Synthesis, characterization and X-ray crystal structures of cyclam derivatives. Part VI. Proton binding studies of a pyridine-strapped 5,12-dioxocyclam based macrobicycle†‡§
Michel
Meyer
a,
Laurent
Frémond
a,
Alain
Tabard
a,
Enrique
Espinosa
a,
Guy Yves
Vollmer
a,
Roger
Guilard
*a and
Yves
Dory
b
aLaboratoire d’Ingénierie Moléculaire pour la Séparation et les Applications des Gaz (LIMSAG, CNRS UMR 5633), Université de Bourgogne, Faculté des Sciences, 6 boulevard Gabriel, 21100, Dijon, France. E-mail: roger.guilard@u-bourgogne.fr; Fax: +33 3 80 39 61 17; Tel: +33 3 80 39 61 11
bLaboratoire de Synthèse Supramoléculaire, Département de Chimie, Université de Sherbrooke, 2500 boulevard de l‘Université, Sherbrooke, PQ J1K 2R1, Canada
First published on 14th December 2004
Abstract
The 14-membered cyclic diamide 1,4,8,11-tetraazacyclotetradecane-5,12-dione (5,12-dioxocyclam) can be considered as a trans-autodiprotected tetraazamacrocycle and provides a convenient starting material for the preparation of macrobicyclic receptors. As an example, the secondary amine nitrogen atoms located at the 1 and 8 positions were cross-bridged with a 1,3-pyridyl strap, affording the constrained ansa-dioxocyclam ligand 1,9,12,18,22-pentaazatricyclo[7.6.6.13,7]docosa-3,5,7(22)-triene-13,19-dione (L1). The proton binding properties of this cage-type compound, which possesses a hemispherical cavity, were fully investigated by spectroscopic (IR, NMR, UV, MALDI-TOF MS), quantum chemical, and potentiometric methods. While both bridgehead tertiary amines have their free lone pairs oriented inside the cavity, intramolecular hydrogen bonding was found to play a key role in determining the structural features of the free base and its protonated forms. L1 behaves as a diprotic base in water with log K011 = 8.94(1) and log K012 = 2.32(9), but most interestingly shows slow proton-transfer rates on the NMR timescale.
Introduction
The design and synthesis of macropolycyclic receptors incorporating ligating heteroatoms are actively pursued for the development of functional nanodevices.2–5 Selective molecular recognition6,7 and signalling8–12 properties are sought to provide the major impetus for further progress in this flourishing area of chemistry, while various applications in biochemistry and medicine, catalysis, or material sciences have already opened new horizons for the utilization of this class of compounds.13–17 In this respect, conformationally constrained bicyclic polyamines with bridgehead nitrogen atoms have gained increasing attention over the last three decades because the additional rigidity provided by strapping the macrocyclic framework imparts intriguing anion-18 and metal-binding properties.19 Contemporary to the disclosure of the first cryptands by Lehn’s group,2,3 Simmons and Park showed that protonation of diazabicyclo[k,l,m]alkanes of various sizes [6 ≤ (k,l,m) ≤ 10] occurs outside the cavity in the initial stage, while the inversion of the ammonium nitrogen atom is in the hour time range.20,21 They also evidenced the encapsulation of halide anions in the central cavity, the guest being hold in-between both in,in-diprotonated amines by hydrogen bonds.22 Shorter analogs (i.e., k, l, m = 4) were later shown to behave as “proton sponges”.23–27Reinforced28,29 and cross-bridged17,30–32 cyclic tetraamines such as 1,4,7,10-tetraazacyclododecane (cyclen) and 1,4,8,11-tetraazacyclotetradecane (cyclam), are among the most common and readily accessible polynitrogenated bicyclic architectures.33 They are obtained by linking either two adjacent or two nonadjacent secondary amines of the parent macrocycle. The latter possibility has given rise to a new family of macrobicyclic ansa-ligands, the so-called bowl adamanzanes,34 with a number of these also displaying “proton sponge” properties and very slow protonation kinetics.35,36
In contrast, amide-containing azamacrobicyclic ligands have attracted much less attention,37 although they provide a convenient access to strapped cyclen or cyclam derivatives upon reduction of the protecting carbonyl groups.8,9,38–41 From a synthetic point of view, trans-dioxocyclam (5,12-dioxocyclam or 1,4,8,11-tetraazacyclotetradecane-5,12-dione, compound 1 in Scheme 1)42,43 is an attractive starting material since two out of the four secondary amines of the 14-membered ring, namely those located in positions 4 and 11, take part in two peptidic bonds and thus are masked. Optically active C-functionalized analogs, obtained by photolysis of chromium carbene complexes in the presence of suitable imidazolines, have been described more recently by Hegedus and coworkers.44–47 Provided these trans “autodiprotected” synthons are reacted with the appropriate biselectrophile, hemispherical cage-type molecules are isolated in a single step and in high yield.8,9,38,39,46,48–56 Their dual personality stems from their intermediate structure as they possess all the features of the corresponding saturated polyamines and those of oligocyclopeptides.57,58 However, the physico-chemical consequences of their interplay in a constrained environment remain largely unknown today. Recently, we reported on the unusual properties displayed by the copper(II) complexes formed with ligand L1.1 Depending upon the medium’s acidity, either a red tetra- or a blue pentacoordinated species was isolated, the former possessing one and the latter two N-deprotonated amide groups. The fast pH-triggered interconversion was shown to be accompanied by metal translocation and a simultaneous molecular reorganization of the ligand scaffold reminiscent of a glider motion.
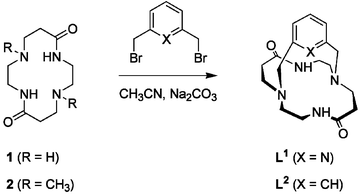 | ||
| Scheme 1 | ||
Although a few o-xylyl,48 α,α′-pyridyl,52,53,55 or polyethylene glycol capped trans-dioxocyclams51 have been structurally characterized, to the best of our knowledge no report describing the solution structure and the acid-base properties of this class of compounds has appeared so far. The structural data indicate that the strap spanning across the dioxocyclam fragment brings the bridgehead nitrogen atoms in close vicinity and enforces an endo orientation of both lone pairs, most probably freezing out the Walden inversion. The resulting ring conformation contrasts markedly with the extended layout adopted by 1 in either its free base or diprotonated form, suggesting that both amines behave independently of each other.43 Indeed, solution studies revealed negligible Coulombic interactions between both basic sites for trans-dioxocyclam, since their protonation gives rise to a weak cooperative effect. Hereafter, the thermodynamic and kinetic proton-uptake properties of an ansa-dioxocyclam macrobicycle are discussed for the first time in light of NMR, potentiometric, spectrophotometric, and quantum-chemical studies of the pyridine derivative L1 (Scheme 1).
Results and discussion
Synthesis
The cage-like macrobicyclic ligand L1 was synthesized according to a [1 + 1] nucleophilic substitution reaction (Scheme 1) as previously described by Guilard and coworkers.38,39 Condensation in refluxing acetonitrile of 2,6-bis(bromomethyl)pyridine (3) in the presence of 5,12-dioxocyclam (1) and a large excess of sodium carbonate as base (Scheme 1) produces exclusively the desired 1,8-cross-bridged perazacryptand L1 in good yield (∼70% after chromatographic purification). The detailed characterization of L1 by mass spectrometry and high-resolution NMR spectroscopy (vide infra), is reported herein for the first time.The steric constraints imposed by the rigid pyridyl handle prevents the chiral tertiary amines from undergoing the Walden inversion at room temperature. Thus, reaction of the dihalogenated biselectrophile 3 with the C2h-symmetric dioxocyclam unit affords a racemic mixture of (R,R) and (S,S) diastereoisomers.52 The analytical purity was checked by elemental CHN analysis, a negative silver nitrate assay, and by MALDI-TOF mass spectroscopy. The mass spectrum of the free base evidenced a featureless baseline with a single peak at m/z = 331.1, showing the predicted isotopic line pattern for the radical-cation [L1]+˙ (calcd m/z = 331.2). The one-mass-unit shifted signal found for the monohydrobromic acid salt crystallized in acetonitrile further confirmed the compound’s identity. It may be noted that none of the possible pyridinyl-bridged bismacrocyclic [2 + 1] or macrotricyclic [2 + 2] cyclization by-products is formed in appreciable amounts if the reaction is carried out at the centimolar concentration level, thus avoiding high-dilution conditions.
Solid-state and solution FTIR spectroscopy
The solid-state infrared spectrum of L1 displays two intense νNH absorption bands at 3224 and 3174 cm−1, which are typical of hydrogen-bonded secondary amides.59 While the N–H stretching vibration is found at 3215 cm−1 for the uncapped model compound 2,60 this band experiences a more than 90 cm−1 shift to higher energies when compared to the parent macrocycle 1 (νNH = 3304 cm−1). The crystal structure of the latter has evidenced the involvement of the amidic NH protons in intermolecular hydrogen bonds interconnecting the channel-forming molecules piled-up in the lattice.43 Thus, it might be inferred that the large bathochromic shift observed for L1 and 2 with respect to 1 reflects intramolecular interactions of the NH protons with the tertiary amine’s lone pairs. This assumption is corroborated by the crystal structures of various C-substituted 5,12-dioxocyclams capped by functionalized pyridines, which are closely related to L1.55 Other diagnostic absorptions such as the amide I (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1654 cm−1) and amide II (δCNH = 1526 cm−1) bands appear in the expected spectral windows and are listed in the Experimental section.59 A weak feature centered at 3057 cm−1 for L1, 3052 cm−1 for 2, and 3091 cm−1 for 1, is assigned either to a combination mode of the amide I and amide II bands or to an overtone of the latter.61,62
O = 1654 cm−1) and amide II (δCNH = 1526 cm−1) bands appear in the expected spectral windows and are listed in the Experimental section.59 A weak feature centered at 3057 cm−1 for L1, 3052 cm−1 for 2, and 3091 cm−1 for 1, is assigned either to a combination mode of the amide I and amide II bands or to an overtone of the latter.61,62
In order to ascertain the formation of intramolecular N–H⋯N hydrogen bonds, spectra were recorded in CDCl3 between 1500 and 3500 cm−1. As far as no shift in energy for any band was detected upon increasing the concentration of L1, it is concluded that the amidic protons are effectively engaged in such interactions both in the solid state and in solution. In addition, solubilization of amorphous L1 in chloroform perturbs only very slightly the vibrational energy levels as evidenced by comparing the KBr pellet and solution spectra. While the carbonyl stretch remains unaffected, the frequency difference for the νNH doublet and the δCNH mode does not exceed 17 cm−1. These results further support the intramolecular nature of the hydrogen bonds and suggest also a close conformational similarity of the molecule in both states. Another piece of structural information is provided by the absence of a lower-energy satellite associated to the strong C–D stretching band arising from the solvent at 2253 cm−1. The appearance of such a band near 2170 cm−1, as observed when increasing amounts of triethylamine are mixed with CDCl3, would be indicative of C–D⋯X (X = N or O) hydrogen bond formation between the solvent and the solute. This criterion can be used to probe the relative orientation of the bridgehead tertiary amine’s lone pairs pointing either inside (in configuration) or outside (out configuration) the cavity.63 Since hydrogen bond formation is only possible when the doublet is exposed outwards the cavity to the bulk, the bridgeheads of L1 adopt an in-in configuration.
NMR spectroscopy
1H NMR spectroscopy provides another means to prove the formation in solution of hydrogen bonds involving the amide protons. It is generally accepted that the resonance for non-hydrogen-bonded amide protons in CDCl3 appears in the 6 ppm region, while those that are hydrogen-bonded give rise to signals above 8 ppm. As expected, the corresponding absorption is observed as a broad singlet at 9.72 ppm in CDCl3 (10.05 ppm in CD3OH), which is close to the position reported for 1 (δH = 9.10 ppm in CDCl3).43 Moreover, the chemical shift is concentration independent, emphasizing the intramolecular character of the hydrogen bonds. At room temperature, the N–H/D exchange rate in D2O is fast enough to preclude a quantitative analysis. Thus, the solvent exposure of the amide protons was investigated by measuring the temperature dependence of the N–H chemical shift. The temperature coefficient (Δδ/ΔT) was taken as the slope of the least-squares-fitted line of the chemical shifts against temperature. For 16 spectra acquired in CDCl3 between 213 and 333 K, the so-called Curie plot was linear with a temperature coefficient of −3.57(2) ppb K−1, which is indicative of hydrogen bonding. No significant change occurred when the experiment was performed in a more dissociating solvent such as CD3OH [Δδ/ΔT = 3.83(1) ppb K−1 for 30 spectra recorded between 180 and 335 K]. For peptides, Δδ/ΔT values in the range of 3–4 ppb K−1 are consistent with involvement in an intrachain hydrogen bond such as in an α-helix or a β-sheet, while non-hydrogen-bonded peptide protons exposed to solvent tend to display higher Δδ/ΔT values.64,65The upper panel in Fig. 1 shows the aliphatic region of the high-resolution 1H NMR spectrum of L1 recorded at room temperature. Like the 13C NMR spectrum, it displays only a single set of signals corresponding to one-half of the number of atoms, indicating that the molecular cage assumes a time-averaged C2-symmetric structure on the NMR timescale. However, all methylenic protons are diastereotopic and give rise to complex geminal and vicinal coupling patterns because of the magnetic anisotropy induced by the pyridyl moiety.
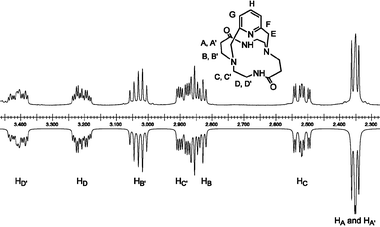 | ||
| Fig. 1 Experimental (upper panel) and gNMR66 simulated (lower panel) aliphatic part of the 1H NMR (500 MHz) spectrum of L1. Solvent: CDCl3; T = 20 °C. | ||
The methylenic hydrogen atoms in the α position to the aromatic nucleus appear as two doublets, typical of an AB spin system (2J = 16.6 Hz) at 3.78 and 3.97 ppm. Interestingly, L1 gives rise to a more pronounced anisochrony of these two pairs of protons (Δν = 95 Hz) compared to L22 (Δν = 45 ppm).60 Spectroscopic results obtained in solution together with crystallographic data for the latter diazacryptand argue for a more rigid framework where the m-xylyl linker is maintained through a three-center hydrogen bond almost perpendicular to the dioxocyclam subunit. Therefore, the higher Δν value found for L1 might reflect a more tilted orientation of the pyridine versus the benzene ring, as expected if the electrostatic repulsion between the three nitrogen lone pairs converging in the center of the cavity is taken into account. The dynamics of the back-and-forth flipping of the 2,6-pyridinedimethylene fragment has been investigated by variable temperature experiments. Cooling a sample down to 213 K in CDCl3 did not freeze out the rocking motion, which would have doubled the total number of resonances. Hence, the activation barrier for the interchange between the various conformational arrangements adopted by the macrocyclic scaffold is well below 40 kJ mol−1 at temperatures as low as −50 °C. Similarly, the lineshape remained unchanged upon heating up to 100 °C.
Two-dimensional homonuclear (1H–1H COSY) and heteronuclear (1H–13C HETCOR) experiments allowed the unequivocal assignment of all proton and carbon resonances (see ESI), although their relative exo or endo orientation with respect to the aromatic cross-bridge could not be determined by these techniques. The most up-field shifted triplet in the 1H NMR spectrum arises from the diastereotopic methylenic protons HA and HA′ located in α positions with respect to the carbonyl group.43 The spectral pattern can be analyzed in terms of coincidentally isochronous nuclei coupled with the neighboring pair of HB (2.84 ppm) and HB′ (3.03 ppm) protons, since they appear as a two resolved multiplets in D2O. The fact that two distinct peaks are also observed in the case of ligand L22 further supports this view.60 The splitting into five lines of the signal assigned to proton HB′ suggests that both vicinal 3J scalar coupling constants take almost identical values and that these should be very close to one-half of |2JB,B′|. Likewise, similar values for 3JB,A and 3JB,A′ (3JB,A = 3JB,A′ < 1/2 × |2JB,B′|) are expected in order to explain the doublet of triplets attributed to HB. The set of ethylenediamine proton resonances is readily identified by a COSY experiment. While HC and HC′ resonate respectively at 2.52 and 2.89 ppm as doublets of doublets of doublets giving rise to eight-line patterns, protons labelled HD and HD′ appear as complex 12-line manifolds indicating additional coupling with the adjacent amide proton (broad singlet at 9.72 ppm). Scalar coupling between the HD and N–H protons is further confirmed by a cross peak in the COSY correlation. Assignment of the well-resolved experimental 1H NMR spectrum was confirmed by numerical simulation using the gNMR software (lower panel in Fig. 1).66 Iterative refinement of the entire spectrum by full lineshape analysis further enabled an accurate determination of the chemical shifts of L1, as well as those of the geminal and vicinal coupling constants, which are reported in detail in the Experimental section.
Potentiometric and UV spectroscopic studies of the protonation
The cumulative protonation constants (β01h) of the triprotic base L1 were determined separately by conventional potentiometry and UV-absorption spectrophotometry at 25.0(1) °C and 0.1 M ionic strength using KNO3 and KCl as supporting electrolytes, respectively. The stepwise equilibrium constants (K01h) related to equilibrium (1) are defined by eqn. (2) and were deduced from the refined β01h values by relation (3):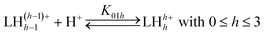 | (1) |
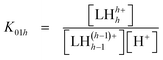 | (2) |
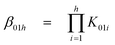 | (3) |
L1 was titrated from low to high p[H] (p[H] = −log[H3O+]) in the range 2.1–11.8 and vice versa. The neutralization curve of an acidic solution shows a first and sharp inflection point around p[H] 6 after the addition of one equivalent of base, followed by a buffering region that extends until the second equivalent is reached. Thus, L1 possesses at least two well-separated protonation constants, one close to 9 and one below 3. Nonlinear least squares refinement of four independent data sets with HYPERQUAD67 indicated that only two out of the three protonation constants can be measured by glass-electrode potentiometry. The average values are log K011 = 8.94(1) and log K012 = 2.32(9), the numbers in parenthesis corresponding to the standard deviations of four replicates expressed as the last significant digit.
Compared to 5,12-dioxocyclam [log K011 = 7.60(1) and log K012 = 7.44(3)],43 the cross-bridging of the secondary amines profoundly affects the proton affinity since the value of the first protonation constant of L1 is about two orders of magnitude higher. Assuming an in,in orientation of both electron doublets belonging to the bridgehead nitrogen atoms, as suggested by the spectroscopic properties and the DFT calculations, the basicity increase encountered by L1 indicates that the first added proton is encrypted and tightly bound inside the macrocyclic cavity. The more acidic nature of 2,6-dimethylpyridine (log K011 = 6.73; I = 0; T = 25 °C)68 also corroborates this assumption and clearly evidences a so-called cryptand effect.63,69
Replacement of the dioxocyclam fragment by a cyclam unit further increases the ligand’s basicity. Indeed, the first protonation constant of the corresponding macrobicyclic cage was found to be too high to be measured by glass-electrode potentiometry and thus the ligand was considered as a proton sponge.39 The electron-withdrawing properties of amide groups might explain the weaker hydrogen ion affinity of L1 compared to its cyclam-based analog. In both cases, however, the stabilization brought by the bridging group strongly suggests that all three donor atoms are involved in the proton binding, probably via intramolecular hydrogen bonds, although the precise localization from a microscopic point-of-view cannot be inferred at this stage.
The presence of a cross-linker also exerts a strong influence on the second protonation constant. Whereas both secondary amines of 5,12-dioxocyclam behave independently of each other, minimizing the repulsive Coulombic interactions between positive charges,43 the addition of a second proton to L1 is accompanied by a high energetic cost as evidenced by the very large gap between K011 and K012 (6.6 log units). The confinement inside the cavity provided by a rather rigid cage might explain this difference in binding strength, which is much larger than that expected from a purely statistical point of view.70 Thus, factors such as hydrogen bond breakup, conformational reorganization or charge accumulation, which increase the repulsion energy, might be invoked to explain the low value of K012. This difficulty to protonate a second site also suggests an inside location of the additional hydrogen atom.20,21,71,72
In order to get a more precise picture of the acid-base properties displayed by L1, the UV absorption changes taking place upon protonation of the bridgehead tertiary amines and/or pyridine core have been investigated in the 230–300 nm range. Since nitrate salts strongly absorb in this region, spectroscopically “silent” inert electrolyte and acid solutions such as KCl and HCl had to be used. The electronic spectrum of the free base tails out below 250 nm and shows the so-called pyridine B band (π → π*) centered at 263.5 nm with an extinction coefficient (εmax = 2930 M−1 cm−1) that is close to the value found for pyridine in water (εmax = 2750 M−1 cm−1 at λmax = 257 nm) but slightly lower to that of 2-methylpyridine (εmax = 3560 M−1 cm−1 at λmax = 262 nm).59 Acidification of the medium down to p[H] ∼ 7 (Fig. 2) induces severe perturbations of the spectra: the broad B band undergoes a slight bathochromic shift (λmax = 267 nm), accompanied by a strong hyperchromic effect resulting in the doubling of the apparent extinction coefficient. Concomitantly, the intensity of the high-energy absorption tail decreases as a consequence of a blue shift of the corresponding absorption maximum. Further addition of acid causes, between p[H] 4 and 2.5, a strong decrease in intensity of the major π → π* transition, which resolves into two peaks (λmax = 262 and 268 nm) and shows a low-energy shoulder near 257 nm (Fig. 2). Lowering the p[H] below 2.5 did not result in additional spectral changes.
![Spectrophotometric titration of L1 by HCl in water. [L1]tot = 1.95 mM; I = 0.1 (KCl); T = 25 °C; l = 0.1 cm. Spectra 1–36: p[H] = 2.52, 2.54, 2.56, 2.59, 2.62, 2.65, 2.68, 2.71, 2.75, 2.78, 2.83, 2.87, 2.93, 2.99, 3.05, 3.13, 3.23, 3.35, 3.51, 3.76, 4.38, 7.40, 8.15, 8.46, 8.68, 8.87, 9.04, 9.21, 9.40, 9.61, 9.85, 10.11, 10.33, 10.49, 10.62, 10.73.](/image/article/2005/NJ/b410260f/b410260f-f2.gif) | ||
| Fig. 2 Spectrophotometric titration of L1 by HCl in water. [L1]tot = 1.95 mM; I = 0.1 (KCl); T = 25 °C; l = 0.1 cm. Spectra 1–36: p[H] = 2.52, 2.54, 2.56, 2.59, 2.62, 2.65, 2.68, 2.71, 2.75, 2.78, 2.83, 2.87, 2.93, 2.99, 3.05, 3.13, 3.23, 3.35, 3.51, 3.76, 4.38, 7.40, 8.15, 8.46, 8.68, 8.87, 9.04, 9.21, 9.40, 9.61, 9.85, 10.11, 10.33, 10.49, 10.62, 10.73. | ||
Factor analysis based on singular-value decomposition of the entire multiwavelength data set, recorded between p[H] 2.52–10.73 and comprising 36 spectra with 448 evenly distributed wavelengths ranging between 230 and 300 nm (Fig. 2), indicated that three eigenvectors are sufficient to describe the observed features above the noise level. Subsequent nonlinear least-squares treatment of the experimental absorbance values with the program SPECFIT73–75 confirmed the hypothesized chemical model, comprising only the three absorbing species previously detected by potentiometry. The best fit converged to the final values log K011 = 8.98(2) and log K012 = 2.40(8) with an overall residual standard deviation of 1.25 × 10−4 absorbance unit. Identical results within experimental error were also obtained by HYPERQUAD when the potentiometric and spectrophotometric data were refined together. Moreover, both protonation constants and their associated errors are in good agreement with those determined by glass-electrode measurements alone [log K011 = 8.94(1) and log K012 = 2.32(9)].
Fig. 3(a) displays the calculated absorption spectra of L1 (λmax = 263.5 nm; εmax = 2700 M−1 cm−1), [H(L1)]+ (λmax = 267 nm; εmax = 5725 M−1 cm−1), and [H2(L1)]2+ (λmax = 262 nm; εmax = 3350 M−1 cm−1 and λmax = 268 nm; εmax = 2657 M−1 cm−1), which are in excellent agreement with the spectra recorded after dissolution of L1 in basic or strongly acidic aqueous media. It is well-known that the formation of a pyridinium cation induces a strong hyperchromic effect on the π → π* transition, while protonation of an amine engages the lone pair of the nitrogen atom and therefore moves the n(N) → π* charge-transfer (CT) transition to higher energy.59,76 Hence, the spectral changes observed upon addition of the first equivalent of hydrochloric acid are consistent with the protonation of the pyridine nitrogen atom. The close resemblance of the limiting high- and low-pH absorption spectra of 2,6-bis(bromomethyl)pyridine with those exhibited by L1 and [H(L1)]+ further support this assignment.
![(a) Calculated electronic absorption spectra of L1 and its protonated forms. (b) Comparison of the absorption spectra of [H2(L1)]2+ and of the monoprotonated form of N,N′-dimethyl(pyridine-2-yl)methylamine. I = 0.1 (KCl); T = 25 °C.](/image/article/2005/NJ/b410260f/b410260f-f3.gif) | ||
| Fig. 3 (a) Calculated electronic absorption spectra of L1 and its protonated forms. (b) Comparison of the absorption spectra of [H2(L1)]2+ and of the monoprotonated form of N,N′-dimethyl(pyridine-2-yl)methylamine. I = 0.1 (KCl); T = 25 °C. | ||
The remarkably strong hypochromic effect observed upon formation of the diprotonated species is associated to the disappearance of the absorption due to the pyridinum chromophore and suggests a redistribution of the added hydrogen ions between both types of binding sites. Thus, regioselective protonation is expected to occur at both bridgehead tertiary amines, together with a concomitant deprotonation of the pyridine nitrogen atom (Scheme 2).
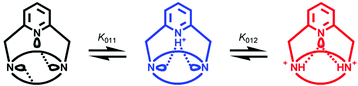 | ||
| Scheme 2 | ||
Additional stabilization of the [H2(L1)]2+ species might be provided by hydrogen bonding if the ligand takes up a conformation in which the lone pair of the pyridine nitrogen atom interacts with one or possibly both ammonium protons, hence reducing the Coulombic repulsion between positive charges in the center of the cavity. The rearrangement of the hydrogen bonding pattern induced by the second protonation is also consistent with the spectral similarity of [H2(L1)]2+ and the monoprotonated form of N,N′-dimethyl(pyridine-2-yl)methylamine [Fig. 3(b)]. The slight shift of ca. 5 nm between both spectra is ascribed to the different substitution level of the aromatic ring for each substance.
NMR spectroscopic studies of the protonation
Further details on the protonation processes at the microscopic level were derived from 1H and 13C NMR studies.77 The changes in proton and carbon chemical shifts experienced by nuclei remote from the protonation sites were recorded in D2O solutions for the free base and each deuterated species, [D(L1)]+ and [D2(L1)]2+, by adjusting the p[D] (p[D] = −log [D+]) to 11.8, 5.4, and 1.7, respectively (Table 1). Complete assignment of the individual resonances relies on the interpretation of two-dimensional 1H–1H COSY and 1H–13C HETCOR correlation charts. As in CDCl3, the time-averaged C2-symmetric structure is also preserved in D2O, since a single set of signals was observed for each spectrum. However, the signals arising from the HC and HD methylene groups could not be assigned unambiguously by a COSY experiment because of the rapid H/D exchange of the amide proton in D2O. However, a comparison of the 13C NMR spectra of L1 recorded at high p[D] in D2O and in CDCl3 showed only slight solvent-induced shifts, enabling the final identification of both CC and CD signals, and consequently those related to HC and HD with the help of the HETCOR chart.| 1H NMR | 13C NMR | |||||
|---|---|---|---|---|---|---|
| Signal | L1 | [D(L1)]+ | [D2(L1)]2+ | L1 | [D(L1)]+ | [D2(L1)]2+ |
| A | 2.24 | 2.29 | 2.91 | 33.8 | 35.3 | 31.2 |
| 2.33 | 2.55 | |||||
| B | 2.84 | 2.78 | 3.69 | 50.7 | 51.9 | 51.5 |
| 2.92 | 3.04 | 4.13 | ||||
| C | 2.45 | 2.64 | 3.40 | 54.1 | 53.2 | 59.9 |
| 2.84 | 2.96 | 3.91 | ||||
| D | 3.01 | 3.30 | 3.23 | 38.7 | 36.7 | 36.0 |
| 3.22 | 3.62 | 4.32 | ||||
| E | 3.87 | 4.14 | 4.83 | 58.3 | 53.7 | 57.6 |
| F | 159.6 | 153.1 | 148.6 | |||
| G | 7.13 | 7.79 | 7.62 | 120.7 | 124.8 | 124.2 |
| H | 7.66 | 8.41 | 8.13 | 138.7 | 147.0 | 142.1 |
| CO |
176.0 | 175.3 | 174.9 | |||
The NMR data confirm the protonation scheme deduced from the spectrophotometric UV titration. Formation of a pyridinium cation between p[D] 11.8 and 5.4 is evidenced by the following observations: (i) the aromatic protons HG (Δδ = 0.66 ppm) and HH (Δδ = 0.75 ppm) are strongly deshielded, (ii) in CDCl3 containing traces of acid, a broad lowfield-shifted resonance appears at 13.55 ppm in the 1H NMR spectrum, which is characteristic for a hydrogen-bonded pyridinium proton.78,79 As the [D(L1)]+ species is converted into the [D2(L1)]2+ one, relocation of the deuteron from the pyridinum to the amine sites causes an upfield shift of the aromatic signals (Δδ = 0.17 and 0.28 ppm for the HG doublet and HH triplet, respectively), while the methylenic HE protons from the handle resonate at much lower field (Δδ = 0.69 ppm). Moreover, the occurrence of an off-diagonal cross-peak that correlates the HB and HC signals in the COSY spectrum of [D2(L1)]2+ implies a four-bond coupling through the bridgehead nitrogen atoms, which further argues in favor of their deuterated state. Deuteration of both tertiary amines is also corroborated by the upfield shift of the resonances assigned to the carbon atoms CA and CD located in β positions with respect to the bridgehead nitrogen atom (Table 1).80,81
Fig. 4 shows a stack plot of pertinent 300 MHz 1H NMR spectra recorded over the p[D] range 1.7–11.8. The most striking features are the progressive disappearance of the free base resonances upon acidification and the simultaneous build-up of the signals due to the monodeuterated species with a concomitant line broadening observed as long as some free base is present (p[D] > 7). Conversion below p[D] 5 of [D(L1)]+ to [D2(L1)]2+ occurs in a similar way. Identical spectral changes could be observed when the titration was followed by 13C NMR spectroscopy. This unexpected result is a clear indication that the deuteration kinetics of L1 is slow on the NMR timescale, whereas potentiometric studies revealed a fast protonation process in the second time range.
![Stack plot of representative 1H NMR spectra of L1 recorded at 300 MHz in D2O as a function of p[D]; T = 25 °C.](/image/article/2005/NJ/b410260f/b410260f-f4.gif) | ||
| Fig. 4 Stack plot of representative 1H NMR spectra of L1 recorded at 300 MHz in D2O as a function of p[D]; T = 25 °C. | ||
Using the mass-balance eqn. (4) and the corresponding expressions of the successive equilibrium constants given by relation (2), the theoretical fractional concentration of each species in solution is described by eqns. (5) to (7), where KD01i represents the ith deuteration constant. Experimental values of the relative concentration for a given species were readily obtained by integration of the 1H NMR signals corresponding to the aromatic HG and HH protons (Fig. 5). Deuteration constants were adjusted by nonlinear minimization of the sum of the squared deviations between the theoretical and observed concentration ratios using a Microsoft Excel spreadsheet. The solid lines in Fig. 5 correspond to the calculated species distribution curves drawn with the optimized values of log KD011 = 9.36 and log KD012 = 3.10. Uncertainties are not reported since a single titration was performed. Estimates of the protonation constants in water, corrected for the deuterium effect according to the empirical correlation proposed by Delgado et al.82 (log KH011 = 8.66 and log KH012 = 2.66), are in fair agreement with those determined by potentiometric and spectrophotometric experiments, considering the error introduced in the absence of supporting electrolyte by the evolving ionic strength during the NMR titration.
| [L1]tot = [L1](1 + KD011[D+] + KD011KD012[D+]2) | (4) |
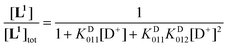 | (5) |
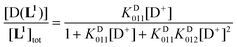 | (6) |
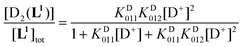 | (7) |
There are only a very few chemical systems that undergo slow protonation or deprotonation, the dynamics of such reactions being diffusion limited in the majority of cases. Typical examples are found among proton sponges.25 For the compounds belonging to the prototypical class of N-alkylated 1,8-diaminonaphthalene, the slow proton-transfer rates might be ascribed to the strong intramolecular N–H⋯N hydrogen bonding interaction.83–85 Tricyclic tetraamines, also called adamanzanes, exhibit analogous properties and were found to be very inert with respect to the proton entrapped in the cavity.34–36 Of particular relevance with respect to the present work are the macrobicyclic diazacycloalkanes20–22,26 and cryptands,63,71,86 in which the proton is tightly concealed inside an often rigid cavity. Bridged bicyclic structures of this type contain medium to large rings (at least 7-membered) in which the lone pairs on bridgehead nitrogen atoms can point either inward or outward, thus allowing inside or outside protonation.87 The slow insertion rate of a proton into the cage of the extraordinarily strong base 1,6-diazabicyclo[4.4.4]tetradecane (pKa ∼ 25) is believed to be the consequence of an indirect process involving a 1,2- or 1,5-transfer of an α-methylenic hydrogen atom.23,24 For larger representatives, internal protonation was shown to occur according to a sequence of concerted prototropic reactions and nitrogen atom inversion starting from the rapidly formed exo-protonated state.20–22 Similarly, the endo-endo conformation is the thermodynamically most stable form for the most constrained [1.1.1] cryptand and its protonated species. The slow protonation rates result from the high activation energy barrier (ca. 110 kJ mol−1) for the incoming proton, whereas nitrogen inversion followed by outside protonation is a much faster process.71 Conversion of the exo to the endo orientation of the ammonium group constitutes the rate-limiting step and most probably occurs via a nitrogen inversion pathway, which implies a deprotonation-inversion-reprotonation sequence. Because the associated rates in water are slow at room temperature, the reaction was followed as a function of time by integrating the respective signals assigned to both states of the molecule.
![Relative concentrations of L1 (black dots), [D(L1)]+ (blue circles), and [D2(L1)]2+ (red squares) determined by integration of the 1H NMR spectra. The nonlinear least squares distribution curves calculated according to eqns. (5)–(7) are represented as solid lines. Solvent: D2O; T = 25 °C.](/image/article/2005/NJ/b410260f/b410260f-f5.gif) | ||
| Fig. 5 Relative concentrations of L1 (black dots), [D(L1)]+ (blue circles), and [D2(L1)]2+ (red squares) determined by integration of the 1H NMR spectra. The nonlinear least squares distribution curves calculated according to eqns. (5)–(7) are represented as solid lines. Solvent: D2O; T = 25 °C. | ||
This is in contrast with the present investigation as no time dependence of the recorded spectra could be detected over a period extending up to one month. Internal protonation of L1 occurs obviously much faster since any exo conformer could never be observed by NMR spectroscopy. Assistance of the pyridine moiety might be invoked to explain the accelerated and direct entry of the protons in the cavity of L1. Assuming that the internal hydrogen-bonding network, which stabilizes both [H(L1)]+ and [H2(L1)]2+ species, is preserved in water, the pyridyl nitrogen site can act as a proton relay and slow reorganization on the NMR timescale of the three-center hydrogen bond takes place upon proton binding.
Molecular modelling
Although single crystals of L1 could be grown from aqueous or hydroalcoholic solutions, their low quality did not allow to collect enough X-ray diffraction data, in particular at high scattering angles. Since only unacceptably low-resolution structures could be obtained after several attempts, molecular modelling of L1 and its protonated forms was undertaken in an effort to ascertain the proton-binding scheme deduced from potentiometric and spectroscopic solution studies, and to account for the proton redistribution encountered upon addition of the second equivalent of acid. The equilibrium geometries were fully optimized using molecular orbital methods implemented in GAMESS (version 2002 on Apple Xserve™).88 Low-energy conformers generated by semi-empirical methods (AM1) were further optimized using density functional theory (DFT) with a 6-31G* basis set (DFT/B3LYP/6-31G*).As expected, the lowest energy structure computed for L1 [Fig. 6(a)] shows an inward orientation of both tertiary amine lone pairs with a transannular N⋯N separation of 4.42 Å, which is in good agreement with the distance measured on the crystal structure of L22 (4.64 Å).60 The slightly bent dioxocyclam ring has both amidic hydrogen atoms directed towards the center of the cavity, one interacting with the nitrogen atom belonging to the tilted pyridine ring (N⋯Npy = 2.96 Å, N–H⋯Npy = 157°) and the other with the neighboring amine located 2.26 Å away (N⋯N = 3.05 Å, N–H⋯N = 134°). Hence, the molecular modelling calculations carried out on L1 reproduce all the key structural features displayed by closely related pyridine-capped C-substituted dioxocyclams.55
![DFT-optimized molecular structures of (a) L1 and (b) [H(L1)]+. Possible hydrogen bonds are visualized with dashes. For the sake of clarity, only the pertinent hydrogen atoms are represented.](/image/article/2005/NJ/b410260f/b410260f-f6.gif) | ||
| Fig. 6 DFT-optimized molecular structures of (a) L1 and (b) [H(L1)]+. Possible hydrogen bonds are visualized with dashes. For the sake of clarity, only the pertinent hydrogen atoms are represented. | ||
The reliability of the minimization procedure was checked by examining the planarity of both amide groups, which is conveniently expressed by the OC–N–H torsion angles, which are 166 and 169°. Further validation was provided by computing estimates of the NMR 3JαNH coupling constants corresponding to the H–N–C–H dihedral angles (ϕ) using a revised Karplus equation.89 For the axial methylenic proton adjacent to the amide group (ϕ = −76 and −83.3°), 3JαNH averages 6.7(5) Hz, which is in fair agreement with the value derived from the full lineshape analysis of the C2-symmetric time-averaged 1H NMR spectrum recorded at room temperature in CDCl3. The magnitude of the vicinal coupling constant with the equatorial hydrogen atom (ϕ = 167.9 and 161.2°) is expected to be smaller [3JαNH = 2.5(5) Hz] as experimentally observed (3JαNH = 3.9 Hz).
It is therefore clear that the DFT calculations are fully supported by a vast collection of experimental and spectroscopic data in this particular case. This reliable tool can now be used efficiently to describe the effect of protonation on the geometry of L1. Thus, from a structural point of view, formation of the monoprotonated species breaks the intramolecular hydrogen-bond network and results in a ca. 90° rotation of both almost planar amide groups (OC–N–H =
−176 and −179°), affording a cone-shaped molecular cavity delimited by both carbonyl groups oriented on the same side as the aromatic ring [Fig. 6(b)]. In turn, protonation has only a limited influence on the distance between the bridgehead nitrogen atoms, which shrinks by only 0.07 Å, but enforces a less tilted orientation of the capping pyridine moiety. The pyridinium proton points in-between both amines in an almost perpendicular fashion with respect to the dioxocyclam skeleton, forming a three-center hydrogen bond with them (NpyH⋯N = 2.22 and 2.32 Å, sum of NHN angles = 359.6°) as already found in the free-base structure of L22.60 Furthermore, molecular modelling of the diprotonated species confirmed that the in,in-diammonium salt was the most stable conformer (see ESI).
Conclusion
5,12-Dioxocyclam was used as a precursor for the preparation of a hemispherical macrobicyclic ligand obtained by cross-bridging the 1 and 8 positions with a 2,6-pyridyl group. Structural characterization of the free base and its mono- and diprotonated forms was obtained from NMR, UV and IR spectroscopic studies, as well as from theoretical computations carried out with DFT. These investigations revealed the predominant role played by intramolecular hydrogen bonds in determining both the conformation and the proton affinities of the receptor. The first proton encrypted inside the cavity is tightly bound to the pyridine ring and stabilized through a three-center interaction with both bridgehead nitrogen donors. Redistribution occurs after inclusion of the second proton, as both are localized on the tertiary amines and are involved in hydrogen bonds with the free pyridyl nitrogen atom. Slow kinetic processes on the NMR timescale characterize proton uptakes and transfers. In order to unravel the influence of the cross-bridging unit, further studies involving analogous ligands that lack a coordinating pyridine group are underway and will be reported in due course.Experimental
Preparation of compounds
Unless otherwise noted, all chemicals and starting materials were obtained commercially and used without further purification. 5,12-Dioxocyclam (1) was synthesized according to literature procedures.42,43 Aluminum oxide 90 standardized (Merck) was used for column chromatography. Organic solvents and mineral acids were of reagent grade and were used as supplied. Water was deionized and further purified by passage through an Elgastat UHQII (Elga) ion-exchange cartridge system (resistivity > 18 MΩ cm).O), 1574 (νCC), 1526 (δCNH), 1449 (νCC), 1424 (νCN Ar). Anal. calcd for C17H25N5O2: C, 61.61; H, 7.60; N, 21.13; found: C, 61.41; H, 7.71; N, 21.10.
Spectroscopic measurements
1H and 13C NMR spectra were recorded on a Bruker Avance 300 or Avance DRX 500 superconducting Fourier transform spectrometer operating at 300.13 and 500.13 MHz, respectively, at the “Centre de Spectroscopie Moléculaire de l′Université de Bourgogne”. All chemical shifts were referenced either to the solvent peak or to CH3CN added as an internal standard to D2O solutions (δH = 2.06 ppm, δC = 1.47 ppm).90 MALDI-TOF mass spectra were obtained on a Bruker PROFLEX III spectrometer using dithranol as matrix. Electron impact (EI) mass spectra were recorded on a Kratos Concept 32 spectrometer. Microanalyses were performed on a Fisons EA 1108 CHNS instrument. Infrared spectra were measured on a Bruker IFS 66v Fourier transform spectrophotometer either as KBr pellets from 4000 to 400 cm−1 or in CDCl3 solution from 4000 to 1000 cm−1 using a variable path length cell from Perkin–Elmer equipped with IRTRAN-2 windows.Solution thermodynamics
All 0.1 M titrant solutions were prepared from Merck concentrates (Titrisol®) diluted with doubly deionized, argon-purged boiled water and stored under an atmosphere of purified argon using Ascarite II (Acros, 20–30 mesh) scrubbers in order to prevent absorption of carbon dioxide. The carbonate-free KOH solution was standardized by titrating against oven-dried (120 °C for 2 h) potassium hydrogenphthalate (Aldrich, 99.99%), while 0.1 M HNO3 and HCl solutions were titrated against oven-dried (120 °C for 2 h) TRIS buffer (Aldrich, 99.9%) using a combined Ag/AgCl glass semimicroelectrode (Mettler-Toledo) filled with a solution saturated with KCl and AgCl. Equivalent points were calculated by the second-derivative method. The concentration of the standardized solutions corresponded to the average of at least five replicates and was known with a relative precision of less than 0.15%.Titrations were performed using a T110 (Schott) piston burette equipped with a calibrated TA10 interchangeable unit of 10 mL and a TR250 (Schott) data acquisition unit, both instruments being controlled by TR600 software (release 5.2) running on an IBM-compatible PC computer. The volume of the burette-delivered aliquots was corrected according to a linear calibration function obtained by weighing known quantities of water and by taking into account the buoyancy effect.91 Magnetically stirred solutions were maintained under an argon atmosphere and at constant temperature [25.0(1) °C] by using a water-jacketed titration vessel fitted to a Lauda RE 106 water circulator.
Potentiometric titrations
Ligand protonation constants were determined by glass-electrode potentiometric measurements at constant ionic strength (I = 0.1, KNO3) by using a XG100 (Radiometer-Tacussel) glass-bulb electrode together with a XR110 (Radiometer-Tacussel) KCl-saturated calomel electrode. The reference electrode was separated from the bulk of the solution by a sintered glass bridge filled with a 0.1 M KNO3 solution. Before each titration, the electrochemical cell was calibrated to read hydronium ion concentrations (p[H] = −log[H3O+]) by titrating 4.000 mL of standardized HNO3 diluted in 25 mL of 0.100 M KNO3 with 9.010 mL of standardized KOH in 0.12 mL increments. The titration data comprised about 68 points collected between p[H] 1.86 to 2.65 and 10.97 to 11.90. The sum of the unweighed square residuals on the observed potential readings (Emes) were minimized according to the modified Nernst equation (Emes = E0 + Slog[H3O+] + Ja[H3O+] + JbKw[H3O+]−1) by using the Solver routine implemented in Microsoft Excel. The minimization procedure allowed the simultaneous refinement of the standard cell potential (E0), the Nernst slope (S), the correction terms accounting for the changes in liquid junction potential in strongly acid (Ja) or alkaline (Jb) media,92 together with the base concentration factor (γ). The ionic product of water was maintained constant at the literature value (pKw = 13.78 in 0.1 M KNO3 at 25 °C).68,93 Calibration data were rejected when the standard deviation of the residuals exceeded 0.1 mV, indicating a carbonate contamination of the base usually higher than 0.5%.Protonation constants were measured by titrating ca. 0.1 mmol of ligand dissolved in 25 mL of supporting electrolyte (0.1 M KNO3). The electrochemical cell was allowed to equilibrate for at least 30 s after each addition of a 0.03 mL increment of titrating solution. The collected potential readings were converted into p[H] values, spanning the range 2.1–11.8, with the help of an Microsoft Excel spreadsheet by iterative solving of the rearranged Nernst equation: p[H] = (E0 − Emes + Ja[H3O+] + JbKw[H3O+]−1)/S. The titration data were analyzed by the weighted nonlinear least-squares procedure implemented in the HYPERQUAD 2000 program.67 The selected weighing scheme, derived from the error propagation law,94 seeks to reduce the weight of the less accurate measurements (i.e., points located in the steep region of a titration curve). On the basis of the instrument’s calibration, the uncertainties associated with the experimental p[H] values (σpH) and the volumes delivered by the piston burette (σv) were estimated as 0.003 pH unit (or ∼0.1 mV) and 0.005 mL, respectively. In the final refinement step, the total amounts of titrated ligand and initially added base or acid were also allowed to vary. The fit between calculated and experimental emf data was evaluated through the squared sum of residuals (1.0 < σ < 1.8) and an approximately normal distribution of the residues.94 The thermodynamic reversibility was checked by cycling from low to high p[H] and vice versa. Data from forward and backward titrations afforded statistically identical values of the adjusted thermodynamic parameters, pointing out the chemical stability of the ligand over the explored pH range. For each data set, the standard deviations of the log K01h values were derived from the full variance/covariance matrix of the refined global constants (log β01h).95 The extradiagonal terms, which correspond to the correlation coefficients between the stepwise protonation constants,95 were close to 0.5. The final values are reported as the arithmetic mean of four independent measurements together with their standard deviations, which were systematically higher compared to those derived for a single experiment from the full variance/covariance matrix.
Spectrophotometric titrations
Potentiometric measurements coupled with spectrophotometric detection were carried out using the same titration cell and electrode calibration procedure as described above. Since nitrate salts strongly absorb light below 320 nm, 0.1 M KCl (Merck, >99%) instead of KNO3 solutions were used as the supporting electrolyte (50 mL). In order to facilitate the dissolution of the titrated ligand ([L1]tot ∼10−3 M), known amounts of a standard 0.1 M HCl solution were added until the p[H] reached a value of ∼2. The standardized 0.1 M KOH solution was then delivered manually with the help of a Gilmont micropipette (2 µL resolution). The glass-electrode potential of the electrochemical cell was measured with a PHM240 (Radiometer-Tacussel) pH-meter, the UV absorption spectra being recorded in situ with a Varian Cary 50 Probe spectrophotometer equipped with an immersion cell (Hellma, reference 661.202) of 0.1 cm path length made of quartz (SUPRASIL® 300). Both 2 m long built-in light guides of the dip probe were directly connected through SMA plugs to the coupling device, fitted inside the sample compartment of the spectrophotometer. Prior to the titration, the reference spectrum of the electrolyte solution was acquired in the 230–300 nm range. The standard deviation of the measured absorbance for the baseline was constant over the entire wavelength region and did not exceed 0.001 absorbance unit. After the addition of each base increment, enough time was allowed to reach equilibrium. The potential drift criterion was set at dE/dt < 0.1 mV min−1. Absorption data collection was repeated with 2 min delays between two consecutive measurements until superimposable spectra were obtained with optical densities not exceeding 1.1 units. At each p[H] value, the pH-meter readings were stable in less than 2 min and no more than two spectral recordings were required.The multiwavelength data sets were decomposed into their principal components by factor analysis with the SPECFIT program.73–75 The protonation constants and the extinction coefficients corresponding to each species were subsequently adjusted to the reduced data sets by the Marquardt nonlinear least-squares algorithm. Furthermore, the data from each titration were also processed by the program HYPERQUAD 2000, which allows the simultaneous treatment of potentiometric and spectrophotometric data.67 While SPECFIT uses the measured p[H] values to calculate directly the free H3O+ concentration, the mass-balance equations for all components, including the hydronium ion, are solved at each titration point by the program HYPERQUAD. Since the precision of spectrophotometric and potentiometric measurements is different, it is of prime importance to define a proper weighing scheme relying on the error propagation rule, which gives to each type of observation an approximately equal contribution to the residual sum of squares. Realistic parameter estimates of the quadratic absorbance error function (σA = a + bA + cA2) were evaluated by computing, for each recorded wavelength of 50 replicates of the ligand spectrum, the average absorbance and its associated standard deviation.67,94
NMR titrations
In a jacketed cell maintained at 25.0(1) °C and held under an argon atmosphere, a ca. 15 × 10−3 M solution of L1 dissolved in D2O (SDS, D > 99.90%) was titrated by addition of small amounts of a ca. 3.7% diluted DCl solution (Aldrich, 37% by wt, D > 99.5%) in D2O with a Gilmont micropipette. After each titrant addition, a ca. 0.5 mL aliquot was taken from the titration vessel. Apparent p[H] values, noted p[H]*, were measured with a combined Ag/AgCl glass semimicroelectrode (Mettler–Toledo) filled with a saturated KCl/AgCl solution in water and fitted to a PHM240 (Radiometer-Tacussel) pH-meter. The electrode was calibrated in water in terms of hydronium ion concentration according to the procedure described above. The corrected p[D] values were calculated according to the formula p[D] = p[H]* + 0.4.96 Deuteration constants were determined by nonlinear least-squares refinement using the Solver routine available in Excel (Microsoft) and corrected for the isotopic effect according to the correlation found by Delgado et al.: log KD = 0.32 + 1.044![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log KH.82
log KH.82
Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique (CNRS) and the Conseil Régional de Bourgogne. L. F. thanks the Ministère de l’Education Nationale, de la Recherche et de la Technologie for a Ph.D. scholarship.References
- M. Meyer, L. Frémond, E. Espinosa, R. Guilard, Z. Ou and K. M. Kadish, Inorg. Chem., 2004, 43, 5572–5587 CrossRef CAS.
- B. Dietrich, P. Viout and J. M. Lehn, Aspects de la chimie des composés macrocycliques, InterEditions/Editions du CNRS, Paris, 1991 Search PubMed.
- J. M. Lehn, La Chimie Supramoléculaire. Concepts et Perspectives, De Boeck Université, Paris, 1997 Search PubMed.
- J. Nelson, V. McKee and G. Morgan, Prog. Inorg. Chem., 1998, 47, 167–316 CAS.
- J. D. Chartres, L. F. Lindoy and G. V. Meehan, Coord. Chem. Rev., 2001, 216–217, 249–286 CrossRef CAS.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Poggi and A. Taglietti, Coord. Chem. Rev., 2001, 219–221, 821–837 CrossRef CAS.
- H. Plenio and C. Aberle, Chem. Commun., 1998, 2697–2698 RSC.
- H. Plenio, C. Aberle, Y. A. Shihadeh, J. M. Lloris, R. Martinez-Manez, T. Pardo and J. Soto, Chem.-Eur. J., 2001, 7, 2848–2861 CrossRef CAS.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef.
- L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS.
- L. Fabbrizzi, M. Licchelli, G. Rabaioli and A. Taglietti, Coord. Chem. Rev., 2000, 205, 85–108 CrossRef CAS.
- V. Alexander, Chem. Rev., 1995, 95, 273–342 CrossRef CAS.
- P. Virta and H. Lönnberg, J. Org. Chem., 2003, 68, 8534–8538 CrossRef CAS.
- X. Liang and P. J. Sadler, Chem. Soc. Rev., 2004, 33, 246–266 RSC.
- T. J. Hubin, J. M. McCormick, S. R. Collinson, M. Buchalova, C. M. Perkins, N. W. Alcock, P. K. Kahol, A. Raghunathan and D. H. Busch, J. Am. Chem. Soc., 2000, 122, 2512–2522 CrossRef CAS.
- T. J. Hubin, Coord. Chem. Rev., 2003, 241, 27–46 CrossRef CAS.
- B. Dietrich, B. Dilworth, J. M. Lehn, J. P. Souchez, M. Cesario, J. Guilhem and C. Pascard, Helv. Chim. Acta, 1996, 79, 569–587 CrossRef CAS.
- M. Formica, V. Fusi, M. Micheloni, R. Pontellini and P. Romani, Coord. Chem. Rev., 1999, 184, 347–363 CrossRef CAS.
- H. E. Simmons and C. H. Park, J. Am. Chem. Soc., 1968, 90, 2428–2429 CrossRef CAS.
- C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2429–2431 CrossRef CAS.
- C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS.
- R. W. Alder and R. B. Sessions, J. Am. Chem. Soc., 1979, 101, 3651–3652 CrossRef CAS.
- R. W. Alder, A. Casson and R. B. Sessions, J. Am. Chem. Soc., 1979, 101, 3652–3653 CrossRef CAS.
- R. W. Alder, Chem. Rev., 1989, 89, 1215–1223 CrossRef CAS.
- R. W. Alder, Tetrahedron, 1990, 46, 683–713 CrossRef CAS.
- R. W. Alder, E. Heilbronner, E. Honegger, A. B. McEwen, R. E. Moss, E. Olefirowicz, P. A. Petillo, R. B. Sessions, G. R. Weisman, J. M. White and Z. Z. Yang, J. Am. Chem. Soc., 1993, 115, 6580–6591 CrossRef CAS.
- R. Kowallick, M. Neuburger, M. Zehnder and T. A. Kaden, Helv. Chim. Acta, 1997, 80, 948–959 CAS.
- P. V. Bernhardt and P. C. Sharpe, Inorg. Chem., 2000, 39, 2020–2025 CrossRef CAS.
- M. Micheloni, M. Formica, V. Fusi, P. Romani, R. Pontellini, P. Dapporto, P. Paoli, P. Rossi and B. Valtancoli, Eur. J. Inorg. Chem., 2000, 51–57 CrossRef CAS.
- C. Bazzicalupi, A. Bellusci, A. Bencini, E. Berni, A. Bianchi, A. Ciattini, C. Giorgi and B. Valtancoli, J. Chem. Soc., Dalton Trans., 2002, 2151–2157 RSC.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K.-C. Lam, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 10561–10572 CrossRef CAS.
- A. Ingham, M. Rodopoulos, K. Coulter, T. Rodopoulos, S. Subramanian and A. McAuley, Coord. Chem. Rev., 2002, 233–234, 255–271 CrossRef CAS.
- J. Springborg, J. Chem. Soc., Dalton Trans., 2003, 1653–1665 RSC.
- J. Springborg, B. Nielsen, C. E. Olsen and I. Sotofte, J. Chem. Soc., Perkin Trans. 2, 1999, 2701–2706 RSC.
- J. Springborg, B. Nielsen, C. E. Olsen and I. Sotofte, J. Am. Chem. Soc., 2002, 124, 5084–5090 CrossRef CAS.
- M. Rodopoulos, T. Rodopoulos, J. N. Bridson, L. I. Elding, S. J. Rettig and A. McAuley, Inorg. Chem., 2001, 40, 2737–2742 CrossRef CAS.
- F. Denat, S. Lacour, S. Brandès and R. Guilard, Tetrahedron Lett., 1997, 38, 4417–4420 CrossRef CAS.
- S. Brandès, F. Denat, S. Lacour, F. Rabiet, F. Barbette, P. Pullumbi and R. Guilard, Eur. J. Org. Chem., 1998, 2349–2360 CrossRef CAS.
- F. Denat, S. Brandès and R. Guilard, Synlett, 2000, 561–575 CAS.
- C. Hu, T. D. Nguyen, P. S. Wagenknecht and L. C. Nathan, Inorg. Chem., 2003, 42, 742–749 CrossRef CAS.
-
D. A. Tomalia and L. R. Wilson, US Pat. 4517122, 1985 Search PubMed.
- L. Frémond, E. Espinosa, M. Meyer, F. Denat, R. Guilard, V. Huch and M. Veith, New J. Chem., 2000, 24, 959–966 RSC.
- C. Betschart and L. S. Hegedus, J. Am. Chem. Soc., 1992, 114, 5010–5017 CrossRef CAS.
- L. S. Hegedus and W. H. Moser, J. Org. Chem., 1994, 59, 7779–7784 CrossRef CAS.
- Y. Hsiao and L. S. Hegedus, J. Org. Chem., 1997, 62, 3586–3591 CrossRef CAS.
- L. S. Hegedus, M. M. Greenberg, J. J. Wendling and J. P. Bullock, J. Org. Chem., 2003, 68, 4179–4188 CrossRef CAS.
- T. J. Hubin, N. Tyryshkin, N. W. Alcock and D. H. Busch, Acta Crystallogr., Sect. C, 1999, 55, 1888–1889 CrossRef.
- S. Dumas, E. Lastra and L. S. Hegedus, J. Am. Chem. Soc., 1995, 117, 3368–3379 CrossRef CAS.
- E. Kuester and L. S. Hegedus, Organometallics, 1999, 18, 5318–5321 CrossRef CAS.
- K. Puntener, M. D. Hellman, E. Kuester and L. S. Hegedus, J. Org. Chem., 2000, 65, 8301–8306 CrossRef CAS.
- T. Wynn and L. S. Hegedus, J. Am. Chem. Soc., 2000, 122, 5034–5042 CrossRef CAS.
- T. A. Brugel and L. S. Hegedus, J. Org. Chem., 2003, 68, 8409–8415 CrossRef CAS.
- L. S. Hegedus, M. J. Sundermann and P. K. Dorhout, Inorg. Chem., 2003, 42, 4346–4354 CrossRef CAS.
- M. Achmatowicz, L. S. Hegedus and S. David, J. Org. Chem., 2003, 68, 7661–7666 CrossRef CAS.
- G. Trippé, F. Le Derf, M. Mazari, N. Mercier, D. Guilet, P. Richomme, A. Gorgues, J. Becher and M. Sallé, C. R. Chimie, 2003, 6, 573–580 Search PubMed.
- E. Kimura, J. Coord. Chem., 1986, 15, 1–28 CrossRef CAS.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, John Wiley and Sons, New York, 5th edn., 1991 Search PubMed.
- L. Frémond, Ph.D Thesis, Université de Bourgogne, Dijon, France, 2002 Search PubMed.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Chapman and Hall, London, 3rd edn., 1975, vol. 1 Search PubMed.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules. Advances in Infrared Group Frequencies, Chapman and Hall, London, 2nd edn., 1980, vol. 2 Search PubMed.
- J. Cheney, J. P. Kintzinger and J. M. Lehn, Nouv. J. Chim., 1978, 2, 411–418 Search PubMed.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1982, 21, 512–523 CrossRef.
- C. N. Kirsten and T. H. Schrader, J. Am. Chem. Soc., 1997, 119, 12061–12068 CrossRef CAS.
- P. H. M. Budzelaar, gNMR, version 4.1, Cherwell Scientific Publishing, Oxford, 1995 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS.
- R. M. Smith, A. E. Martell and R. J. Motekaitis, NIST Critically Selected Stability Constants of Metal Complexes Database, version 6.0, NIST Standard Reference Data No. 46, NIST, Gaithersburg, MD, 2001 Search PubMed.
- J. M. Lehn and J. P. Sauvage, J. Am. Chem. Soc., 1975, 97, 6701–6707.
- B. Perlmutter-Hayman, Acc. Chem. Res., 1986, 19, 90–96 CrossRef.
- P. B. Smith, J. L. Dye, J. Cheney and J. M. Lehn, J. Am. Chem. Soc., 1981, 103, 6044–6048 CrossRef CAS.
- P. G. Potvin and M. H. Wong, Can. J. Chem., 1988, 66, 2914 CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 95–101 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 251–264 CrossRef.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 1133–1139 CrossRef CAS.
- C. Saudan, V. Balzani, P. Ceroni, M. Gorka, M. Maestri, V. Vicinelli and F. Vögtle, Tetrahedron, 2003, 59, 3845–3852 CrossRef CAS.
- J. L. Sudmeir and C. N. Reilley, Anal. Chem., 1964, 36, 1707–1712 CrossRef CAS.
- C. Dolain, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2003, 42, 2738–2740 CrossRef CAS.
- E. Kolomiets, V. Berl, I. Odriozola, A. M. Stadler, N. Kyritsakas and J. M. Lehn, Chem. Commun., 2003, 2868–2869 RSC.
- J. E. Sarneski, H. L. Surprenant, F. K. Molen and C. N. Reilley, Anal. Chem., 1975, 47, 2116–2124 CrossRef.
- H. L. Surprenant, J. E. Sarneski, R. R. Key, J. T. Byrd and C. N. Reilley, J. Magn. Reson., 1980, 40, 231–243 CAS.
- R. Delgado, J. J. R. Frausto Da Silva, M. T. S. Amorim, M. F. Cabral, S. Chaves and J. Costa, Anal. Chim. Acta, 1991, 245, 271–282 CrossRef CAS.
- R. W. Alder, P. S. Bowman, W. R. S. Steele and D. R. Winterman, J. Chem. Soc., Chem. Commun., 1968, 723–724 RSC.
- F. Hibbert, J. Chem. Soc., Perkin Trans. 2, 1974, 1862–1866 RSC.
- P. Hodgson, G. C. Lloyd-Jones, M. Murray, T. M. Peakman and R. L. Woodward, Chem.-Eur. J., 2000, 6, 4451–4460 CrossRef CAS.
- B. G. Cox, N. V. Truong and H. Schneider, J. Chem. Soc., Perkin Trans. 2, 1983, 515–521 RSC.
- R. W. Alder and S. P. East, Chem. Rev., 1996, 96, 2097–2111 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, S. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- G. W. Vuister and A. Bax, J. Am. Chem. Soc., 1993, 115, 7772–7777 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- R. Battino and A. G. Williamson, J. Chem. Educ., 1984, 61, 51–52.
- A. Avdeef and J. J. Bucher, Anal. Chem., 1988, 50, 2137–2142.
- I. Kron, S. L. Marshall, P. M. May, G. Hefter and E. Königsberger, Monatsh. Chem., 1995, 126, 819–837 CAS.
- P. Gans, Data Fitting in the Chemical Sciences, Wiley, Chichester, U.K., 1992 Search PubMed.
- K. N. Raymond and J. M. McCormick, J. Coord. Chem., 1998, 46, 51–57 CAS.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
Footnotes |
| † For the previous paper in this series see ref. 1. |
| ‡ Electronic supplementary information (ESI) available: 1H–1H NMR COSY and 1H–13C NMR HETCOR charts for L1 (Figs. S1–S3); DFT-calculated energies of L1, [H(L1)]+, and [H2(L1)]2+ in different geometries (Tables S1–S3); Cartesian x, y, z coordinates of the DFT-minimized lowest-energy conformers of L1, [H(L1)]+, and [H2(L1)]2+ structures (Tables S4–S6). See http://www.rsc.org/suppdata/nj/b4/b410260f/ |
| § This paper is dedicated to Dr Jean-Pierre Sauvage on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |