DOI:
10.1039/B415818K
(Paper)
New J. Chem., 2005,
29, 90-98
Anion binding by Ag(I) complexes of urea-substituted pyridyl ligands
Received
(in Montpellier, France)
12th October 2004
, Accepted 10th November 2004
First published on 9th December 2004
Abstract
A series of Ag(I) complexes of ureidopyridyl ligands 1 and 2 have been prepared from oxo-anion salts. In all cases the new materials contain the AgL2+ cation interacting with oxo-anions via the urea moiety. The complexes containing the para ligand 2: [Ag(2)2]CF3SO3·2H2O (3), [Ag(2)2]CH3CO2·1.33H2O·MeOH (4) and [Ag(2)2]NO3·H2O (5), all exhibit remarkably similar chain-like structures based around a linear Ag(I) centre, despite the change in the counter-ion. A recurring R22(8) hydrogen-bonding ring motif between the urea group and the oxo-anion is observed in almost all cases. An exception to this trend is the anhydrous nitrate structure [Ag(2)]NO3 (6) in which the nitrate is coordinated in a bridging position between two silver centres, which adopt distorted trigonal pyramidal geometries. Structures containing the ligand 1, [Ag(1)2]CF3SO3·0.5H2O (7), [Ag(1)2]CF3SO3·H2O·MeCN (8), [Ag(1)2]2SO4 (9), [Ag(1)2]NO3·MeOH (10) and [Ag(1)2]NO3·0.5MeOH·0.5MeNO2 (11), display very different geometries, although the R22(8) is observed to persist throughout. The most notable of these structures are 10 and 11 in which the nitrate anion is chelated within a ‘pincer’ arrangement by the silver complex. The nitrate anion is situated asymmetrically within the cavity of the host complex. This discrete nitrate complex persists in solution with strong nitrate binding by the [Ag(1)2]+ host compared to other anions being observed.
Introduction
The binding and detection of anionic species by synthetic receptors is currently an area of significant interest.1–3 The intrinsic challenges of anion binding, compared to cationic or neutral guests, is compensated for by the relevance and benefits of such systems in biological4 and environmental sensing5 applications. Although the majority of anion receptors have been based around organic scaffolds, there has been a recent and growing trend towards the use of metal-based assemblies as host species. Hosts assembled around metal centres can be either inert or covalent entities6 or can themselves be formed under thermodynamic control in solution.7–10 Labile host systems can be advantageous, in that the presence of certain guest species may template the formation of the host and stabilise the self-assembled complex.
We now present a simple, self-assembling host based around a urea-substituted pyridyl ligand that shows remarkable shape selectivity for nitrate in the solid state and in solution. We contrast this discrete species with a variety of extended hydrogen-bonded polymers linked via host⋯anion interactions. Part of this work has previously been communicated.11
Results and discussion
Extended hydrogen-bonded systems
We have previously reported the ureidopyridyl ligands 1 and 2 in the context of solid-state networks involving hydrogen bonds to metal-bound chloride.12 The ligand 2 has also been used within systems displaying marked metal distortion due to the strength of multiple hydrogen bonds.13 Urea is well-known in its ability to interact well with oxo-anions as such a pairing offers the possibility of forming an R22(8) hydrogen-bonded ring (Fig. 1).10,14,15 A search of the Cambridge Structural Database (CSD)16 reveals many structures involving urea in this type of interaction with a variety of oxo-anions. The R22(8) geometry of the interaction is expected from Etter’s rules since the urea group and the oxo-anions represent, respectively, the strongest hydrogen-bond donors and acceptors in the crystal.17 It is therefore expected that the combination of Ag(I) salts with 1 and 2 would produce systems in which the complex-anion interaction occurs via such a motif.
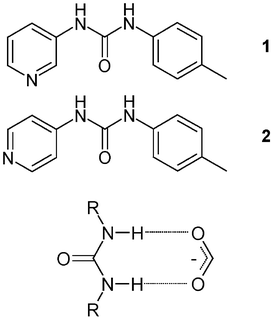 |
| | Fig. 1 The R22(8) hydrogen bond motif that can form between urea and an oxo-anion. | |
Ligands 1 and 2 were reacted with AgNO3, AgCF3SO3, AgCH3CO2 and Ag2SO4, resulting in seven new materials. Four species (Table 1) were obtained using ligand 2, namely [Ag(2)2]CF3SO3·2H2O (3), [Ag(2)2]CH3CO2·1.33H2O·MeOH (4), [Ag(2)2]NO3·H2O (5) and [Ag(2)2]NO3 (6). The new compounds were characterised by X-ray crystallography while bulk composition was confirmed by elemental analysis. The crystal structures of 3–5 all contain the [Ag(2)2]+ cation, which adopts an essentially linear coordination geometry around the Ag(I) centre. The N–Ag–N angle is slightly removed from 180° in all cases with the exact deviation dependant upon the proximity of other potentially ligating solvent groups. The linear geometry of the cationic complex gives rise to chain-like motifs running through these three structures.
| |
3
|
4
|
5
|
6
|
| Formula |
C27H30AgF3N6O7S1 |
C29H35AgN6O6.25 |
C26H28AgN7O6 |
C13H13AgN4O4 |
|
M
|
747.50 |
676.86 |
642.42 |
397.14 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P-1 |
P-1 |
C2/c |
P21/c |
|
a/Å |
10.2106(9) |
9.3295(5) |
38.1880(16) |
7.0590(14) |
|
b/Å |
11.4626(11) |
12.9275(6) |
10.1919(4) |
8.5990(17) |
|
c/Å |
14.2081(14) |
13.1198(6) |
13.7940(5) |
22.610(5) |
|
α/° |
72.229(2) |
83.850(2) |
90 |
90 |
|
β/° |
89.484(2) |
76.896(2) |
98.755(3) |
94.14(3) |
|
γ/° |
69.943(2) |
75.806(2) |
90 |
90 |
|
U/Å3 |
1479.0(2) |
1491.86(13) |
5306.2(4) |
1368.9(5) |
|
Z
|
2 |
2 |
8 |
4 |
|
μ/mm−1 |
0.828 |
0.729 |
0.815 |
1.498 |
| Unique reflections |
4253 |
6807 |
3801 |
3113 |
| Observed reflections |
3181 |
5592 |
3436 |
2160 |
|
R
int
|
0.0688 |
0.0365 |
0.0201 |
0.1085 |
|
wR
2 (all data) |
0.0814 |
0.0950 |
0.0686 |
0.1814 |
|
R [I
≥ 2σ(I)] |
0.0403 |
0.0419 |
0.0316 |
0.0800 |
The structure of 3 involves a triflate anion interacting with one of the urea groups via two of the sulfonate oxygen atoms in an R22(8) motif (Fig. 2). The third oxygen atom receives a hydrogen bond from one of the two enclathrated water molecules within the structure. This water molecule itself receives hydrogen bonds from the remaining urea group in an R21(6) motif. The second water molecule is weakly coordinated to the silver centre, resulting in a distortion from linearity of the N–Ag–N axis, with an angle of 168.7(2)°. This solvent molecule also bridges two urea oxygen atoms in neighbouring molecules (Fig. 3). The sheets of [Ag(2)2]+ in the structure are stacked above each other through multiple face-to-face π-π interactions.
![The immediate environment around one [Ag(2)2]+ complex in the structure of 3 with a weak silver-oxygen interaction, Ag(1)–O(7), of 2.685(4) Å. Hydrogen bond data are shown in Table 2.](/image/article/2005/NJ/b415818k/b415818k-f2.gif) |
| | Fig. 2 The immediate environment around one [Ag(2)2]+ complex in the structure of 3 with a weak silver-oxygen interaction, Ag(1)–O(7), of 2.685(4) Å. Hydrogen bond data are shown in Table 2. | |
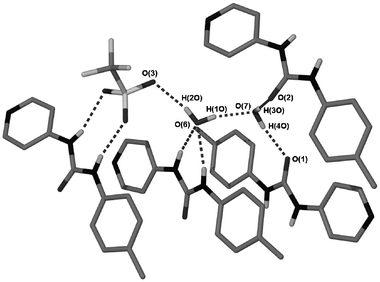 |
| | Fig. 3 Hydrogen bonding involving enclathrated water within the structure of 3. Some ligands are omitted for clarity. Hydrogen bond data are shown in Table 2. | |
Table 2 Hydrogen bond data for structures 3–6
| |
Symmetry equivalent |
d(D–H)/Å |
d(H⋯A)/Å |
d(D⋯A)/Å |
∠(D–H⋯A)/° |
| Complex 3 |
| N(2)–H(2N)⋯O(6) |
x, y, z |
0.91(5) |
1.94(5) |
2.814(6) |
162(4) |
| N(3)–H(3N)⋯O(6) |
x, y, z |
0.78(4) |
2.22(4) |
2.937(6) |
153(4) |
| N(5)–H(5N)⋯O(5) |
x, y, z |
0.74(4) |
2.27(4) |
2.992(5) |
163(4) |
| N(6)–H(6N)⋯O(4) |
x, y, z |
0.83(5) |
2.11(5) |
2.927(5) |
169(4) |
| O(7)–H(4O)⋯O(1) |
−x, −y + 1, −z |
0.81(5) |
1.94(5) |
2.747(5) |
172(5) |
| O(7)–H(3O)⋯O(2) |
−x, −y, −z + 1 |
0.92(9) |
1.82(9) |
2.735(5) |
171(8) |
| O(6)–H(2O)⋯O(3) |
−x, −y, −z + 1 |
0.82(6) |
2.10(6) |
2.878(5) |
159(6) |
| O(6)–H(1O)⋯O(7) |
x, y + 1, z |
0.82(7) |
1.91(7) |
2.713(5) |
167(7) |
| Complex 4 |
| N(2)–H(2N)⋯O(3) |
x, y, z |
0.78(4) |
2.05(4) |
2.826(3) |
173(4) |
| N(3)–H(3N)⋯O(4) |
x, y, z |
0.78(3) |
2.11(3) |
2.888(3) |
173(3) |
| N(5)–H(5N)⋯O(6) |
x, y, z |
0.79(4) |
2.12(4) |
2.889(3) |
163(3) |
| N(6)–H(6N)⋯O(6) |
x, y, z |
0.77(3) |
2.40(3) |
3.109(4) |
154(3) |
| O(5)–H(1O)⋯O(4) |
x, y, z
− 1 |
0.77(4) |
1.99(5) |
2.747(3) |
166(5) |
| O(6)–H(2O)⋯O(3) |
−x + 2, −y + 1, −z |
0.795(10) |
2.10(2) |
2.878(4) |
164(6) |
| O(6)–H(3O)⋯O(1) |
−x + 1, −y + 1, −z |
0.75(5) |
2.12(6) |
2.841(3) |
164(6) |
| Complex 5 |
| N(2)–H(2N)⋯O(6) |
x, y, z |
0.79(4) |
2.06(4) |
2.838(4) |
165(3) |
| N(3)–H(3N)⋯O(5) |
x + 1/2, y
− 1/2, z |
0.73(4) |
2.33(4) |
2.977(4) |
148(4) |
| N(5)–H(5N)⋯O(4) |
x, y, z |
0.78(3) |
2.17(4) |
2.950(4) |
174(4) |
| N(6)–H(6N)⋯O(3) |
x, y, z |
0.77(3) |
2.18(3) |
2.926(4) |
163(3) |
| O(6)–H(1O)⋯O(3) |
x + 1/2, y
− 1/2, z |
0.76(4) |
2.04(5) |
2.792(4) |
173(5) |
| O(6)–H(2O)⋯O(1) |
x, −y, z
− 1/2 |
0.88(5) |
1.98(5) |
2.858(4) |
176(5) |
| Complex 6 |
| N(2)–H(2N)⋯O(4) |
x, −y
− 1/2, z + 1/2 |
0.77(9) |
2.24(10) |
3.006(10) |
173(9) |
| N(3)–H(3N)⋯O(2) |
x, −y
− 1/2, z + 1/2 |
0.87(9) |
2.19(9) |
3.039(10) |
168(8) |
The acetate containing structure, 4, closely resembles that of 3, with the acetate anion residing in the same position as occupied by the triflate (Fig. 4). The remaining urea site is once more occupied by an enclathrated water molecule. Methanol is also included within the structure and engages in a weak interaction with the Ag(I) centre [Ag–O, 2.621(2) Å], again causing a distortion from a linear coordination geometry, with an N–Ag–N angle of 168.1(1)°. There is also a partial occupancy water molecule enclathrated within the asymmetric unit. As with the triflate complex, the cationic complexes are π-stacked with the enclathrated water occupying a bridging position between neighbouring molecules and also hydrogen bonding to the acetate anions. The methanol is also involved in hydrogen-bond donation to the acetate.
![The immediate environment around one [Ag(2)2]+ unit in the structure of 4 with a weak silver-methanol interaction, Ag(1)–O(5), of 2.621(2) Å. Hydrogen bond data are shown in Table 2.](/image/article/2005/NJ/b415818k/b415818k-f4.gif) |
| | Fig. 4 The immediate environment around one [Ag(2)2]+ unit in the structure of 4 with a weak silver-methanol interaction, Ag(1)–O(5), of 2.621(2) Å. Hydrogen bond data are shown in Table 2. | |
The structure of [Ag(2)2]+ in the nitrate complex 5 has the same general form as complexes 3 and 4 and consists of a linear Ag(2)2+ cation binding the anion to one urea group in an R22(8) motif. Differences arise in the nature of the enclathrated solvent in the structure. No interaction between the solvent and the silver centre is observed. Instead, the solvent occupies a position where it bridges urea protons of one molecule and a urea carbonyl oxygen on another. The water molecule also interacts with the nitrate anion forming an R33(10) pattern (Fig. 5).
![The environment around one [Ag(2)2]+ unit in 5. Hydrogen bond data are shown in Table 2.](/image/article/2005/NJ/b415818k/b415818k-f5.gif) |
| | Fig. 5 The environment around one [Ag(2)2]+ unit in 5. Hydrogen bond data are shown in Table 2. | |
A second nitrate structure was also obtained, complex 6, which does not have any solvent enclathrated within it and which adopts a 1∶1 Ag∶2 stoichiometry. The only difference between the formation conditions of these two materials was the solvent used: acetonitrile and methanol for 5 and 6, respectively. The structure of complex 6 is radically different to that of the hydrated 1∶2 structure. Rather than the more common linear geometry the Ag(I) centre exists in a distorted trigonal geometry coordinated to one pyridyl and two nitrato ligands.18 This unit forms a silver–nitrate–silver coordination polymer (Fig. 6). The polymeric chains are connected together via urea⋯nitrate hydrogen bonding with the R22(8) motif that is so prevalent within this series of structures (Fig. 7). The structure of 6 has the nitrate positioned in the correct geometry to form a bidentate interaction but at a long Ag–O distance of 2.739(6) Å. A number of other silver nitrate coordination polymers are known with nitrate adopting both bidentate and monodentate coordination modes.19,20
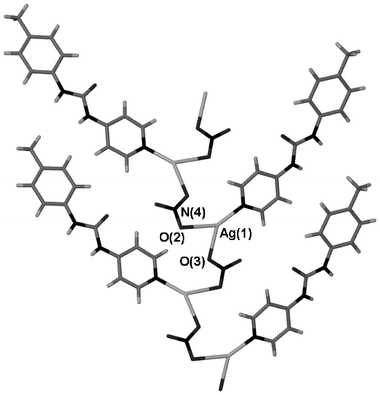 |
| | Fig. 6 A silver-nitrate coordination polymer running through the structure of 6. Selected distances (Å): Ag(1)–O(2), 2.510(6); Ag(1)–O(3), 2.334(7). | |
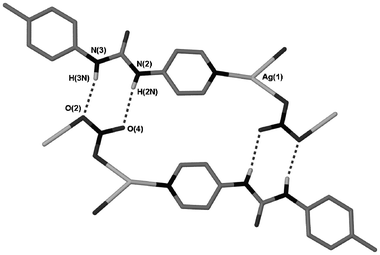 |
| | Fig. 7 Hydrogen bonding between coordination polymer chains in 6. Hydrogen bond data is given in Table 2. | |
The para substitution of the urea upon the pyridyl ring in 2 allows for chain-like architectures to form readily within Ag(I) complexes of this ligand, as seen in 3–5. However, when the urea group is situated in the meta position, as in ligand 1, very different structures, both from those discussed above and from each other, are observed (Table 3), although crucially, the urea⋯anion R22(8) motif is retained in all but one of the structures (that of 8). The structure of the triflate complex [Ag(1)2]CF3SO3·0.5H2O (7) is the most akin to those formed using 2 and displays a geometry in which the ligands are rotated 180° away from each other around a linearly coordinated silver atom, with the urea protons facing away from the metal centre (Fig. 8). The triflate anions bridge urea groups, once more forming both R22(8) and R21(6) rings. All of the urea groups are therefore involved in hydrogen bonding with triflate anions. The sulfonate group is disordered between these two ring-forming positions. The long-range structure is composed of discrete, helical trimers that are held together by Ag–O and CH⋯O interactions (Fig. 9).
| |
7
|
8
|
9
|
10
|
11
|
| Formula |
C27H26.67AgF3N6O5.33S |
C28H31AgN7O6F3S |
C52H52Ag2N12O8S |
C27H30AgN7O6 |
C27H27.2AgN7.6O6.6 |
|
M
|
716.80 |
770.54 |
1220.86 |
656.45 |
671.63 |
| Crystal system |
Triclinic |
Monoclinic |
Tetragonal |
Monoclinic |
Monoclinic |
| Space group |
P-1 |
P21 |
P
-421c |
C2/c |
C2/c |
|
a/Å |
7.1774(5) |
10.2326(11) |
17.4146(6) |
49.675(4) |
30.599(3) |
|
b/Å |
16.7930(15) |
15.6739(17) |
17.4146(6) |
13.4895(10) |
13.8115(12) |
|
c/Å |
19.6054(18) |
10.4685(11) |
8.2655(6) |
33.477(3) |
32.625(3) |
|
α/° |
107.586(3) |
90 |
90 |
90 |
90 |
|
β/° |
93.998(3) |
106.780(2) |
90 |
122.132(7) |
91.3120(10) |
|
γ/° |
91.066(4) |
90 |
90 |
90 |
90 |
|
U/Å3 |
2245.2(3) |
1607.5(3) |
2506.7(2) |
18996(3) |
13767(2) |
|
Z
|
3 |
2 |
2 |
28 |
20 |
|
μ/mm−1 |
0.810 |
0.763 |
0.892 |
0.799 |
0.792 |
| Unique reflections |
5223 |
7300 |
2878 |
20860 |
11431 |
| Observed reflections |
3996 |
6857 |
2766 |
18601 |
9242 |
|
R
int
|
0.0531 |
0.0351 |
0.0288 |
0.0613 |
0.0352 |
|
wR
2 (all data) |
0.1419 |
0.0850 |
0.0706 |
0.2750 |
0.1261 |
|
R [I
≥ 2σ(I)] |
0.0566 |
0.0350 |
0.0288 |
0.0660 |
0.0519 |
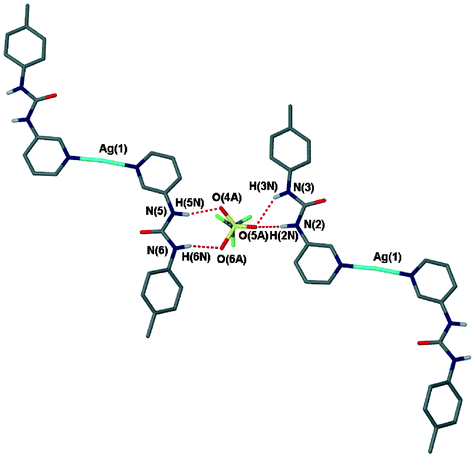 |
| | Fig. 8 The hydrogen bonding around one disordered triflate anion in 7. Hydrogen bond data is given in Table 4. | |
![A discrete trimeric assembly within the structure of 7 held together by long-range Ag–O interactions [2.851(5) and 2.967(6) Å] and weak CH⋯O interactions.](/image/article/2005/NJ/b415818k/b415818k-f9.gif) |
| | Fig. 9 A discrete trimeric assembly within the structure of 7 held together by long-range Ag–O interactions [2.851(5) and 2.967(6) Å] and weak CH⋯O interactions. | |
Table 4 Hydrogen bond data for structures 7–11
| |
Symmetry equivalent |
d(D–H)/Å |
d(H⋯A)/Å |
d(D⋯A)/Å |
∠(D–H⋯A)/° |
|
NH hydrogen positions are geometrically fixed.
Only data for one crystallographically independent [Ag(1)2]NO3 complex shown as an example for brevity.
|
| Complex 7a |
| N(2)–H(2N)⋯O(5A) |
x + 1, y + 1, z |
0.88 |
1.91 |
2.72(4) |
152.1 |
| N(2)–H(2N)⋯O(5) |
x + 1, y + 1 ,z |
0.88 |
2.15 |
2.929(13) |
147.8 |
| N(3)–H(3N)⋯O(4) |
x + 1, y + 1, z |
0.88 |
2.19 |
3.043(16) |
164.7 |
| N(3)–H(3N)⋯O(5A) |
x + 1, y + 1, z |
0.88 |
2.20 |
2.97(4) |
145.8 |
| N(5)–H(5N)⋯O(4A) |
x, y, z |
0.88 |
2.10 |
2.96(3) |
163.4 |
| N(5)–H(5N)⋯O(6) |
x, y, z |
0.88 |
2.10 |
2.905(15) |
151.5 |
| N(6)–H(6N)⋯O(6) |
x, y, z |
0.88 |
1.98 |
2.783(12) |
150.8 |
| N(6)–H(6N)⋯O(6A) |
x, y, z |
0.88 |
2.13 |
2.95(3) |
154.5 |
| N(8)–H(8N)⋯O(7) |
x, y, z |
0.88 |
2.22 |
3.033(17) |
153.9 |
| N(8)–H(8N)⋯O(9) |
−x + 2, −y, −z |
0.88 |
2.36 |
3.153(13) |
150.7 |
| N(9)–H(9N)⋯O(7) |
x, y, z |
0.88 |
2.04 |
2.881(16) |
159.3 |
| Complex 8 |
| N(2)–H(2N)⋯O(5) |
x, y, z |
0.77(4) |
2.48(4) |
3.154(4) |
146(4) |
| N(3)–H(3N)⋯O(5) |
x, y, z |
0.68(4) |
2.19(4) |
2.861(4) |
175(5) |
| N(5)–H(5N)⋯O(7) |
x, y, z |
0.85(4) |
2.11(4) |
2.919(4) |
160(4) |
| N(6)–H(6N)⋯O(7) |
x, y, z |
0.80(5) |
2.23(5) |
2.974(4) |
156(4) |
| C(1)–H(1)⋯O(4) |
x, y, z |
0.93 |
2.97 |
3.715(4) |
138.2 |
| O(7)–H(2O)⋯O(4) |
x, y, z
− 1 |
0.77(5) |
2.14(5) |
2.906(5) |
171(4) |
| C(5)–H(5)⋯O(3) |
x, y, z
− 1 |
0.93 |
2.78 |
3.591(4) |
146.4 |
| C(14)–H(14)⋯O(3) |
x, y, z
− 1 |
0.93 |
2.45 |
3.296(4) |
150.9 |
| O(7)–H(1O)⋯O(1) |
x
− 1, y, z
− 1 |
0.795(4) |
2.049(4) |
2.791(4) |
155.3(4) |
| Complex 9 |
| N(2)–H(2N)⋯O(2) |
x, y, z |
0.84(3) |
2.18(3) |
3.005(2) |
168(3) |
| N(3)–H(3N)⋯O(2) |
−y + 1, x, −z + 1 |
0.77(2) |
2.08(3) |
2.844(3) |
174(2) |
| Complex 10ab |
| N(11)–H(11N)⋯O(106) |
x, y, z |
0.88 |
2.21 |
2.995(11) |
148.0 |
| N(12)–H(12N)⋯O(106) |
x, y, z |
0.88 |
2.16 |
2.922(9) |
144.8 |
| N(14)–H(14N)⋯O(107) |
x, y, z |
0.88 |
2.08 |
2.928(10) |
162.1 |
| N(15)–H(15N)⋯O(108) |
x, y, z |
0.88 |
2.06 |
2.940(9) |
176.6 |
| C(53)–H(53A)⋯O(107) |
x, y, z |
0.93 |
2.52 |
3.317(13) |
142.0 |
| Complex 11b |
| N(2)–H(2N)⋯O(7B) |
x, y, z |
0.83(6) |
2.26(6) |
3.02(2) |
151(5) |
| N(3)–H(3N)⋯O(7B) |
x, y, z |
0.74(5) |
2.16(6) |
2.86(3) |
158(6) |
| N(5)–H(5N)⋯O(6B) |
x, y, z |
0.73(4) |
2.39(4) |
3.084(9) |
159(4) |
| N(6)–H(6N)⋯O(8B) |
x, y, z |
0.77(4) |
2.15(5) |
2.89(3) |
162(4) |
| C(14)–H(14)⋯O(6B) |
x, y, z |
0.95 |
2.32 |
3.185(12) |
150.4 |
A second triflate structure was obtained in which both water and acetonitrile are enclathrated: [Ag(1)2]CF3SO3·H2O·MeCN (8), in the chiral space group P21. This difference in the enclathrated solvent, brought about due to different reaction conditions, results in a different conformation of the Ag(1)2+ cation and therefore very different intermolecular interactions from those in the structure of 7. Whereas in the structure of 7 the urea NH groups are facing away from the central silver atom, the structure of 8 has these groups facing inwards, although the arms are still rotated 180° away from each other about the N–Ag–N axis (Fig. 10). The triflate anion within 8 shows no disorder, arising from the fact that it is held in place by six hydrogen bonds, with each oxygen atom receiving two. The donor groups are a urea group, with the recurring R21(6) motif, one hydrogen bond from the enclathrated water (which is involved in a bridging position with a urea oxygen atom) and three pyridyl CH⋯O interactions (Fig. 11). This arrangement makes the triflate anion hydrogen-bond saturated.
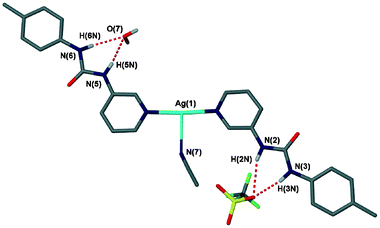 |
| | Fig. 10 The asymmetric unit of 8, showing the urea groups in R21(6) interactions with both the triflate anion and water. CH hydrogen atoms not shown for clarity. Hydrogen bond data shown in Table 4. | |
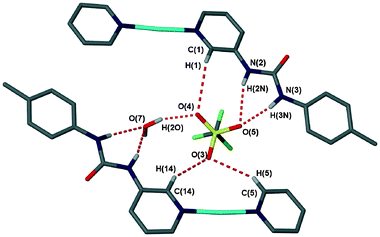 |
| | Fig. 11 The hydrogen bonding environment around the triflate anion in the structure of 8. Coordinated acetonitrile omitted for clarity. | |
The sulfate complex [Ag(1)2]2SO4 (9) forms helical, interdigitating stacks in the solid-state structure (Fig. 12). These chains are held together by virtue of long-range Ag–O interactions [3.217(2) Å] utilising the urea oxygen atom. The sulfate anion, as in the previous structures, is bound to the urea protons in an R22(8) motif on the ‘outside’ of the complex. Each sulfate anion is surrounded by four urea donors, making the oxygen atoms hydrogen-bond saturated. The structure bears a close resemblance to previous sulfate-containing structures using the same ligand coordinated to octahedral metal centres.13
![Part of a helical chain in the structure of 9 held together by Ag–O interactions [Ag–O = 3.217(2) Å]. Hydrogen atoms omitted for clarity.](/image/article/2005/NJ/b415818k/b415818k-f12.gif) |
| | Fig. 12 Part of a helical chain in the structure of 9 held together by Ag–O interactions [Ag–O = 3.217(2) Å]. Hydrogen atoms omitted for clarity. | |
Discrete structures
The most interesting complexes from an anion binding standpoint, however, are those containing the [Ag(1)2]NO3 complex. Two solvates of this complex were obtained, [Ag(1)2(MeOH)]NO3 (10), and a co-crystal, 2[Ag(1)2(MeOH)]NO3·3[Ag(1)2(MeNO2)]NO3 (11). Unlike the other structures reported in this work, the urea hydrogen atoms are orientated ‘inwards’ and converge on a central pocket in which the nitrate anion is positioned in an fashion that is co-planar with the ligands, chelated between the two arms of the cationic complex. The structures are not, therefore, hydrogen-bonded polymers akin to the other species reported in this work, but instead are comprised of discrete entities, with the exception of hydrogen-bonded interactions to urea carbonyl moieties from ligated methanol in both cases. The [Ag(1)2]+ host system displays a remarkably good shape match for the nitrate anion.
Within the methanol solvate, 10, the solvent is bound to the silver centre, resulting in a distorted trigonal geometry. The anion is not positioned symmetrically within the host complex, which would result in two R22(8) rings. Instead, the skewed orientation allows for a CH⋯O interaction to the nitrate, in addition to the four hydrogen bonds from the more acidic urea protons (Fig. 13). This off-centre position is a consistent feature of the four independent molecules in 10 and the three closely related independent molecules in 11, although disorder is present in some complexes such that the nitrate can lie towards either the left- or right-hand side of the complex. The urea groups interact with the nitrate anion by means of both R22(8) and R21(6) motifs, as seen in the structure of 7. The crystal packing of 10 is also remarkable, with 3.5 independent molecules contained in the asymmetric unit.21 The half molecule displays an apparently linear Ag⋯O–N vector due to its position on a rotation axis, although this is merely a disorder average over two asymmetric orientations as adopted by the other three independent molecules. The long-range structure consists of discrete π-stacked heptamers held together by hydrogen bonding between the methanol and urea carbonyl (Fig. 14).
![Close size and shape complementarity of the nitrate anion with [Ag(1)2]+ in the structure of 10.](/image/article/2005/NJ/b415818k/b415818k-f13.gif) |
| | Fig. 13 Close size and shape complementarity of the nitrate anion with [Ag(1)2]+ in the structure of 10. | |
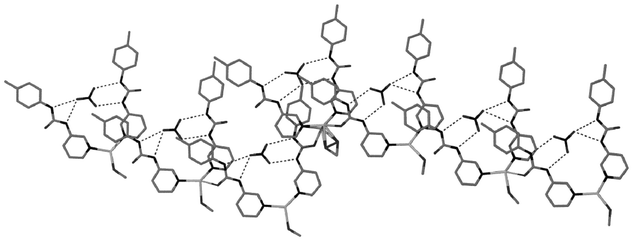 |
| | Fig. 14 A discrete heptameric assembly within the structure of 10. | |
The MeOH/MeNO2 solvate 11 also displays unusual crystal packing. The structure adopts the same space group as that of 10 (C2/c) but with two and a half independent molecules per asymmetric unit. Each asymmetric unit contains one [Ag(1)2]NO3 complex with nitromethane occupying the third coordination site, one complex with a 50∶50 occupancy of nitromethane and methanol and the half complex is a methanol complex situated on the twofold rotation axis. As with 10 the nitrate anions are chelated by both urea groups in an offset fashion (with some disorder), allowing for CH⋯O as well as NH⋯O interactions. It is noteworthy that this discrete ML2(NO3) unit persists with both MeOH and NO2Me co-ligands. Moreover, the bidentate ligation of the Ag(I) centre by NO2Me is highly unusual (Fig. 15), with only one previous example of this coordination mode to a transition metal, also silver(I), recorded in the CSD.22
![The [Ag(1)2(MeNO2)]NO3 complex within the structure of 11. Selected bond lengths (Å): Ag(1)–O(14), 2.623(4); Ag(1)–O(15), 2.717(4). Only one disordered nitrate position shown.](/image/article/2005/NJ/b415818k/b415818k-f15.gif) |
| | Fig. 15 The [Ag(1)2(MeNO2)]NO3 complex within the structure of 11. Selected bond lengths (Å): Ag(1)–O(14), 2.623(4); Ag(1)–O(15), 2.717(4). Only one disordered nitrate position shown. | |
Anion binding behaviour
The exact size and shape fit of the nitrate anion in the structures of 10 and 11 indicates that an assembly of this type may persist in solution. As a result we have undertaken a survey of the solution anion-binding properties of ligands 1 and 2 and their silver(I) complexes. The unbound ligands 1 and 2 themselves interact well with oxo-anions despite their lack of charge. 1H NMR titrations were carried out in acetone on the uncoordinated ligands, with the oxo-anions added as tetrabutylammonium salts. Association constants were calculated for 1 and 2 using the program HypNMR23 (Table 5).
Table 5 Binding constants (M−1) obtained via1H NMR titrations for the free ligands 1 and 2 and the complex [Ag(1)2]+
| Anion |
1
|
2
|
[Ag(1)2]+ |
|
K
11
|
K
11
|
K
11
|
K
12
|
K
13
|
|
Binding constants are too high/low to be accurately determined.
Acetate does not form an identifiable 1∶2 complex with [Ag(1)2]+, see text.
For nitrate K13 represents the ligation of the anion to the metal centre, whereas for acetate K11 represents this process, see text.
Modelled as simultaneous binding of two anions, i.e., K12
×
K13.
|
| NO3− |
956 |
3500 |
30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200c 200c |
2900 |
550c |
| CH3O2− |
>105a |
>105a |
4.97 × 105c |
—
b |
5.31 × 106cd |
| ReO4− |
<10a |
<10a |
<10a |
<10a |
<10a |
These results show that nitrate and particularly acetate are both able to hydrogen bond strongly to the urea functionalities of the ligands. Upon addition of one equivalent of acetate to a solution of either 1 or 2 remarkable chemical shift changes of up to Δδ = 4.0 ppm are observed. Binding constants with this guest are above 105 M−1, the limit of detection by NMR methods. This is attributable to the high basicity of the acetate anion. The spectra with acetate present also show a splitting of the urea NH proton signals into two species, believed to represent two separate binding modes, possibly with a dimeric structure forming, a phenomenon that is not observed for the binding of nitrate or perrhenate. Ligand 2 displays stronger anion binding than 1, which may be due to the inductive effects of the pyridyl N atom on urea acidity.
The tweezer-like conformation of the [Ag(1)2]NO3 complex that exists in the solid-state structures of 10 and 11 is also observed to persist in solution. A 1H NMR titration of [Ag(1)2]CF3SO3 with nitrate (added as the tetrabutylammonium salt) clearly shows two separate binding events occurring when following the signal of the para-pyridyl proton (Fig. 16). Whilst the signals corresponding to the urea NH protons steadily move downfield during the experiment, the para pyridyl resonance displays a clear inflection point after the addition of one equivalent of nitrate, corresponding to a change in the binding mode. The first of these associations is believed to result in a 1∶1 host–guest complex analogous to the solid-state structure with the subsequent binding event giving rise to a 1∶2 stoichiometry. A Job plot confirms that this is indeed the case. The values of the association constants (K11 and K12) are 30200 and 2900 M−1 for the 1∶1 and 1∶2 complexes, respectively. In a preliminary communication we reported slightly lower values.11 The present data is of higher precision and was obtained at lower concentration, eliminating precipitation problems. In addition to these two binding constants that are associated with the urea ligands there is also a third, weaker binding process that is apparent during the modelling of the data (K13 = 550 M−1). This is attributed to a direct coordination of the nitrate anion to the silver ion when a significant excess of the guest is present. Evidence for this process comes from the much stronger interaction of the metal centre with acetate (vide infra).
![The chemical shift change of the para-pyridyl proton during the titration of [Ag(1)2]CF3SO3 + TBA-NO3, showing two different processes occurring.](/image/article/2005/NJ/b415818k/b415818k-f16.gif) |
| | Fig. 16 The chemical shift change of the para-pyridyl proton during the titration of [Ag(1)2]CF3SO3 + TBA-NO3, showing two different processes occurring. | |
In the presence of the triflate anion there is apparently little or no chelation of the anion within the tweezer cavity, whereas the presence of the nitrate anion brings about a change in conformation, resulting in the chelated geometry observed in the crystal structure. This structure persists until addition of more than one equivalent of nitrate, at which point the chelated 1∶1 structure gradually converts to a situation in which nitrate anions bind separately to the two ligands on the ‘outside’ of the complex in a fashion analogous to anion binding by free 1 (Fig. 17). Such a conformational change would significantly alter the environment of the para-pyridyl proton, with it facing towards the urea oxygen atom in the 1∶1 complex and closer to a urea hydrogen atom in the 1∶2 complex, giving rise to the observed change. The much higher value of K11 compared to K12 supports the formation of the chelated geometry observed crystallographically.
![The two proposed conformations adopted by the [Ag(1)2]+ host in solution with either one anion coordinated between arms or with the arms binding independently.](/image/article/2005/NJ/b415818k/b415818k-f17.gif) |
| | Fig. 17 The two proposed conformations adopted by the [Ag(1)2]+ host in solution with either one anion coordinated between arms or with the arms binding independently. | |
Titration of [Ag(1)2]CF3SO3 with acetate produced a notably different response to that of nitrate. Very little change in the chemical shift values of the NH protons was observed until after the addition of one equivalent of acetate. This behaviour is attributed to the ligation of the Ag(I) centre by the first equivalent of acetate, unlike nitrate, which does not ligate until an excess of the guest is present (Fig. 18). The addition of further aliquots of TBA-acetate produced significant binding, although there was no evidence of two separate binding events as observed for nitrate. Hence, the behaviour of the complex was modelled as 1∶1 and 1∶3 aggregates, with values of K11 and K12
×
K13 found to be 4.97 × 105 and 5.31 × 106 M−1, respectively. It is speculated that either a complex in which one acetate anion binds to each arm forms, or, the ligation of the acetate causes a break-up of the [Ag(1)2]+ complex and the arms act independently. The acetate anion is not of the correct shape to template the formation of the tweezer geometry of the complex and therefore a very different conformation may result.
![Titration plots of [Ag(1)2]CF3SO3 with TBA-NO3 (squares) and TBA-CH3CO2 (diamonds) following the urea NH resonance adjacent to the pyridyl ring. The first equivalent of acetate coordinates to the silver before binding occurs with increasing guest concentration, whereas with nitrate binding occurs prior to coordination.](/image/article/2005/NJ/b415818k/b415818k-f18.gif) |
| | Fig. 18 Titration plots of [Ag(1)2]CF3SO3 with TBA-NO3 (squares) and TBA-CH3CO2 (diamonds) following the urea NH resonance adjacent to the pyridyl ring. The first equivalent of acetate coordinates to the silver before binding occurs with increasing guest concentration, whereas with nitrate binding occurs prior to coordination. | |
Binding of perrhenate by [Ag(1)2]+ was almost negligible, as expected for this labile anion with diffuse charge. An upfield shift is observed for the urea proton signals, contrary to the downfield shifts observed for acetate and nitrate. This suggests that the perrhenate anion is hydrogen bonded less strongly than the triflate anion that it displaces.
Unfortunately, the solution behaviour of the [Ag(2)2]+ complex could not be ascertained due to its insolubility. However, this complex is not expected to show cooperative binding between the ligands due to the position of the urea group on the pyridyl ring, as evidenced in the crystal structures obtained.
Conclusions
The complexes obtained with various anions using both the [Ag(1)2]+ and [Ag(2)2]+ cations show that the urea functionality has a strong tendency to form R22(8) motifs with oxo-anions. When this affinity is harnessed into a host assembly with the correct geometry for a specific guest, as in the case of [Ag(1)2]NO3, then significant binding persists in solution with the binding of the templating anion markedly enhanced compared to the free ligand. In the presence of strongly ligating anions such as acetate, however, the self-assembly process is disrupted by direct binding of the anion to the metal centre.
Experimental
The syntheses of the ligands 1 and 2 have previously been reported.12 All commercial starting materials and solvents were used as purchased with no further purification or drying. The syntheses of 3–11 were aimed toward the formation of crystalline samples and generally proceeded in good yield. Elemental analyses were carried out at the University of Durham using an Exeter Analytical E-440 instrument.
1H NMR titration experiments were conducted in acetone-d6 using a Varian Mercury 400-BB spectrometer with TMS as an internal reference. An initial concentration of 0.034 mol dm−3 of 1 or 2 was used in all cases, with the guest anions added as tetrabutylammonium salts in 10 µl aliquots (corresponding to 0.1 equivalents of the guest).
Synthesis
[Ag(2)2]CF3SO3·2H2O (3).
Crystals grown from a solution of 2 (0.050 g, 0.22 mmol) and AgCF3SO3 (0.057 g, 0.22 mmol) in water–methanol (50∶50 v/v; 5 ml) at room temperature. Anal. calcd for C27H30N6O7SF3Ag: C, 43.38; H, 4.05; N, 11.24%; found: C, 43.36; H, 4.05; N, 11.27%.
[Ag(2)2]CH3CO2·1.25H2O·MeOH (4).
Crystals grown from a solution of 2 (0.050 g, 0.22 mmol) and AgCH3CO2 (0.037 g, 0.22 mmol) in water–methanol (50∶50 v/v; 5 ml) at room temperature. Anal. calcd for C29H34.5N6O6.25Ag: C, 51.53; H, 5.26; N, 12.44%; found: C, 51.55; H, 5.27; N, 12.52%.
[Ag(2)2]NO3·H2O (5).
Crystals grown from a solution of 2 (0.050 g, 0.22 mmol) and AgNO3 (0.038 g, 0.22 mmol) in wet acetonitrile (5 ml) at room temperature. Anal. calcd for C26H30N7O6Ag: C, 48.61; H, 4.39; N, 15.26%; found: C, 48.35; H, 4.37; N, 15.32%.
[Ag(2)]NO3 (6).
Crystals grown from a solution of 2 (0.050 g, 0.22 mmol) and AgNO3 (0.038 g, 0.22 mmol) in methanol (5 ml) at room temperature. Anal. calcd for C13H13N4O4Ag: C, 39.32; H, 3.30; N, 14.11%; found: C, 39.25; H, 3.28; N, 14.16%.
[Ag(1)2]CF3SO3·0.5H2O (7).
Crystals grown from a solution of 1 (0.025 g, 0.11 mmol) and AgCF3SO3 (0.0094 g, 0.04 mmol) in cyclohexane–methanol (50∶50 v/v; 5 ml) at room temperature. Anal. calcd for C27H27N6O5.5SF3Ag: C, 45.00; H, 3.75; N, 11.67%; found: C, 45.23; H, 3.77; N, 11.60%.
[Ag(1)2]CF3SO3·H2O·MeCN (8).
Crystals grown from a solution of 1 (0.025 g, 0.11 mmol) and AgCF3SO3 (0.0094 g, 0.04 mmol) in water–acetonitrile (50∶50 v/v; 5 ml) at room temperature. Anal. calcd for C29H31N7O6SF3Ag: C, 45.19; H, 4.02; N, 12.73%; found: C, 45.26; H, 4.12; N, 12.62%.
[Ag(1)2]2SO4 (9).
Crystals grown from a solution of 1 (0.025 g, 0.11 mmol) and AgSO4 (0.011 g, 0.04 mmol) in acetonitrile–water (50∶50 v/v; 5 ml) at room temperature. Anal. calcd for C52H52N12O8Ag2S: C, 51.15; H, 4.26; N, 13.77%; found: C, 51.01; H, 4.21; N, 13.83%.
[Ag(1)2]NO3·MeOH (10).
Crystals grown from a solution of 1 (0.025 g, 0.11 mmol) and AgNO3 (0.006 g, 0.04 mmol) in toluene–methanol (50∶50 v/v; 5 ml) at room temperature. Anal. calcd for C27H30N7O6Ag: C, 49.39; H, 4.57; N, 14.94%; found: C, 48.93; H, 4.27; N, 15.16%.
[Ag(1)2]NO3·0.4MeOH·0.6MeNO2 (11).
Crystals grown from a solution of 1 (0.025 g, 0.11 mmol) and AgNO3 (0.006 g, 0.04 mmol) in nitromethane–methanol (50∶50 v/v; 5 ml) at 4 °C. Anal. calcd for C27H29.5N7.5O6.5Ag: C, 48.32; H, 4.40; N, 15.66%; found: C, 48.05; H, 4.43; N, 15.48%.
Crystallography†
X-Ray diffraction data were collected using either Bruker 3-circle diffractometers with SMART 1000 (3–6, 10) or Apex (8, 9 and 11) CCD detectors or a Nonius KappaCCD diffractometer (7), with monochromated Mo-Kα radiation in all cases. All data were collected at 120 K as maintained by Oxford Cryosystems open-flow N2 cryostats. The structures were solved by direct methods using SHELXS-9724 and refined by full-matrix least squares against F2 of all reflections using SHELXL-97.25 All C–H bond lengths were set to fixed X-ray distances, as were N–H bonds in lower quality data sets (0.880 Å), and allowed to ride. Hydrogen atoms attached to oxygen or nitrogen were located experimentally from difference Fourier maps, except in the cases of 7 and 10 for which the data quality was not sufficient to permit this. All hydrogen atoms are treated with isotropic atomic displacement parameters.
Structure 7 contains one triflate anion that has an SO3 group disordered over two positions (50∶50 occupancies) between two urea groups. The half triflate per asymmetic unit exists in the same site as the half occupancy enclathrated water molecule, refined as separate parts. Structure 10 contains disordered methanol ligands on two of the independent molecules. Structure 11 contains disordered nitrate anions (two and a half per asymmetric unit) across two positions, centred around the nitrogen atom, of 50∶50 occupancies.
Acknowledgements
We thank the EPSRC for studentships (DRT, ECS) and for an advanced fellowship (JAKH). Special thanks to Dr Len Barbour, University of Stellenbosch, South Africa, for the SHELX 25 interface program X-Seed (www.x-seed.net).26
References
- P. A. Gale, Coord. Chem. Rev., 2003, 240, 191 CAS.
- P. A. Gale, Coord. Chem. Rev., 2001, 213, 79 CrossRef CAS.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS.
- T. S. Snowden and E. V. Anslyn, Curr. Opin. Chem. Biol., 1999, 6, 740 CrossRef.
-
C. F. Mason, Biology of Freshwater Pollution, Longman, Harlow, Essex, 2nd edn., 1991 Search PubMed.
- K. J. Wallace, R. Daari, W. J. Belcher, L. Abouderbala, M. G. Boutelle and J. W. Steed, J. Organomet. Chem., 2003, 666, 63 CrossRef CAS.
- X. L. Chi, A. J. Guerin, R. A. Haycock, C. A. Hunter and L. D. Sarson, J. Chem. Soc., Chem. Commun., 1995, 2563 RSC.
- C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77 CAS.
- C. R. Bondy, P. A. Gale and S. J. Loeb, Chem. Commun., 2001, 729 RSC.
- C. R. Bondy, P. A. Gale and S. J. Loeb, J. Am. Chem. Soc, 2004, 126, 5030 CrossRef CAS.
- D. R. Turner, E. C. Spencer, J. A. K. Howard, D. A. Tocher and J. W. Steed, Chem. Commun., 2004, 1352 RSC.
- D. R. Turner, B. Smith, A. E. Goeta, I. Radosavljevic-Evans, D. A. Tocher, J. A. K. Howard and J. W. Steed, CrystEngComm, 2004 Search PubMed , submitted.
- D. R. Turner, M. B. Hursthouse, M. E. Light and J. W. Steed, Chem. Commun., 2004, 1354 RSC.
- M. Albrecht, J. Zauner, R. Burget, H. Rottele and R. Frohlich, Mater. Sci. Eng. C, 2001, 18, 185 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 33, 143.
-
(a)
CSD Version 5.25, CCDC, Cambridge, Nov. 2003 Search PubMed;
(b) F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380 CrossRef.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
-
F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochman, Advanced Inorganic Chemistry, John Wiley and Sons, New York, 1999 Search PubMed.
- S. C. Blackstock and J. K. Kochi, J. Am. Chem. Soc., 1987, 109, 2484 CrossRef CAS.
- Y. A. Simonov, M. D. Mazus, S. T. Malinovsky, E. S. Levchenko, S. V. Pavlova and L. I. Budarin, Kristallografiya, 1987, 32, 93 CAS.
- J. W. Steed, CrystEngComm, 2003, 169 RSC.
- N. Schulthiess, D. R. Powell and E. Bosch, Inorg. Chem., 2003, 42, 8886 CrossRef CAS.
-
P. Gans, HypNMR, Protonic Software, University of Leeds, Leeds, 2000 Search PubMed.
-
G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
-
G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 Search PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.