Monophasic and biphasic hydrosilylations of enones and ketones using a fluorous rhodium catalyst that is easily recycled under fluorous–organic liquid–liquid biphasic conditions
Long V.
Dinh
and
John A.
Gladysz
*
Institut für Organische Chemie, Friedrich-Alexander Universität Erlangen-Nürnberg, Henkestraße 42, 91054, Erlangen, Germany. E-mail: GLADYSZ@ORGANIK.UNI-ERLANGEN.DE
First published on 9th December 2004
Abstract
Additions of PhMe2SiH to 2-cyclohexen-1-one, 2-cyclopenten-1-one, 4,4-dimethyl-2-cyclohexen-1-one, and cyclohexanone (11) are catalyzed by the rhodium complexes ClRh[P(CH2CH2(CF2)n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −1CF3)3]3
(1; n
= 6, 8), which have very high affinities for fluorous liquid phases. Reactions of the enones are conducted with 0.80 mol%
1 at 60 °C under biphasic conditions in CF3C6F11–toluene or monophasic conditions in CF3C6F11–hexanes. The reactions are faster under the latter conditions and the catalyst and products are efficiently separated at lower temperatures under biphasic conditions. Distillations of the non-fluorous phases give mixtures of 1,4- and 1,2-hydrosilylation products (>99–92∶<1–8) in 93–88% yields. The catalyst-containing CF3C6F11 phases are recycled two times with similar results. Reactions of 11 are studied under analogous conditions (0.2 mol%
1) at 28 and 40 °C and give cyclohexyl dimethylphenylsilyl ether. Rate profiles show induction periods for the first cycle, indicating an irreversible transformation of 1, but not for subsequent cycles. Only minor decreases in activity are observed over four cycles, consistent with little catalyst leaching or decomposition. When catalyst loadings are decreased to 0.02 mol%, TON of 4400 can be obtained.
−1CF3)3]3
(1; n
= 6, 8), which have very high affinities for fluorous liquid phases. Reactions of the enones are conducted with 0.80 mol%
1 at 60 °C under biphasic conditions in CF3C6F11–toluene or monophasic conditions in CF3C6F11–hexanes. The reactions are faster under the latter conditions and the catalyst and products are efficiently separated at lower temperatures under biphasic conditions. Distillations of the non-fluorous phases give mixtures of 1,4- and 1,2-hydrosilylation products (>99–92∶<1–8) in 93–88% yields. The catalyst-containing CF3C6F11 phases are recycled two times with similar results. Reactions of 11 are studied under analogous conditions (0.2 mol%
1) at 28 and 40 °C and give cyclohexyl dimethylphenylsilyl ether. Rate profiles show induction periods for the first cycle, indicating an irreversible transformation of 1, but not for subsequent cycles. Only minor decreases in activity are observed over four cycles, consistent with little catalyst leaching or decomposition. When catalyst loadings are decreased to 0.02 mol%, TON of 4400 can be obtained.
Introduction
Fluorous catalysts contain varying numbers of “pony tails” of the formula (CH2)m(CF2)n−1CF3 or (CH2)mRfn.1 These labels or “phase tags” allow catalyst recovery by a number of protocols, most commonly involving a fluorous liquid or solid phase.2 The first report, by Horváth and Rabai, featured rhodium catalysts with fluorous phosphine ligands.3 These showed excellent activity for the hydroformylation of alkenes. Due to the large number of pony tails or high “fluorous content”, they were easily separated from the products under fluorous–organic liquid–liquid biphasic conditions. Cartoons that illustrate such procedures are presented in the Results section. Since this time, fluorous catalysts have been developed for a number of metal-catalyzed reactions.4
We have had a long-standing interest in fluorous rhodium catalysts or precatalysts of the formula ClRh[P(CH2CH2Rfn)3]3, with n = 6 (1-Rf6) or 8 (1-Rf8).5 These can be regarded as aliphatic analogs of Wilkinson’s catalyst. They are easily synthesized from [Rh(μ-Cl)(COD)]2 and the corresponding fluorous phosphines,5 and exhibit high fluorous phase affinities. In the case of 1-Rf6, the partition coefficient between CF3C6F11 [perfluoro(methylcyclohexane)] and toluene is 99.86∶0.14 (696∶1) at 27 °C. That for 1-Rf8 is at least 99.88∶0.12 (811∶1). As detailed in two previous full papers, 1-Rf6 and 1-Rf8 afford effective and easily recycled catalysts for the hydroboration and hydrogenation of alkenes.5,6 However, it should be emphasized that what is actually being recycled is the catalyst rest state—a species that is usually distinct from the precatalyst.
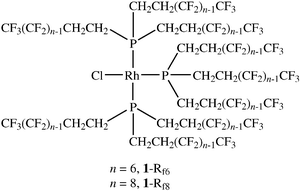
As communicated earlier, 1-Rf6 and 1-Rf8 also afford excellent catalysts for the hydrosilylation of carbonyl compounds,7 a reaction of considerable importance in fine chemical synthesis.8 The catalysts were efficiently recycled under fluorous–organic liquid–liquid biphasic conditions. Since that time, alternative protocols have been developed that are not dependent upon fluorous solvents.2,9–12 Hence, the original results have taken on added significance as a benchmark for newer studies. This has prompted us to present and document the full details of this work in this full paper, together with new, unreported experiments. Additional observations are collected elsewhere.13 Other fluorous rhodium hydrosilylation catalysts have since been described,14 and these complementary studies are treated in the discussion section.
Results
1. Hydrosilylations under biphasic conditions
As summarized in Fig. 1, mixtures of 2-cyclohexen-1-one (3; 1.30 mmol), PhMe2SiH (1.1 equiv.), and toluene (1.0 ml) were treated with CF3C6F11 solutions of 1-Rf6 or 1-Rf8 (1.00 ml; 0.80 mol%). The samples were heated to 60 °C. Toluene and CF3C6F11 are miscible in all proportions only above 88.6 °C,15 so it was not surprising that the phases did not mix.16 Nonetheless, hydrosilylation went to completion over the course of 10 h, as assayed by GC. The samples were cooled to room temperature and the upper toluene layers carefully separated by syringe. The remaining CF3C6F11 layers, which contain the catalyst rest states, were extracted with toluene (1.0 ml). | ||
| Fig. 1 Hydrosilylation of 2-cyclohexen-1-one (3) under biphasic conditions (CF3C6F11–toluene, 60 °C). | ||
Distillations of the toluene phases gave 92∶8 mixtures of the isomeric 1,4-hydrosilylation product 4 and 1,2-hydrosilylation product 5 as clear liquids in 90–88% yields, as summarized in Fig. 1. This corresponds to TON values of 113–100. Product 4 was characterized by NMR and the data were in good agreement with those reported earlier.17 Isomer 5 exhibited a SiOCHR21H NMR signal at ca. δ 3.90, very close to the HOCHR2 signal of the corresponding allylic alcohol, and CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH signals at δ 5.55–5.68 ppm (CDCl3).18 The CF3C6F11 catalyst solutions were recharged with 3, PhMe2SiH, and toluene. A second cycle gave 4 and 5 in 88–85% yields (TON 110–107) and a third cycle gave similar results (Fig. 1).
CH signals at δ 5.55–5.68 ppm (CDCl3).18 The CF3C6F11 catalyst solutions were recharged with 3, PhMe2SiH, and toluene. A second cycle gave 4 and 5 in 88–85% yields (TON 110–107) and a third cycle gave similar results (Fig. 1).
As summarized in Fig. 2, analogous three-cycle sequences were conducted with 2-cyclopenten-1-one (6) and 1-Rf6 or 1-Rf8. Both the 1,4-hydrosilylation product 7 and a minor species believed to be the 1,2-hydrosilylation product 8 formed.18c However, as with 3, the former greatly dominated. Distillations gave 98∶2 mixtures in 93–90% yields (TON 116–113). The NMR properties of 7 were in good agreement with those reported earlier.17 A comparable series of experiments with 4,4-dimethyl-2-cyclohexen-1-one (9) gave only the known 1,4-addition product 10,17,19 and essentially identical yields.
 | ||
| Fig. 2 Hydrosilylation of additional enones under the conditions of Fig. 1. | ||
2. Partition coefficients
In order to better analyze the preceding reactions, quantitative data on reactant and product fluorophilicities were sought. Thus, CF3C6F11–toluene partition coefficients were determined for PhMe2SiH, the enones 3, 6, 9, cyclohexanone (11), and representative silyl ethers under standard conditions at 24 °C.20 Data are summarized in Table 1. The silyl ethers showed very low fluorous phase affinities (<1∶>99) and thus should be efficiently isolated under the conditions of Fig. 1. The carbonyl compounds showed marginally higher fluorous phase affinities (1.7–2.2∶98.3–97.8), as did PhMe2SiH (3.6∶96.4). All of these values are in the expected ranges based upon measurements of other monofunctional organic compounds.20| Compound | Partition coefficient (CF3C6F11–toluene, 24 °C) | |
|---|---|---|
| PhMe2SiH | 3.6∶96.4 | |
| 3 |

|
1.7∶98.3 |
| 6 |

|
1.9∶98.1 |
| 9 |

|
1.7∶98.3 |
| 10 |

|
0.6∶99.5 |
| 11 |
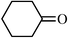
|
2.2∶97.8 |
| 12 |
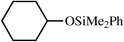
|
0.8∶99.2 |
The low fluorous phase affinities of the carbonyl compounds and PhMe2SiH mean that under the conditions of Fig. 1, the educts and catalyst reside predominantly in different liquid phases. With monophasic conditions, much faster reactions would normally be expected. Accordingly, we sought to replace toluene with non-fluorous solvents that would become miscible with CF3C6F11 at lower temperatures.
3. Hydrosilylations under monophasic conditions
Hexane and ether are among the few solvents miscible with CF3C6F11 at room temperature, consistent with their low polarities and small volumes.15b However, two phases are obtained at 0 °C or below, providing a means for catalyst/product separation. Thus, the hydrosilylations of 3, 6 and 9 were repeated with 1-Rf6 and 1-Rf8 in CF3C6F11–hexanes. As summarized in Fig. 3, reactions occurred smoothly under monophasic conditions at 60 °C. GC analyses showed product formation to be complete on time scales of 1–2 h as opposed to 4–10 h for Figs. 1 and 2. The CF3C6F11 and hexanes phases were separated at −30 °C, the temperature of a conveniently located freezer. The hydrosilylation products were isolated in yields comparable to those in Figs. 1 and 2, and the catalysts could be used for a minimum of three cycles. | ||
| Fig. 3 Hydrosilylation of enones under monophasic conditions (CF3C6F11–hexanes, 60 °C). | ||
For the purposes of assaying rates and catalyst recovery (below), a substrate that can give only one type of addition product was desired. Thus, as summarized in Fig. 4, cyclohexanone (11; 1.30 mmol) and PhMe2SiH (1.1 equiv.) were reacted in the presence of tridecane as an internal standard. As more fully analyzed elsewhere, higher catalyst loadings can be used to mask poor catalyst recoverabilities.2,21 Hence, the amount of 1-Rf6 was reduced to 0.20 mol%. Two solvent systems, CF3C6F11–hexanes and CF3C6F11–ether, were screened under monophasic conditions at 28 °C. After 8 h, the phases were separated as in Fig. 3. GC analyses indicated 96–97% yields of cyclohexyl dimethylphenylsilyl ether (12),22 corresponding to TON values of 485–481. A total of four cycles could be conducted, with similar results for both solvent systems and only modest decreases in yields (Fig. 4).
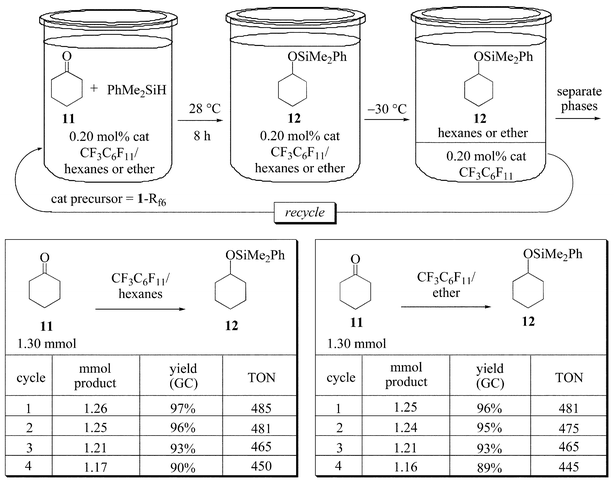 | ||
| Fig. 4 Hydrosilylation of cyclohexanone (11) under monophasic conditions (CF3C6F11–hexanes or CF3C6F11–ether, 28 °C). | ||
A preparative reaction was conducted in CF3C6F11–hexanes with 13.0 mmol of 11. The catalyst loading was reduced still further to 0.02 mol%. After 72 h, distillation gave 12 in 88% yield, corresponding to a TON value of 4400. However, the reaction times for 11 under these monophasic conditions were considered longer than optimum, especially for rate studies. Hence, temperatures were increased to 40 °C for the experiments in the following section.
4. Rates of hydrosilylation
As analyzed elsewhere, rates constitute one of the best criteria for catalyst recovery.2,21 Thus, the reaction sequence conducted in CF3C6F11–hexanes in Fig. 4 was repeated at 40 °C. Aliquots were removed every 0.5 h for GC analyses. Yields of 12 reached maximum values after 3 h. The reaction profiles for four cycles are illustrated in Fig. 5.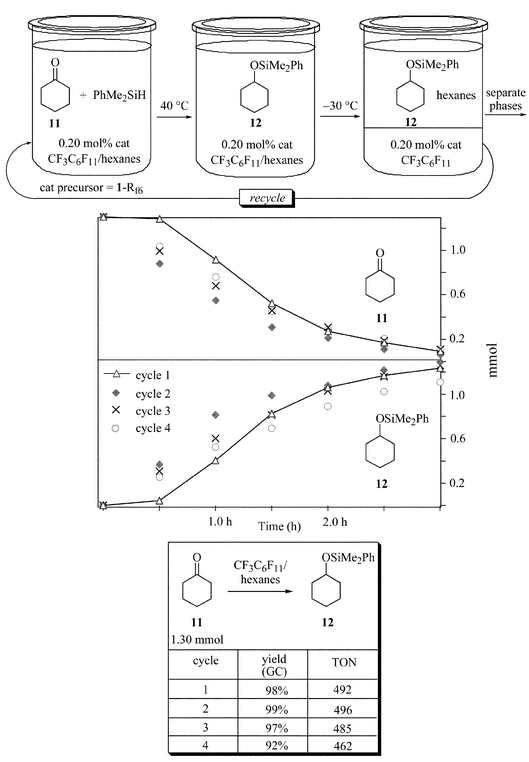 | ||
| Fig. 5 Rate profile for the hydrosilylation of cyclohexanone (11) under monophasic conditions (CF3C6F11–hexanes, 40 °C). | ||
Importantly, the first cycle exhibited a induction period (10–15 min), indicating some irreversible transformation needed to convert 1-Rf6 to the active catalyst. However, subsequent cycles showed no induction periods, indicating that the active catalyst—not just a catalyst precursor23—is recycled. The rate of formation of 12 or consumption of 11 slowed somewhat in the third cycle and more noticeably in the fourth cycle. The implications are analyzed in the discussion section.
For comparison, an analogous set of experiments were conducted under biphasic conditions in CF3C6F11–toluene, as summarized in Fig. 6. As noted for Fig. 3vs.Figs. 1 and 2 above, the hydrosilylations became slower. However, the product yields were similar. In this experiment, the tridecane internal standard can partition between two phases. Fortunately, the CF3C6F11–toluene partition coefficient (2.4∶97.6)15b is similar to those of 11 and 12 (Table 1), so the small errors approximately cancel. The first cycle again showed a brief induction period and there was a similar drop in activity in the third and fourth cycles.
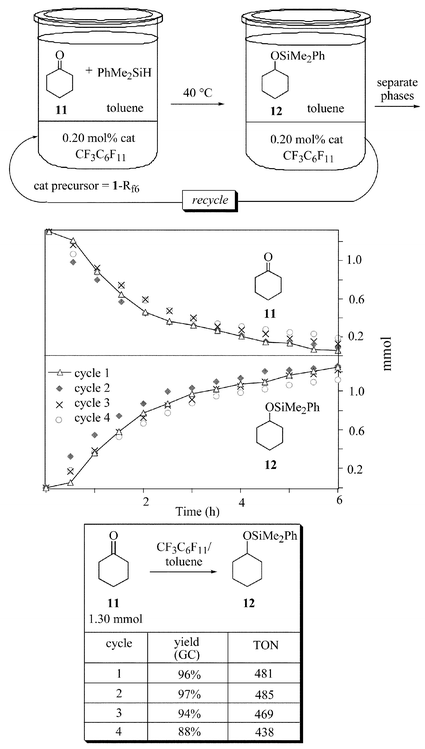 | ||
| Fig. 6 Rate profile for the hydrosilylation of cyclohexanone (11) under biphasic conditions (CF3C6F11–toluene, 40 °C). | ||
Discussion
Figs. 1–5 demonstrate that 1-Rf6 and 1-Rf8 are highly effective catalyst precursors for hydrosilylations of enones and ketones. Naturally, a variety of other rhodium(I) hydrosilylation catalysts are known. In the most relevant earlier work, Ojima and Kogure reported that ClRh(PPh3)3 (Wilkinson's catalyst) effects hydrosilylations of enones as summarized in Scheme 1.24 In general, monohydrosilanes afforded 1,4-additions to give silyl enol ethers, whereas dihydrosilanes afforded 1,2-additions to give silyl ethers of allylic alcohols. Chan obtained comparable results with the more active catalyst HRh(PPh3)4.17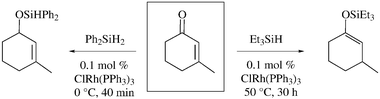 | ||
| Scheme 1 Typical regioselectivies for rhodium(I)-catalyzed enone hydrosilylations. | ||
The monohydrosilane that we used with 1-Rf6 and 1-Rf8, PhMe2SiH, gives similar regioselectivity. Scouting reactions with the dihydrosilane Ph2SiH2 were also conducted. With 2-cyclohexen-1-one (3), GC analyses indicated complete conversion to a 1,2-addition product analogous to 5. Multiple cycles could be conducted. However, technical difficulties were encountered with initial preparative syntheses and the isolation of the corresponding allylic alcohol, and this chemistry was not further pursued. Nonetheless, it is significant that the fluorous catalysts 1-Rf6 and 1-Rf8 give the same regioselectivities as ClRh(PPh3)3 in Scheme 1. The fluorous medium and phosphine ligands apparently have no effect.
Thus, the major advantage of 1-Rf6 and 1-Rf8 over ClRh(PPh3)3 is their recyclability. Over the course of three (Figs. 1–3) or four (Figs. 4–6) cycles, product yields are essentially constant. However, as analyzed elsewhere,2,21 yields are normally misleading measures of catalyst recovery. Rates are much more accurate indicators and the profiles in Figs. 5 and 6 establish very high levels of recovery. The modest declines in activities reflect the aggregate losses due to catalyst deactivation and leaching.
Although leaching was not specifically assayed in this study, measurements have been carried out for similar hydrosilylations using other fluorous recycling protocols, showing that rhodium losses are minor.12 For catalytic hydroborations conducted under comparable conditions, only four rhodium atoms from 1-Rf6
(or two from 1-Rf8) are leached for every 1000000 product molecules.5 However, some leaching of the fluorous phosphines occurs.12 We suggest this is connected to the induction period that generates the active catalyst. Also, it is quite probable that only two phosphine ligands remain bound during the catalytic cycle. Based upon currently accepted mechanisms,8,17 possibilities for the other ligands of the catalyst rest state include hydride, silyl, α-silyloxyalkyl, alkoxide and ketone moieties. A rest state can also depend upon which of two reactants are in excess; representative possibilities include species of the types 13–15.
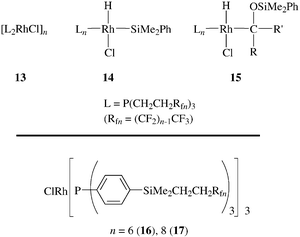
Following our communication,7 the fluorous rhodium(I) complexes 16 and 17 were employed as catalyst precursors for the hydrosilylation of alkenes.14 These contain fluorous triarylphosphine ligands and are therefore closer analogs of Wilkinson's catalyst. They are also likely effective catalyst precursors for hydrosilylations of carbonyl compounds. Many other strategies have been applied to the immobilization and/or recovery of rhodium(I) hydrosilylation catalysts,25 all of which have various merits or advantages.
No catalyst recovery method is without drawbacks. The liquid/liquid biphasic product/catalyst separations in Figs. 1–6 require fluorous solvents, which are expensive. However, this is less of an issue with laboratory scale or fine chemical synthesis than with commodity chemical synthesis. Also, it should be possible to carry out the preceding reactions with FC-72, a mixture of perfluorinated hexanes that is the cheapest fluorous solvent available. For this study, the isomerically homogeneous solvent CF3C6F11 was selected in the interest of maximizing reproducibility. It should also be noted that small equilibrium amounts of fluorous solvents remain in organic solvents under biphasic conditions (and vice versa).15b Thus, there is some leaching of fluorous solvents into the organic phases under the conditions of Figs. 1–6.
There are active ongoing efforts to address these issues,9–11 and the next paper in this series will feature alternative protocols that allow the catalysts in Figs. 1–6 to be recovered without recourse to fluorous solvents.12 In the interim, this work provides another convincing demonstration of the convenience, efficiency, and generality with which appropriately designed fluorous catalysts can be recovered via liquid/liquid biphasic methods. It furthermore extends the repertoire of fluorous reactions to an important laboratory scale transformation, the hydrosilylation of carbonyl compounds.
Experimental
General
Reactions were conducted under inert atmospheres. Hexanes, ether and toluene were distilled from Na/benzophenone and then freeze-pump-thaw degassed (3×); CF3C6F11 (ABCR) was distilled from CaH2 and analogously degassed. 2-Cyclohexen-1-one (3; Aldrich, 99%), 2-cyclopenten-1-one (6; Fluka, ≥99%), 4,4-dimethyl-2-cyclohexen-1-one (9; Fluka, 96%), cyclohexanone (11; Fluka, ≥99%), PhMe2SiH (Aldrich, 99%) and tridecane (Aldrich, ≥99%) were freeze-pump-thaw degassed (3×). Complexes 1-Rf6 and 1-Rf8 were prepared as described earlier.5 NMR spectra were recorded on 300 or 400 MHz instruments at ambient probe temperature and referenced to residual internal CHCl3 (1H, δ 7.27) or CDCl3 (13C, δ 77.2). Gas chromatography was conducted on a ThermoQuest Trace GC 2000 instrument.Hydrosilylations under biphasic conditions (CF3C6F11–toluene, 60 °C; Figs. 1 and 2)26
Product characterization
NMR samples were prepared under conditions slightly different from Figs. 1 and 2.Hydrosilylations under monophasic conditions (CF3C6F11–hexanes, 60 °C; Fig. 3)26
Hydrosilylations under monophasic conditions (CF3C6F11–hexanes or –ether, 28 °C; Fig. 4)
Hydrosilylation rate profiles
Partition coefficients
The following are representative.Acknowledgements
We thank the US Department of Energy (DOE) and the Bundesministerium für Bildung und Forschung (BMBF) for support. The initial portion of this study was conducted at the University of Utah.References
- General reference: Handbook of Fluorous Chemistry, eds. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004 Search PubMed.
- J. A. Gladysz and R. C. da Costa, in Handbook of Fluorous Chemistry, eds. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004, pp. 24–40 Search PubMed.
- (a) I. T. Horváth and J. Rábai, Science, 1994, 266, 72 CrossRef CAS; (b) I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rabái and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133 CrossRef CAS; (c) I. T. Horváth, in Handbook of Fluorous Chemistry, eds. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004, pp. 5–10 Search PubMed.
- (a) R. C. da Costa and J. A. Gladysz, in Transition Metals for Organic Synthesis, eds. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2004, pp. 527–543 Search PubMed; (b) See also ch. 10.8–10.14 in ref. 1..
- J. J. J. Juliette, D. Rutherford, I. T. Horváth and J. A. Gladysz, J. Am. Chem. Soc., 1999, 121, 2696 CrossRef CAS.
- D. Rutherford, J. J. J. Juliette, C. Rocaboy, I. T. Horváth and J. A. Gladysz, Catal. Today, 1998, 42.
- L. V. Dinh and J. A. Gladysz, Tetrahedron Lett., 1999, 40, 8995 CrossRef CAS.
- Reviews covering rhodium-catalyzed hydrosilylations of carbonyl compounds: (a) H. Brunner, in Transition Metals for Organic Synthesis, eds. M. Beller and C. Bolm, Wiley-VCH, Weinheim and New York, 1998, vol. 2, ch. 1.4.2 Search PubMed; (b) I. Ojima, Z. Li and J. Zhu, in The Chemistry of Organic Silicon Compounds, eds. Z. Rappoport and Y. Apeloig, Wiley, New York, 1998, vol. 2, ch. 29 Search PubMed.
- (a) M. Wende, R. Meier and J. A. Gladysz, J. Am. Chem. Soc., 2001, 123, 11490 CrossRef CAS; (b) M. Wende and J. A. Gladysz, J. Am. Chem. Soc., 2003, 125, 5861 CrossRef CAS.
- (a) K. Ishihara, S. Kondo and H. Yamamoto, Synlett., 2001, 1371 CrossRef CAS; (b) K. Ishihara, A. Hasegawa and H. Yamamoto, Synlett., 2002, 1299 CAS; (c) J. Otera, Acc. Chem. Res., 2004, 37, 288 CrossRef CAS; (d) G. Maayan, R. H. Fish and R. Neumann, Org. Lett., 2003, 5, 3547 CrossRef CAS.
- (a) C. C. Tzschucke, C. Markert, H. Glatz and W. Bannwarth, Angew. Chem., Int. Ed., 2002, 41, 4500 CrossRef CAS; (b) C. C. Tzschucke, C. Markert, H. Glatz and W. Bannwarth, Angew. Chem., 2002, 114, 4678 CrossRef; (c) A. Biffis, M. Zecca and M. Basato, Green Chem., 2003, 5, 170 RSC.
- L. V. Dinh and J. A. Gladysz, J. Am. Chem. Soc. Search PubMed , submitted.
- L. V. Dinh, Ph.D. Thesis, University of Utah, Salt Lake City, Utah, 2004 Search PubMed.
- (a) E. de Wolf, E. A. Speets, B. J. Deelman and G. van Koten, Organometallics, 2001, 20, 3686 CrossRef CAS; (b) J. van den Broeke, F. Winter, B.-J. Deelman and G. van Koten, Org. Lett., 2002, 4, 3851 CrossRef CAS; (c) E. de Wolf, E. Riccomagno, J. J. M. de Pater, B.-J. Deelman and G. van Koten, J. Combinat. Chem., 2004, 6, 363 Search PubMed.
- (a) J. H. Hildebrand and D. R. F. Cochran, J. Am. Chem. Soc., 1949, 71, 22 CrossRef; (b) J. A. Gladysz and C. Emnet, in Handbook of Fluorous Chemistry, eds. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004, pp. 11–23 Search PubMed.
- Furthermore, solutes often raise the temperatures at which mixing occurs.
- T. H. Chan and G. Z. Zheng, Organometallics, 1995, 14, 70 CrossRef CAS.
- (a) For another reaction that yields a 4/5 mixture (both isomers were isolated), see: M. Koreeda and S. Koo, Tetrahedron Lett., 1990, 31, 831 Search PubMed; (b) The properties of 5 also compare well with those of a related silyl ether: M. D. Cavanaugh, B. T. Gregg and A. R. Cutler, Organometallics, 1996, 15, 2764 Search PubMed; (c) The 4/5 and 7/8 ratios were determined by integrating the olefinic proton of 4 or 7versus the SiOCHR2 proton of 5 or 8; (d) For the trimethylsilyl analog of 8, see: R. Noyori, S. Murata and M. Suzuki, Tetrahedron, 1981, 37, 3899 Search PubMed.
- J. M. Blackwell, D. J. Morrison and W. M. Piers, Tetrahedron, 2002, 58, 8247 CrossRef CAS.
- J. A. Gladysz, C. Emnet and J. Rábai, in Handbook of Fluorous Chemistry, eds. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004, pp. 56–100 Search PubMed.
- J. A. Gladysz, Pure Appl. Chem., 2001, 73, 1319.
- I. Ojima, R. J. Donovan and N. Clos, Organometallics, 1991, 10, 2606 CrossRef CAS.
- C. Rocaboy and J. A. Gladysz, New J. Chem., 2003, 27, 39 RSC.
- I. Ojima and T. Kogure, Organometallics, 1982, 1, 1390 CrossRef CAS.
- (a) I. Ojima, in The Chemistry of Organic Silicon Compounds, eds. S. Patai and Z. Rappoport, Wiley, New York, 1989, vol. 1, part 2, pp. 1491–1492 Search PubMed; (b) Z. Lu, E. Lindner and H. A. Mayer, Chem. Rev., 2002, 102, 3543 CrossRef CAS; (c) D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3640.
- Experiments with 1-Rf8 were conducted analogously.
- Additional details are given in ref. 13.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |