyuono and M. T. Hamann, J. Am. Chem. Soc., 2003, 125, 13382 CrossRef CAS.
- M. Tsuda and J. Kobayashi, Heterocycles, 1997, 46, 765 CAS.
- H. Nakamura, S. Deng, J. Kobayashi, Y. Ohizumi, Y. Tomotake, T. Matsuzaki and Y. Hirama, Tetrahedron Lett., 1987, 28, 621 CrossRef CAS.
- N. Kasanah, K. V. Rao, M. Yousaf, D. E. Wedge and M. T. Hamann, Tetrahedron Lett., 2003, 44, 1291 CrossRef CAS.
- J. L. Frahn and D. F. O'Keefe, Aust. J. Chem., 1971, 24, 2189 CAS.
- A. Jossang, P. Jossang, H. A. Hadi, T. Sevénet and B. Bodo, J. Org. Chem., 1991, 56, 6527 CrossRef CAS.
- L. K. Larsen, R. E. Moore and G. M. L. Patterson, J. Nat. Prod., 1994, 57, 419 CrossRef CAS.
- S. W. Dantale and B. C. G. Söderberg, Tetrahedron, 2003, 59, 5507 CrossRef CAS.
- J. Ishida, H. K. Wang, M. Oyama, M. L. Cosentino, Q. H. Chang and K. H. Lee, J. Nat. Prod., 2001, 64, 958 CrossRef CAS.
- J. Ishida, H. K. Wang, K. F. Bastow, Q. H. Chang and K. H. Lee, Bioorg. Med. Chem. Lett., 1999, 9, 3319 CrossRef CAS.
- R. S. Kusurkar, S. K. Goswami and S. M. Vyas, Tetrahedron Lett., 2003, 44, 4761 CrossRef CAS.
- A. Pouilhés, Y. Langlois and A. Chiaroni, Synlett, 2003, 1488 CAS.
- A. Deiters, K. Chen, C. T. Eary and S. F. Martin, J. Am. Chem. Soc., 2003, 125, 4541 CrossRef CAS.
- J.-A. Duan, I. D. Williams, C.-T. Che, R.-H. Zhou and S.-X. Zhao, Tetrahedron Lett., 1999, 40, 2593 CrossRef CAS.
- T. Putkonen, A. Tolvanen, R. Jokela, S. Caccamese and N. Parrinello, Tetrahedron, 2003, 59, 8589 CrossRef CAS.
- S. Luo, C. A. Zificsak and R. P. Hsung, Org. Lett., 2003, 5, 4709 CrossRef CAS.
- L. Polz, J. Stöckigt, H. Takayama, N. Uchida, N. Aimi and S. Sakai, Tetrahedron Lett., 1990, 31, 6693 CrossRef CAS.
- P. D. Bailey, P. D. Clingan, T. J. Mills, R. A. Price and R. G. Pritchard, Chem. Commun., 2003, 2800 RSC.
- J.-L. Pousset, M.-T. Martin, A. Jossang and B. Bodo, Phytochemistry, 1995, 39, 735 CrossRef CAS.
- T. H. M. Jonckers, B. U. W. Maes, G. L. F. Lemiére, G. Rombouts, L. Pieters, A. Haemers and R. A. Dommisse, Synlett, 2003, 615 CAS.
- J. A. May, R. K. Zeidan and B. M. Stoltz, Tetrahedron Lett., 2003, 44, 1203 CrossRef CAS.
- F. Yamada, Y. Makita, T. Suzuki and M. Somei, Chem. Pharm. Bull., 1985, 33, 2162 CAS.
- A. S. Ratnayake, W. Y. Yoshida, S. L. Mooberry and T. K. Hemsheidt, J. Org. Chem., 2001, 66, 8717 CrossRef CAS.
- M. Tsuda, T. Mugishima, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami, M. Shiro, M. Hirai, Y. Ohizumi and J. Kobayashi, Tetrahedron, 2003, 59, 3227 CrossRef CAS.
- R. Veluri, I. Oka, I.-W. Döbler and H. Laatsch, J. Nat. Prod., 2003, 66, 1520 CrossRef CAS.
- E. M. K. Wijeratne, T. J. Turbyville, Z. Zhang, D. Bigelow, L. S. Pierson III, H. D. VanEtten, L. Whitesell, L. M. Canfield and A. A. L. Gunatilaka, J. Nat. Prod., 2003, 66, 1567 CrossRef CAS.
- S. P. Gunasekera, P. J. McCarthy and M.-K. Borges, J. Nat. Prod., 1994, 57, 1437 CrossRef CAS.
- T. Kawasaki, T. Kouko, H. Totsuka and K. Hiramatsu, Tetrahedron Lett., 2003, 44, 8849 CrossRef CAS.
- R. Talpir, Y. Benayahu, Y. Kashman, L. Pannell and M. Schleyer, Tetrahedron Lett., 1994, 35, 4453 CrossRef CAS.
- J. E. Coleman, E. D. de Silva, F. Kong, R. J. Andersen and T. M. Allen, Tetrahedron, 1995, 51, 10653 CrossRef CAS.
- P. Crews, J. J. Farias, R. Emrich and P. A. Keifer, J. Org. Chem., 1994, 59, 2932 CrossRef CAS.
- W. R. Gamble, N. A. Durso, R. W. Fuller, C. K. Weatergaard, T. R. Johnson, D. L. Sackett, E. Hamel, J. H. Cardellina and M. R. Boyd, Bioorg. Med. Chem., 1999, 7, 1611 CrossRef CAS.
- J. A. Nieman, J. E. Coleman, D. J. Wallace, E. Piers, L. Y. Lim, M. Roberge and R. J. Andersen, J. Nat. Prod., 2003, 66, 183 CrossRef CAS.
- C. Campagnuolo, E. Fattorusso and O.-T. Scafati, Eur. J. Org. Chem., 2003, 284 CrossRef CAS.
- W.-L. Li, Y.-H. Yi, H.-M. Wu, Q.-Z. Xu, H.-F. Tang, D.-Z. Zhou, H.-W. Lin and Z.-H. Wang, J. Nat. Prod., 2003, 66, 146 CrossRef CAS.
- K. L. Erickson, K. R. Gustafson, D. J. Milanowski, L. K. Pannell, J. R. Klose and M. R. Boyd, Tetrahedron, 2003, 59, 10231 CrossRef CAS.
- J. E. Leet, W. Li, H. A. Ax, J. A. Matson, S. Huang, R. Huang, J. L. Cantone, D. Drexler, R. A. Dalterio and K. S. Lam, J. Antibiot., 2003, 56, 232 CAS.
- W. Li, J. E. Leet, H. A. Ax, D. R. Gustavson, D. M. Brown, L. Turner, K. Brown, J. Clark, H. Yang, J.-F. Tomc and K. S. Lam, J. Antibiot., 2003, 56, 226 CAS.
- H. Suzuki, H. Morita, S. Iwasaki and J. Kobayashi, Tetrahedron, 2003, 59, 5307 CrossRef CAS.
- Y. Nakao, J. Kuo, W. Y. Yoshida, M. Kelly and P. J. Scheuer, Org. Lett., 2003, 5, 1387 CrossRef CAS.
- G. R. Pettit, R. Tan, Y. Ichihara, M. D. Williams, D. L. Doubek, L. P. Tackett, J. M. Schmidt, R. L. Cerny, M. R. Boyd and J. N. A. Hopper, J. Nat. Prod., 1995, 58, 961 CrossRef CAS.
- A. Napolitano, M. Rodriquez, I. Bruno, S. Marzocco, G. Autore, R. Riccio and L. G. Paloma, Tetrahedron, 2003, 59, 10203 CrossRef CAS.
- E. W. Schmidt and D. J. Faulkner, Tetrahedron, 1998, 54, 7631 CrossRef.
- J. Zhu and D. Ma, Angew. Chem., Int. Ed., 2003, 42, 5348 CrossRef CAS.
- K. Matsuzaki, T. Ogino, T. Sunazuka, H. Tanaka and S. Omura, J. Antibiot., 1997, 50, 236.
- H. Deng, J.-K. Jung, T. Liu, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 9032 CrossRef CAS.
- F. Firooznia, C. Gude, K. Chan, N. Marcopulos and Y. Satoh, Tetrahedron Lett., 1999, 40, 213 CrossRef CAS.
- N. Lindquist, W. Fenical, G. D. V. Duyne and J. Clardy, J. Am. Chem. Soc., 1991, 113, 2303 CrossRef CAS.
- J. Li, A. W. G. Burgett, L. Esser, C. Amezcua and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4770 CrossRef CAS.
- K. C. Nicolaou, P. B. Rao, J. Hao, M. V. Reddy, G. Rassias, X. Huang, D. Y.-K. Chen and S. A. Snyder, Angew. Chem., Int. Ed., 2003, 42, 1753 CrossRef CAS.
- A. W. G. Burgett, Q. L. Q. Wei and P. G. Harran, Angew. Chem., Int. Ed., 2003, 42, 4961 CrossRef CAS.
- J. Khono, Y. Koguchi, M. Nishio, K. Najao, M. Juroda, R. Shimizu, T. Ohnuki and S. Komatsubara, J. Org. Chem., 2000, 65, 990 CrossRef CAS.
- B. K. Albrecht and R. M. Williams, Org. Lett., 2003, 5, 197 CrossRef CAS.
- S. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2002, 41, 512 CrossRef.
- M. Inoue, H. Sakazaki, H. Furuyama and M. Hirama, Angew. Chem., Int. Ed., 2003, 42, 2654 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |