Synthesis, photophysics, electrochemistry and metal ion-binding studies of rhenium(I) complexes with crown ether pendants: selective and specific binding properties for various metal ions
Keith Man-Chung
Wong
,
Wei-Ping
Li
,
Kung-Kai
Cheung
and
Vivian Wing-Wah
Yam
*
Open Laboratory of Chemical Biology of the Institute of Molecular Technology for Drug Discovery and Synthesis, and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, People’s Republic of China
First published on 10th December 2004
Abstract
A series of rhenium(I) diimine complexes with crown ether pendants has been synthesized and the photophysical and electrochemical behaviour studied. By variation of the cavity size and donor atoms of the crown ether, the complexes showed selective and specific binding properties for various metal cations of different sizes and different degrees of hardness and softness. The X-ray crystal structure of one of the complexes has also been determined.
Introduction
Since the pioneering works of Pedersen, Lehn and Cram,1–3 the chemistry and applications of host–guest interactions have been elaborated rapidly and with remarkable results. Numerous kinds of novel crown compounds and their analogues, including aza- and thiacrown compounds, cryptands, optically active compounds and polymeric crown compounds, have been synthesized so far and their specific properties have been investigated.4 Studies on the host–guest interactions of known chemical composition and structure are indeed highly interesting and useful since they can help in elucidating the factors that control receptor-substrate interactions in biological systems, and can also lead to the creation of novel species and invention of novel chemical processes. Selective binding of cations in solution is a distinctive feature of the solution chemistry of macrocyclic polyethers. The design and synthesis of new macrocyclic ligands with different cavity sizes, donor atom types, ring substituents, and so on, have resulted in a large number and variety of compounds. The process of ion-macrocycle association depends on several factors related to characteristic properties of the ligand, reacting ion and solvent. The enhancement of complex stability by a close correspondence between the ionic radius of the metal ion and the radius of the cavity formed by the crown ether ring has been noted.5 As the ring size of the crown ether increases, the larger cations will bind more strongly. The exchange of donor sites in crown ethers, for example, N or S for O, will have even more drastic effects on the complexation behaviour. The complexation of alkali metal ions such as K+ falls dramatically when the ring oxygens are replaced with sulfur or nitrogen, but rises significantly in this order in the case of transition metal ions, such as Ag+.6,7Recently, a series of copper(I),8 ruthenium(II)9, zinc(II)10 gold(I),11 platinum(II)12 and palladium(II)13 complexes containing various crown ether pendants has been synthesized by our group. Their photophysical, spectrochemical and electrochemical properties upon alkali and alkaline earth metal cation binding have been reported. The electronic absorption and emission energies of the complexes were found to be sensitive to the presence of metal cations. The binding constants of the complexes with various metal cations were determined based on the UV-visible spectral changes. It is known that incorporating some soft donor atoms such as sulfur into the crown ether moieties may enhance their complexation ability for transition metal ions.4,14,15 However, relatively little attention has been focused on the incorporation of this type of crown ether units into systems involving transition metal complexes. These reasons, together with our previous interest in the utilization of transition metal complexes containing crown ether pendants to serve as chemosensors of various metal ions, have prompted us to synthesize other related complexes with the oxygen atoms in the crown ether ligands replaced by softer chalcogen atoms such as sulfur or selenium, which may give rise to selective and specific binding to transition metal ions such as silver(I) ion. Variation of the cavity size of the crown ethers will also be performed to achieve selective binding.
As a continuation of our previous studies on the ion-binding properties of a rhenium(I) diimine complex with a crown ether containing ligand, [Re(CO)3(dic)Cl] [1; dic = N-(2-pyridylmethylene)-4-aminobenzo-15-crown-5], towards different alkali or alkaline earth metal ions,16 we report herein the synthesis of a related series of rhenium(I) diimine complexes with crown ether pendants: [Re(CO)3(dic-18-c-6)Cl] (2), [{Re(CO)3Cl}2(ddic)] (3), [Re(CO)3(dic-S)Cl] (4), [Re(CO)3(dic-3S)Cl] (5) and [Re(CO)3(dic-Se)Cl] (6), shown in Scheme 1. The photophysical, electrochemical and cation-binding properties of these systems have been studied. Complexes 2 and 3, with benzo-18-crown-6 and dibenzo-18-crown-6 pendants, respectively, were synthesized to provide a direct comparison of the cation-binding properties between the mononuclear and the dinuclear Re(I) systems as well as with the complex containing the benzo-15-crown-5 pendant, that is 1.16 The selectivities and specific cation-binding properties have also been altered by incorporating different donor atoms such as sulfur and selenium into the crown ether unit in 4–6. Apart from binding studies using alkali and alkaline earth metal cations, other transition metal ions such as Ag(I) were also employed in the investigation of the binding properties. Their stability constants have been determined by both UV-visible spectrophotometric and 1H NMR titrations.
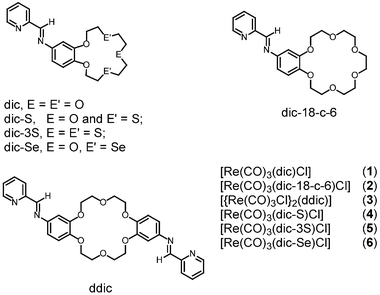 | ||
| Scheme 1 | ||
Experimental
Rhenium(I) pentacarbonyl chloride (Strem), benzo-18-crown-6 (Aldrich), hydrazine hydrate (70%, BDH), silver trifluoromethanesulfonate (99%, Aldrich) and palladium/charcoal (10%, Merck) were used without purification. 2-Pyridinecarboxaldehyde was obtained from Aldrich and distilled under vacuum before use. Sodium perchlorate, potassium acetate, barium perchlorate and tetra-n-butylammonium hexaflurophosphate were recrystallized from hot methanol and dried under vacuum before use. Aminobenzo-18-crown-6,17 diaminobenzo-15-crown-5,18 and trans-4,4′-diaminodibenzo-18-crown-619 were synthesized according to literature procedures. The ligands dic-18-c-6, dic-S, dic-3S and dic-Se were prepared according to the procedures reported previously.8bSyntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 8.7 (m, 2H, pyridyl H). EI-MS: m/z 568 [ddic]+.
CH), 7.1 (dd, 1H, J
= 8.6 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.2 and 1.2 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.0 (d, 1H, J
= 5.2 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2017 (s)
ν(C≡O), 1913 (s)
ν(C≡O), 1895 (s)
ν(C≡O); positive FAB-MS: m/z 677 [M]+, 642 [M
N–), 8.7 (m, 2H, pyridyl H). EI-MS: m/z 568 [ddic]+.
CH), 7.1 (dd, 1H, J
= 8.6 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.2 and 1.2 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.0 (d, 1H, J
= 5.2 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2017 (s)
ν(C≡O), 1913 (s)
ν(C≡O), 1895 (s)
ν(C≡O); positive FAB-MS: m/z 677 [M]+, 642 [M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −Cl]+; Anal. found (%): C 40.40, H 3.80, N 4.11; calcd. for [Re(CO)3(dic)Cl]
(%): C 40.70, H 3.54, N 4.13.
CH), 7.0 (dd, 1H, J
= 8.4 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.6 and 1.2 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.1 (d, 1H, J
= 5.6 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2017 (s)
ν(C≡O), 1914 (s)
ν(C≡O), 1900 (s)
ν(C≡O); positive FAB-MS: m/z 722 [M]+, 687 [M−Cl]+; Anal. found (%): C 41.23, H 4.02, N 3.91; calcd. for [Re(CO)3(dic-18-c-6)Cl]
(%): C 41.57, H 3.88, N 3.88.
CH), 7.0 (dd, 2H, J
= 8.6 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 2H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 2H, J
= 5.6 and 1.2 Hz, pyridyl H), 8.0 (d, 2H, J
= 7.8 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 2H, J
= 7.8 and 1.2 Hz, pyridyl H), 8.8 (s, 2H, –CHN–), 9.1 (d, 2H, J
= 5.6 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2020 (s)
ν(C≡O), 1920 (s)
ν(C≡O), 1905 (s)
ν(C≡O); positive FAB-MS: m/z 1180 [M]+, 1145 [M−Cl]+; Anal. found (%): C 38.22, H 2.96, N 4.57; calcd. for [{Re(CO)3Cl}2(ddic)]
(%): C 38.66, H 2.71, N 4.75.
CH), 7.1 (dd, 1H, J
= 8.6 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.4 and 1.2 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.0 (d, 1H, J
= 5.4 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2014 (s)
ν(C≡O), 1909 (s)
ν(C≡O), 1884 (s)
ν(C≡O); positive FAB-MS: m/z 694 [M]+, 659 [M−Cl]+; Anal. found (%):C 38.86, H 3.33, N 3.78; calcd. for [Re(CO)3(dic-S)Cl]·H2O (%): C 38.77, H 3.65, N 3.93.
CH), 6.9 (dd, 1H, J
= 8.4 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.2 and 1.2 Hz, pyridyl H), 7.9 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN–), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.1 (d, 1H, J
= 5.2 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2015 (s)
ν(C≡O), 1907 (s)
ν(C≡O), 1882 (s)
ν(C≡O); positive FAB-MS: m/z 726 [M]+, 691 [M−Cl]+; Anal. found (%): C 37.14, H 3.13, N 3.60; calcd. for [Re(CO)3(dic-3S)Cl]·H2O (%): C 37.11, H 3.50, N 3.76.
CH), 7.1 (dd, 1H, J
= 8.6 and 2.6 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.6 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.6 and 1.0 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.0 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.0 (d, 1H, J
= 5.6 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2014 (s)
ν(C≡O), 1909 (s)
ν(C≡O), 1883 (s)
ν(C≡O); positive FAB-MS: m/sz 742 [M]+, 707 [M−Cl]+; Anal. found (%): C 37.61, H 3.31, 3.67; calcd. for [Re(CO)3(dic-Se)Cl]
(%): C 37.25, H 3.24, N 3.78.
−Cl]+; Anal. found (%): C 40.40, H 3.80, N 4.11; calcd. for [Re(CO)3(dic)Cl]
(%): C 40.70, H 3.54, N 4.13.
CH), 7.0 (dd, 1H, J
= 8.4 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.6 and 1.2 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.1 (d, 1H, J
= 5.6 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2017 (s)
ν(C≡O), 1914 (s)
ν(C≡O), 1900 (s)
ν(C≡O); positive FAB-MS: m/z 722 [M]+, 687 [M−Cl]+; Anal. found (%): C 41.23, H 4.02, N 3.91; calcd. for [Re(CO)3(dic-18-c-6)Cl]
(%): C 41.57, H 3.88, N 3.88.
CH), 7.0 (dd, 2H, J
= 8.6 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 2H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 2H, J
= 5.6 and 1.2 Hz, pyridyl H), 8.0 (d, 2H, J
= 7.8 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 2H, J
= 7.8 and 1.2 Hz, pyridyl H), 8.8 (s, 2H, –CHN–), 9.1 (d, 2H, J
= 5.6 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2020 (s)
ν(C≡O), 1920 (s)
ν(C≡O), 1905 (s)
ν(C≡O); positive FAB-MS: m/z 1180 [M]+, 1145 [M−Cl]+; Anal. found (%): C 38.22, H 2.96, N 4.57; calcd. for [{Re(CO)3Cl}2(ddic)]
(%): C 38.66, H 2.71, N 4.75.
CH), 7.1 (dd, 1H, J
= 8.6 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.4 and 1.2 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.0 (d, 1H, J
= 5.4 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2014 (s)
ν(C≡O), 1909 (s)
ν(C≡O), 1884 (s)
ν(C≡O); positive FAB-MS: m/z 694 [M]+, 659 [M−Cl]+; Anal. found (%):C 38.86, H 3.33, N 3.78; calcd. for [Re(CO)3(dic-S)Cl]·H2O (%): C 38.77, H 3.65, N 3.93.
CH), 6.9 (dd, 1H, J
= 8.4 and 2.4 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.4 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.2 and 1.2 Hz, pyridyl H), 7.9 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN–), 8.1 (dt, 1H, J
= 7.6 and 1.2 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.1 (d, 1H, J
= 5.2 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2015 (s)
ν(C≡O), 1907 (s)
ν(C≡O), 1882 (s)
ν(C≡O); positive FAB-MS: m/z 726 [M]+, 691 [M−Cl]+; Anal. found (%): C 37.14, H 3.13, N 3.60; calcd. for [Re(CO)3(dic-3S)Cl]·H2O (%): C 37.11, H 3.50, N 3.76.
CH), 7.1 (dd, 1H, J
= 8.6 and 2.6 Hz, aryl H ortho to –NCH), 7.2 (d, 1H, J
= 2.6 Hz, aryl H ortho to –OCH2 and –NCH), 7.6 (dt, 1H, J
= 5.6 and 1.0 Hz, pyridyl H), 8.0 (d, 1H, J
= 7.6 Hz, pyridyl H ortho to –CHN), 8.1 (dt, 1H, J
= 7.6 and 1.0 Hz, pyridyl H), 8.8 (s, 1H, –CHN–), 9.0 (d, 1H, J
= 5.6 Hz, pyridyl H ortho to N). IR (Nujol mull, KBr, cm−1): 2014 (s)
ν(C≡O), 1909 (s)
ν(C≡O), 1883 (s)
ν(C≡O); positive FAB-MS: m/sz 742 [M]+, 707 [M−Cl]+; Anal. found (%): C 37.61, H 3.31, 3.67; calcd. for [Re(CO)3(dic-Se)Cl]
(%): C 37.25, H 3.24, N 3.78.
Physical measurements and instrumentation
UV-visible spectra were obtained with a Hewlett–Packard 8452A diode array spectrophotometer, IR spectra as KBr discs with a Bio-Rad FTS-7 Fourier transform IR spectrophotometer (4000–400 cm−1), and steady-state excitation and emission spectra with a Spex Fluorolog 111 spectrophotometer equipped with a Hamamatsu R-928 photomultiplier tube. All solutions for photophysical studies were prepared under high vacuum in a 10 cm3 round-bottomed flask equipped with a side arm 1 cm fluorescence cuvette and sealed from the atmosphere by a Rotaflo HP6/6 quick-release Teflon stopper. Solutions were rigorously degassed with no fewer than four successive freeze-pump-thaw cycles.1H NMR spectra were recorded with a Bruker DPX-300 FTNMR spectrometer (300 MHz) in CDCl3 or CD2Cl2 at 298 K, and chemical shifts are reported relative to Me4Si. All EI and positive-ion FAB mass spectra were recorded with a Finnigan MAT95 mass spectrometer. Elemental analyses of the new complexes were performed with a Carlo Erba 1106 elemental analyzer at the Institute of Chemistry, Chinese Academy of Sciences.
Cyclic voltammetric measurements were performed by using a CH Instruments, Inc. model CHI 620 Electrochemical Analyzer. Electrochemical measurements were performed in acetonitrile solutions with 0.1 M nBu4NPF6 (TBAH) as supporting electrolyte at room temperature. The reference electrode was a Ag/AgNO3 (0.1 M in acetonitrile) electrode and the working electrode was a glassy carbon electrode (Atomergic Chemetals V25) with a platinum gauze as the counter electrode. The ferrocenium/ferrocene couple (FeCp2+/0) was used as the internal reference. All solutions for electrochemical studies were deaerated with prepurified argon gas just before measurements.
X-Ray crystallography
An orange crystal of 6 with dimensions of 0.40 × 0.10 × 0.05 mm3 mounted on a glass fibre was used for data collection at 28 °C on a Rigaku AFC7R diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) using ω−2θ scans with a ω scan angle (0.63 + 0.35tan θ)° at a scan speed of 86.0 deg min−1 [up to 6 scans for reflections I < 15σ(I)]. Unit cell dimensions were determined based on 25 reflections in the 2θ range of 32.52° to 37.4°. Intensity data (in the range of 2θmax
= 45°; h: −8 to 9; k: 0 to 13; l: −13 to 14 and 3 standard reflections measured after every 300 reflections showed decay of 0.65%), were corrected for Lorentz and polarization effects, and empirical absorption corrections were based on the ψ scan of five strong reflections (minimum and maximum transmission factors 0.649 and 1.000). The space group was used on the basis of a statistical analysis of the intensity distribution and the successful refinement of the structure solved by Patterson methods and expanded by Fourier methods (PATTY22) and refinement by full-matrix least squares using the software package TeXsan23 on a Silicon Graphics Indy computer. Two carbon atoms, C(17) and C(18), were disordered into two sets of positions and refined isotropically. One crystallographic asymmetric unit consists of one molecule.†
Crystal data for 6: ReC23H24N2O7SeCl, Mr
= 741.07, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a
= 8.669(4), b
= 12.752(4), c
= 13.644(4)
Å, α
= 67.57(3)°, β
= 89.21(3)°, γ
= 72.56(3)°, V
= 1321.4(9)
Å3, Z
= 2, μ(Mo-Kα)
= 6.12 cm−1, 3651 reflections measured, 3464 unique (Rint
= 0.015) reflections of which 3037 [I > 3σ(I)] reflections were used for refinement with R
= 0.026 and wR
= 0.032.
(No. 2), a
= 8.669(4), b
= 12.752(4), c
= 13.644(4)
Å, α
= 67.57(3)°, β
= 89.21(3)°, γ
= 72.56(3)°, V
= 1321.4(9)
Å3, Z
= 2, μ(Mo-Kα)
= 6.12 cm−1, 3651 reflections measured, 3464 unique (Rint
= 0.015) reflections of which 3037 [I > 3σ(I)] reflections were used for refinement with R
= 0.026 and wR
= 0.032.
Stability constant determination
The electronic absorption spectral titration for binding constant determination was performed with a Hewlett–Packard 8452A diode array spectrophotometer at 25 °C. Typically, the concentration of rhenium(I) complex solution used for electronic absorption spectral titration was 1.2 × 10−3 mol dm−3, except for 3 with a concentration of 0.6 × 10−4 mol dm−3, in CH3OH or CH3CN. Binding constants for 1∶1 complexation were obtained by a nonlinear least-squares fit12,24 of the absorbance (X) versus the concentration of the metal ion added (Cm) according to eqn. (1): | (1) |
For proton NMR titration experiments, a solution of the rhenium(I) complex was prepared at a concentration typically on the order of 5.5–6.3 × 10−3 mol dm−3 in CD3CN (1 ml). The initial 1H NMR spectrum was recorded and aliquots of silver trifluoromethanesulfonate were added using a microsyringe. The typical concentration of the metal cation was such that a 10 µl addition gave 0.2 equiv. of metal cation. After each addition and mixing, the spectrum was recorded and changes in the chemical shift of certain protons were noted. The results of the experiment were expressed as a plot of chemical shift as a function of the amount of cation added, which was subjected to analysis by curve fitting since the shape of the titration curve is indicative of the stability constant for complex formation. The computer program EQNMR25 was used, which requires a knowledge of the concentration of each component and the observed chemical shift for each data point.
Results and discussion
Crystal structure determination
A perspective drawing of 6 is depicted in Fig. 1. The structure of 6 shows a distorted octahedral geometry with three carbonyl ligands in a facial arrangement, which is commonly observed in other related rhenium(I) tricarbonyl diimine complexes.16,20,26–28 The structure of the crown-free analogue, [Re(CO)3{N-(2-pyridinylmethylene)benzenamine}Cl], has been reported; it showed a similar geometry as well as comparable bond distances and angles.20 The bond distances of C(8)–C(9) and N(2)–C(10) are 1.450(9) and 1.444(7) Å, respectively, indicative of bond character intermediate between a single and a double bond, while the N(2)–C(9) bond with a length of 1.262(8) Å is typical of a double bond. Similar to other related complexes, the N(1)–Re(1)–N(2) bond angle of 74.3(2)°, which is smaller than 90°, is due to the bite distances exerted by the steric requirements of the chelating diimine ligand.16,26–28 | ||
| Fig. 1 Perspective drawing of 6 with the atomic numbering scheme. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. Selected bond distances (Å) and bond angles (deg): Re(1)–Cl(1) 2.472(2), Re(1)–N(1) 2.174(5), Re(1)–N(2) 2.207(5), Re(1)–C(1) 1.903(8), Re(1)–C(2) 1.936(7), Re(1)–C(3) 1.918(8), C(8)–C(9) 1.450(9), N(2)–C(9) 1.262(8), N(2)–C(10) 1.444(7), Cl(1)–Re(1)–C(2) 177.0(2), N(1)–Re(1)–N(2) 74.3(2), N(1)–Re(1)–C(1) 175.3(2), N(2)–Re(1)–C(3) 169.7(2), Re(1)–C(1)–O(1) 177.6(7), Re(1)–C(2)–O(2) 174.1(7), Re(1)–C(3)–O(3) 178.5(7), N(1)–C(8)–C(9) 114.7(6), N(2)–C(9)–C(8) 119.8(6). | ||
Photophysical properties
The electronic absorption spectra of 1–6 in methanol or acetonitrile showed similar patterns with a high-energy absorption band at ca. 272 nm and a low-energy absorption band at ca. 400 nm. The photophysical data for 1–6 are summarized in Table 1. With reference to previous spectroscopic work on rhenium(I) diimine systems,16,20,21,27–39 the low-energy absorption bands at ca. 400 nm are tentatively assigned as a mixed dπ(Re) → π*(diimine) MLCT/π(diimine) → π*(diimine) IL transition. The close resemblance of the electronic absorption energies of the complexes is suggestive of a small influence of the variation of crown ether cavity size and donor atoms on the π*(diimine) orbital energies. This is understandable since a variation of the crown ether unit is too remote to affect the MLCT/IL transition energies. Fig. 2 depicts the electronic absorption spectra of 2 and 3 in MeOH at 298 K. For complex 3, the molar extinction coefficients of both the high-energy and low-energy absorption bands are almost double those of the mononuclear counterparts, 1, 2 and 4–6. This is in accord with the presence of two chromophores in the dinuclear complex. | ||
| Fig. 2 Electronic absorption spectra of 2 (—) and 3 (⋯) in MeOH at 298 K. | ||
| Complex | Medium | λ abs/nm (ε/dm3 mol−1 cm−1) | Corrected λema/nm (τo/μs) |
|---|---|---|---|
| a Measured in dichloromethane solution. | |||
| 1 | CH3OH | 272 (15980), 399 (9150) |
777 (<0.1) |
| 2 | CH3OH | 272 (16080), 400 (9410) |
785 (<0.1) |
| 3 | CH3OH | 272 (31200), 400 (18090) |
790 (<0.1) |
| 4 | CH3OH | 272 (15900), 400 (9440) |
777 (<0.1) |
| CH3CN | 274 (14910), 398 (9000) |
||
| 5 | CH3OH | 272 (16400), 400 (9510) |
785 (<0.1) |
| CH3CN | 272 (15770), 396 (8960) |
||
| 6 | CH3OH | 272 (16390), 400 (9500) |
790 (<0.1) |
| CH3CN | 272 (16020), 400 (9360) |
||
Complexes 1–6 exhibit a rather weak emission band at ca. 777–790 nm in CH2Cl2 solution upon visible light excitation (λ > 350 nm). The emission spectrum of 4 in degassed CH2Cl2 at 298 K is shown in Fig. 3. Although the lifetimes of the complexes are too short to be measured accurately with our instrument (τ < 0.1 µs, thus in the submicrosecond range), the emission is assigned as derived from the dπ(Re)
→
π*(diimine)
3MLCT excited state, which has a lifetime typically in the submicrosecond range. Lower emission energies were observed in 1–6, compared to those of other chlororhenium(I) polypyridyl complexes, [Re(CO)3(N–N)Cl]
(N–N=bpy, phen),27–39 which is attributed to the lower-lying π* orbital energies of the N-(2-pyridinylmethylene)benzenamine ligands. The shortening of emission lifetime in the present system with the comparatively lower-energy 3MLCT excited state is consistent with the prediction from the energy gap law.30 Similar to the electronic absorption spectroscopy, the occurrence of emission bands at similar energies is ascribed to a small influence of the variation of crown ether cavity size and donor atoms on the π*(diimine) orbital energies.
 | ||
| Fig. 3 Corrected emission spectrum of 4 in degassed CH2Cl2 at 298 K. | ||
Electrochemical properties
The electrochemical properties of 1–6 have been investigated by cyclic voltammetric studies, which show very similar electrochemical behaviour. The cyclic voltammograms of 1–6 in acetonitrile show one quasi-reversible reduction couple and an irreversible reduction wave upon reduction scanning, while two irreversible oxidation waves were observed in the oxidation scan. Their electrochemical data are summarized in Table 2. The first quasi-reversible reduction couple for all the complexes occurs at almost the same potential, at ca. −1.01 V vs. SCE, which is assigned as a diimine ligand-centred reduction. A similar reduction potential (−0.94 V vs. SCE) was observed in the crown-free analogue, [Re(CO)3{N-(2-pyridinylmethylene)benzenamine}Cl].20 The relatively greater ease of reduction of the crown-free ligand [N-(2-pyridinylmethylene)benzenamine] than the crown ether containing ligands is in agreement with the lower π* orbital energy of the former than the crown ether containing ligands, which have electron-donating polyether substituents on the aromatic ring. On the other hand, the relatively less negative reduction potential compared with other related chlororhenium(I) diimine complexes, [Re(CO)3(N–N)Cl] (N–N=bpy, phen),28–32,35,36 reveals the lower π* orbital energies of N-(2-pyridinylmethylene)benzenamine ligands, which can be supported by the observation of a lower MLCT transition energy for this series of complexes.
| Complex | Oxidation Epa/V vs. SCEb | Reduction E1/2/V vs. SCEc (ΔEp/mV) |
|---|---|---|
a Working electrode: glassy carbon; scan rate = 100 mV s−1; ΔEp of Fc+/Fc = 69–78 mV.
b
E
pa: anodic peak potential.
c
 , Epa and Epc are anodic and cathodic peak potentials, respectively.
d Cathodic peak potential for the irreversible wave. , Epa and Epc are anodic and cathodic peak potentials, respectively.
d Cathodic peak potential for the irreversible wave.
|
||
| 1 | +1.40 | −1.02 (75.2) |
| +1.58 | −1.46d | |
| 2 | +1.41 | −1.02 (73.8) |
| +1.59 | −1.44d | |
| 3 | +1.39 | −1.04 (78.5) |
| +1.58 | −1.52d | |
| 4 | +1.41 | −1.10 (71.9) |
| +1.59 | −1.42d | |
| 5 | +1.27 | −1.01 (80.2) |
| +1.43 | −1.43d | |
| 6 | +1.13 | −1.01 (72.9) |
| +1.44 | −1.42d | |
The second cathodic wave, which occurs at ca. −1.42 V, is likely to be the Re(I)/Re(0) reduction. Similar assignments have also been suggested in other rhenium(I) diimine complexes.28–32,35,36 The close resemblance of the reduction potentials suggests that the changes in the crown ether moiety, whether a change in the cavity size or replacement of oxygen atoms by other softer donor atoms, will not significantly alter the ease of reduction of the ligands. Similarly, an irreversible anodic wave, which is relatively insensitive to the nature of the crown ether containing ligands, was observed at ca. +1.41 V vs. SCE and is assigned as a Re(I) → Re(II) oxidation. Similar assignments have also been made for the irreversible oxidation wave in other related Re(I) diimine systems.28–32,35,36
Cation-binding properties
Upon addition of alkali and alkaline earth metal ions, such as sodium perchlorate, potassium acetate and barium perchlorate, a blue shift of the MLCT/IL absorption band in methanol solution has been observed for complexes 1–3. Such a shift in absorption energy can be ascribed to encapsulation of metal cations into the crown ether moiety of the complexes as similar shifts are absent in the related crown-free complexes. Other related Cu(I), Ru(II) and Zn(II) systems also exhibited a similar absorption energy shift upon addition of alkali or alkaline earth metal cations.8,9a,10 All the absorption curves show a gradual decrease in absorbance at ca. 400 nm upon increasing the cation concentration, reaching saturation at higher cation concentration. Fig. 4 illustrates the electronic absorption spectral traces of 2 in MeOH at 298 K upon addition of barium ion.![Electronic absorption spectral traces of 2 in methanol at 298 K upon addition of barium perchlorate. The insert shows a plot of absorbance versus
[Ba2+] monitored at λ
= 350 nm (■) and the theoretical fit (—).](/image/article/2005/NJ/b415951a/b415951a-f4.gif) | ||
| Fig. 4 Electronic absorption spectral traces of 2 in methanol at 298 K upon addition of barium perchlorate. The insert shows a plot of absorbance versus [Ba2+] monitored at λ = 350 nm (■) and the theoretical fit (—). | ||
The binding ability of the complexes with the cations can be determined from a knowledge of the stability constants. With the exception of 1 with potassium ion, well-defined isosbestic points were observed at ca. 380–390 nm upon addition of all the three metal cations, suggesting that only two absorbing species in equilibrium are present in the solution: the free and bound species. The observation of well-defined isosbestic points in the UV-visible spectral traces and the satisfactory agreement between experimental data and theoretical fit according to eqn. (1) for sodium, potassium and barium ions with 2 and 3, and for sodium and barium ions with 1, are suggestive of a complexation stoichiometry of 1∶1. The lack of well-defined isosbestic points in the UV-visible spectral traces and the absence of satisfactory agreement of the experimental data to the theoretical fit for potassium ion with 1 are suggestive of a system involving a mixture of 1∶1 and 1∶2 complexation. The stability constants of 1–3 with sodium, potassium and barium cations are summarized in Table 3.
Due to the better size match of the benzo-15-crown-5 cavity with sodium ion than with barium ion, the stability constant for sodium ion was larger than that for barium ion in 1. Similarly, for 2 and 3 with the respective benzo-18-crown-6 and dibenzo-18-crown-6 pendants, the stability constant for potassium ion was larger than those for sodium and barium ions due to the better size match. These results are in accordance with those observed in the related organic systems.1,40–43 Despite the fact that the ionic diameter of sodium ion should better fit the benzo-15-crown-5 cavity than that of crown ethers having 18-membered rings, the stability constants for sodium ion with 2 (log Ks = 4.1) and 3 (log Ks = 3.8), which contain the respective benzo-18-crown-6 and dibenzo-18-crown-6 pendants, are larger than that with 1 (log Ks = 2.4), which contains the benzo-15-crown-5 pendant. A similar trend of log Ks for benzo-15-crown-5 (2.9–3.4) < benzo-18-crown-6 (4.0–4.5) ≈ dibenzo-18-crown-6 (4.4–4.5) in methanol has also been observed.40–43 Such a phenomenon may be due to the larger size of the solvated sodium ion as a result of its good solubility in methanol as well as the fact that the 18-crown-6 is able to form a 18-crown-6–Na+ complex in a folded fashion with one oxygen atom out-of-the-plane in order to obtain a better fit of the sodium ion.44
The cation-binding properties of 4–6 with silver ion (silver trifluoromethanesulfante), which is a soft metal cation and is known to bind to thia- and selenacrown ethers, have also been studied by UV-visible absorption spectrophotometry and 1H NMR spectroscopy. Similar to the titration studies of 1–3 with alkali and alkaline earth metal ions in the electronic absorption spectra, a blue shift in the absorption band of 4–6 was observed upon addition of Ag+ into an acetonitile solution of the complexes. The UV-visible absorption spectral traces of 6 upon sequential addition of silver ions in acetonitrile are shown in Fig. 5. Although the change in the absorbance and the shift of the peak maximum are small, which can be ascribed to the small charge density of the silver ion, a clean isosbestic point was observed in all cases, indicating the presence of two species in equilibrium in such ion-binding experiments. Similarly, the stability constants of 4–6 with silver ion are obtained by the theoretical fits to eqn. (1) and are summarized in Table 4. The close agreement of the experimental data with the theoretical fit is supportive of a 1∶1 stoichiometry. However, no remarkable absorption spectral changes occur upon addition of alkali or alkaline earth metal cations into the solution of complexes 4–6.
![Electronic absorption spectral traces of 6 in acetonitrile at 298 K upon addition of silver trifluoromethanesulfonate. The insert shows a plot of absorbance versus
[Ag+] monitored at λ
= 350 nm (■) and the theoretical fit (—).](/image/article/2005/NJ/b415951a/b415951a-f5.gif) | ||
| Fig. 5 Electronic absorption spectral traces of 6 in acetonitrile at 298 K upon addition of silver trifluoromethanesulfonate. The insert shows a plot of absorbance versus [Ag+] monitored at λ = 350 nm (■) and the theoretical fit (—). | ||
| Complex | UV-Vis absorption spectrophotometrya | 1H NMR spectroscopyb |
|---|---|---|
| a Values in brackets are log Ks values obtained in methanol solution. b In deuterated acetonitrile, errors estimated to be ≤ 2%. | ||
| 4 | 2.6 [4.7] | 2.5 |
| 5 | 3.7 [5.7] | 3.9 |
| 6 | 3.0 [4.9] | 3.1 |
1H NMR studies have also been carried out to investigate the interaction of 4–6 with silver ion. Such results would constitute an independent set of measurements that could be compared to those determined using UV-visible absorption spectrophotometry. The addition of silver ion to deuterated acetonitrile solutions of the complexes resulted in a downfield perturbation of the crown ether protons, especially the methylene protons adjacent to the sulfur or selenium atom (Fig. 6). Typically, such methylene protons display a downfield shift of ca. 0.2 ppm upon addition of 2 equiv. of silver ion. The least-squares fitting program EQNMR25 was used to estimate the stability constants for this 1∶1 complexation from the 1H NMR titration data. By fitting the data using this program, the stability constants were generated after a number of iterations with an estimated error of ≤2% (Table 4). The satisfactory agreement of such theoretical fits is also suggestive of a 1∶1 complexation stoichiometry for the rhenium complex with silver ion. This is consistent with the results from UV-visible absorption spectrophotometry in which a 1∶1 complexation was also revealed.
 | ||
| Fig. 6 1H NMR titration curve showing the perturbation of the chemical shift of the methylene protons adjacent to the sulfur atom on the crown ether unit of 6 upon addition of silver trifluoromethanesulfante in CD3CN. | ||
In contrast, addition of silver ion to complexes 1–3 in CD3CN resulted in no observable shift of the proton signal in the 1H NMR spectrum, suggesting a small binding ability of the complex for silver ion compared to those containing the thia- and selenacrown units. From Table 4, it can be seen that the stability constants obtained using different methods are comparable to each other, indicating the reliability of both methods. As with other related organic systems, introduction of softer donor atoms such as sulfur or selenium into a macrocycle increases the affinity of the macrocycle for Ag+ and hence increases the stability constant. This result is consistent with the fact that the soft donor atoms have high affinity for the soft metal cations, in accord with the hard-soft acid–base principle.
Complex 5 shows the largest stability constant with Ag+, while that of 4 is the smallest amongst the three complexes. The observed difference in the stability constant is reasonable since there are three sulfur atoms to interact with Ag+ in the crown ether moiety of 5 and, therefore, the cation-bound species is more stable or the cation is held more firmly. Because of the higher affinity of selenium atom than sulfur atom for Ag+ ions, it is also understandable that the stability constant with Ag+ for 6 is higher than that for 4. In order to study the solvent effect on such silver ion binding properties of 4–6, another set of stability constants was obtained, using methanol as the solvent, from the UV-visible spectrophotometric titration studies (Table 4). Unlike the case of common alkali or alkaline earth metal cations, for which smaller stability constants are anticipated in methanol compared to in acetonitrile as a result of the higher solvent polarity of methanol, which gives rise to better solvation, the stability constants of 4–6 with silver ion in acetonitrile were found to be smaller than those in methanol. A similar order of the stability constants of cryptand with silver ion in acetonitrile and methanol was reported,41,45 and is ascribed to the much stronger ion-solvent interaction of silver ion with acetonitrile than with methanol.
Concluding remarks
A series of rhenium(I) diimine complexes with crown ether pendants with various cavity sizes and heteroatoms has been synthesized; the photophysical and electrochemical properties were studied. The low-energy absorption band at ca. 400 nm is attributed to a mixed dπ(Re) → π*(diimine) MLCT/π(diimine) → π*(diimine) IL transition, while the solution state emission band is assigned as derived from dπ(Re) → π*(diimine) 3MLCT exited state. The first reduction and oxidation are ascribed to diimine ligand-centred reduction and Re(I) → Re(II) oxidation, respectively. The ion-binding properties of these complexes with various metal ions have also been studied. The UV-visible absorption spectra of the complexes with oxacrown pendants, 1–3, in methanol solution showed a blue shift of the absorption band upon addition of alkali and alkaline earth metal ions. A well-defined isosbestic point in each spectrum was observed and the stability constants of the complexes with various metal cations have been determined by the UV-visible spectroscopic titration studies. Similarly, the thia- or selenacrown ether containing complexes, 4–6, exhibited a blue shift of the absorption bands in acetonitrile upon addition of silver ion. However, no such shift was observed upon addition of alkali and alkaline earth metal cations. Their stability constants for silver ion have also been determined. A considerable downfield shift of the methylene protons attached to the chalcogen atoms has been observed upon addition of silver ion into the deuterated acetonitrile solution. Apart from the UV-visible absorption spectroscopy, 1H NMR spectroscopy was employed to study the silver ion binding of these complexes. The stability constants for silver ion observed from both the UV-visible and 1H NMR experiments were found to be in good agreement with each other, and similar to those obtained for other related organic systems.Acknowledgements
V.W.-W.Y. acknowledges support from the University Development Fund of The University of Hong Kong and the University Grants Committee of Hong Kong Special Administrative Region, China (Project No. AoE/P-10/01). Dr M. J. Hynes is gratefully acknowledged for providing us with the EQNMR program.References
- (a) C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017 CrossRef CAS; (b) C. J. Pedersen and J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 59.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009 CrossRef.
- (a) Y. Inoue and G. W. Gokel, Cation Binding by Macrocycles, Marcel Dekker, New York, 1990 Search PubMed; (b) G. W. Gokel, Crown Ethers and Cryptands, Royal Society of Chemistry, Cambridge, 1994 Search PubMed.
- F. Vögtle, E. Weber and U. Elben, Kontakte (Darmstadt), 1980, 2, 36 Search PubMed.
- R. M. Izatt, R. E. Terry, A. G. Avondet, J. S. Bradshaw, N. K. Dalley, T. E. Jensen, J. J. Christensen and B. L. Haymore, Inorg. Chim. Acta, 1978, 30, 1 CrossRef CAS.
- H. K. Frensdorff, J. Am. Chem. Soc., 1971, 93, 600 CrossRef CAS.
- (a) V. W. W. Yam, K. K. W. Lo and K. K. Cheung, Inorg. Chem., 1995, 34, 279; (b) V. W. W. Yam, Y. L. Pui, W. P. Li, K. K. W. Lo and K. K. Cheung, J. Chem. Soc., Dalton Trans., 1998, 3615 RSC.
- (a) V. W. W. Yam and V. W. M. Lee, J. Chem. Soc., Dalton Trans., 1997, 3005 RSC; (b) V. W. W. Yam, V. W. M. Lee, F. Ke and K. W. M. Siu, Inorg. Chem., 1997, 36, 2124 CrossRef CAS.
- V. W. W. Yam, Y. L. Pui, W. P. Li, K. K. Cheung and N. Zhu, New J. Chem., 2002, 26, 536 RSC.
- (a) V. W. W. Yam, C. L. Chan and C. K. Li, Angew. Chem., Int. Ed., 1998, 37, 2857 CrossRef CAS; (b) V. W. W. Yam, C. L. Chan, C. K. Li and K. M. C. Wong, Coord. Chem. Rev., 2001, 216–217, 173 CrossRef CAS; (c) C. K. Li, X. X. Lu, K. M. C. Wong, C. L. Chan, N. Zhu and V. W. W. Yam, Inorg. Chem., 2004, 43, 7421 CrossRef CAS.
- (a) V. W. W. Yam, R. P. L. Tang, K. M. C. Wong, C. C. Ko and K. K. Cheung, Inorg. Chem., 2001, 40, 571 CrossRef CAS; (b) V. W. W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS; (c) V. W. W. Yam, R. P. L. Tang, K. M. C. Wong, X. X. Lu, K. K. Cheung and N. Zhu, Chem.-Eur. J., 2002, 8, 4066 CrossRef CAS.
- V. W. W. Yam, X. X. Lu and C. C. Ko, Angew. Chem., Int. Ed., 2003, 42, 3385 CrossRef CAS.
- R. M. Izatt, L. D. Hansen, D. J. Eatough, J. S. Bradshaw and J. J. Christensen, in Metal–Ligand Interactions in Organic Chemistry and Biochemistry, eds. B. Pullman and N. Goldblum, Reidel, Dordrecht, 1997, Part I, p. 337 Search PubMed.
- Progress in Macrocyclic Chemistry, eds. R. M. Izatt and J. J. Christensen, Wiley, New York, 1979, vol. 1 Search PubMed.
- V. W. W. Yam, K. M. C. Wong, V. W. M. Lee, K. K. W. Lo and K. K. Cheung, Organometallics, 1995, 14, 4034 CrossRef CAS.
- N. I. Abakumova, I. K. Kolenka and M. I. Kodess, J. Org. Chem., USSR, 1982, 18, 1305.
- (a) R. Ungaro, E. E. Haj and J. Smid, J. Am. Chem. Soc., 1976, 98, 5198 CrossRef CAS; (b) G. E. Pacey, Y. P. Wu and B. P. Bubnis, Synth. Commum., 1981, 11, 323 Search PubMed.
- K. H. Pannell, W. Yee, G. S. Lewandos and D. C. Hambrick, J. Am. Chem. Soc., 1977, 99, 1457 CrossRef CAS.
- R. N. Dominey, B. Hauser, J. Hubbard and J. Dunham, Inorg. Chem., 1991, 30, 4754 CrossRef CAS.
- (a) M. S. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998 CrossRef CAS; (b) M. S. Wrighton, J. Am. Chem. Soc., 1974, 74, 4801; (c) S. M. Fredericks, J. C. Luong and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 7415 CrossRef CAS; (d) R. N. Dominey, B. Hauser, J. Hubbard and J. Dunham, Inorg. Chem., 1991, 30, 4754 CrossRef CAS.
- PATTY: P. T. Beurskens, G. Admiraal, G. Biursken, W. P. Bosman, S. Garcia-Granda, R. O. Gould, J. M. M. Smits and C. Smykalla, The DIRDIF Program System, Technical Report of the Crystallography Laboratory, University of Nijmegen, Nijmegen, The Netherlands, 1992 Search PubMed.
- TeXsan: Crystal Structure Analysis Package, Molecular Structure Corp., The Woodlands, TX, 1985 and 1992 Search PubMed.
- R. R. Buchs, T. L. Netqel, I. Fujita and S. G. Boxer, J. Phys. Chem., 1982, 86, 1947 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- (a) E. Horn and M. R. Snow, Aust. J. Chem., 1980, 33, 2369 CAS; (b) S. A. Moya, J. Guerrero, P. Pastene, R. Schmidt, R. Sariego, R. Startori, J. Sanz-Aparicio, I. Fonseca and M. Martinez-Ripoll, Inorg. Chem., 1994, 33, 2341 CrossRef CAS; (c) J. C. Calabrese and W. Tam, Chem. Phys. Lett., 1987, 133, 244 CrossRef CAS.
- L. Wallace, C. Woods and D. P. Rillema, Inorg. Chem., 1995, 34, 2875 CrossRef CAS.
- V. W. W. Yam, K. M. C. Wong and K. K. Cheung, Organometallics, 1997, 16, 1729 CrossRef CAS.
- L. A. Worl, R. Duesing, P. Chen, L. D. Ciana and T. J. Meyer, J. Chem. Soc., Dalton Trans., 1991, 849 RSC.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952 CrossRef CAS.
- S. V. Wallendael, R. J. Shaver, D. P. Rillema, B. J. Yoblinski, M. Stathis and T. F. Guarr, Inorg. Chem., 1990, 29, 1761 CrossRef.
- L. A. Sacksteder, A. P. Zipp, E. A. Brown, J. Streich, J. N. Demas and B. A. DeGraff, Inorg. Chem, 1990, 29, 4335 CrossRef.
- L. A Sacksteder, M. Lee, J. N. Demas and B. A. DeGraff, J. Am. Chem. Soc., 1993, 115, 8230 CrossRef CAS.
- D. J. Stufkens, Comments Inorg. Chem., 1992, 13, 359 CAS.
- B. P. Sullivan, J. Phys. Chem., 1989, 93, 24 CrossRef CAS.
- (a) G. T. Tapolsky, R. Duesing and T. J. Meyer, J. Phys. Chem., 1989, 93, 3885 CrossRef CAS; (b) G. T. Tapolsky, R. Duesing and T. J. Meyer, Inorg. Chem., 1990, 29, 2285 CrossRef CAS.
- (a) V. W. W. Yam, V. C. Y. Lau and K. K. Cheung, J. Chem. Soc., Chem. Commun., 1995, 259 RSC; (b) V. W. W. Yam, V. C. Y. Lau and L. X. Wu, J. Chem. Soc., Dalton Trans., 1998, 1461 RSC.
- (a) V. W. W. Yam, V. C. Y. Lau and K. K. Cheung, Organometallics, 1995, 14, 2749 CrossRef CAS; (b) V. W. W. Yam, V. C. Y. Lau and K. K. Cheung, Organometallics, 1996, 15, 1740 CrossRef CAS.
- L. A. Worl, R. Duesing, P. Chen, L. D. Ciana and T. J. Meyer, J. Chem. Soc., Dalton Trans., 1991, 849 RSC.
- C. C. Price, in The Chemistry of the Ether Linkage, ed. S. Patai, John Wiley and Sons, London, 1967, ch. 11, pp. 499–523 Search PubMed.
- R. M. Izatt, J. S. Bradshaw, S. A. Nielsen, J. D. Lamb, J. J. Christensen and D. Sen, Chem. Rev., 1985, 85, 271 CrossRef CAS.
- D. E. Fenton, D. Parkin and R. F. Newton, J. Chem. Soc., Perkin Trans. I, 1981, 449 RSC.
- I. Ikeda, H. Emura, S. Yamamura and M. Okahara, J. Org. Chem., 1982, 47, 5150 CrossRef CAS.
- M. Dobler, J. D. Dunitz and P. Seiler, Acta Crystallogr., Sect. B, 1974, 30, 2741 CrossRef.
- B. G. Cox, J. Garcia-Rosas and H. Schneider, J. Am. Chem. Soc., 1981, 103, 1384 CrossRef CAS.
Footnote |
| † CCDC reference numbers 252682. See http://www.rsc.org/suppdata/nj/b4/b415951a/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |